
Abstract
A 51-year-old man with Behçet's disease complained of fever, dry cough and dyspnea during exertion. Chest CT showed ground glass opacities with interstitial septal thickening in both lungs. Bronchoalveolar lavage (BAL) revealed amorphous and lipoproteinaceous material that was periodic acid-Schiff (PAS) stain positive. Transbronchial biopsy specimen demonstrated PAS positive alveolar eosinophilic material consistent with pulmonary alveolar proteinosis. Serum anti-granulocyte-macrophage colony stimulating factor (GM-CSF) antibody was negative. Recent studies have reported anti-GMCSF not present in the the serum of patients with secondary pulmonary alveolar proteinosis (PAP) but they have not reported so in patients with idiopathic PAP. We report a case of alveolar proteinosis in the setting of Behçet's disease with spontaneous remission.
Riassunto
Un paziente maschio di 51 anni con malattia di Behçet riferiva febbre, tosse secca e dispnea da sforzo. Alla TAC torace si evidenziavano opacità a vetro smeriglio con inspessimento dei setti interstiziali in entrambi i polmoni. Il lavaggio broncoalveolare ha recuperato materiale amorfo, lipoproteinaceo, positivo alla colorazione con acido periodico-Schiff (PAS). La biopsia transbronchiale dimostrava materiale alveolare eosinofilo PAS positivo, compatibile con una proteinosi endoalveolare polmonare. Il tasso serico di anticorpi anti fattore stimolante la colonia dei granulociti e macrofagi umani (GMCSF) era negativo. Studi recenti hanno riportato l’assenza di anti-GM-CSF nei pazienti con proteinosi endoalveolare secondaria, a differenza di quanto avviene nelle forme idiopatiche. Viene qui descritto un caso di proteinosi alveolare in corso di malattia di Behçet a remissione spontanea.
Similar content being viewed by others


Introduction
Pulmonary alveolar proteinosis (PAP) is a rare disorder characterized by abundant accumulation of surfactant within the alveoli [1]. The disease presents clinically in one of three forms: congenital, primary acquired (idiopathic), or secondary [2, 3]. The acquired or idiopathic form accounts for more than 90% of all cases and is believed to be caused by autoantibodies targeting granulocyte-macrophage colony stimulating factor (GM-CSF) [4, 5]. Secondary PAP develops in association with hematological malignancies, chronic pulmonary infections, inhalation exposure, and HIV infection [2, 3]. Recent studies have reported the detection of neutralizing autoantibodies against GM-CSF in the serum as well as BAL fluid of patients with idiopathic PAP which suggests that GM-CSF may be a serologic marker for idiopathic PAP [4, 5]. Spontaneous remission may occur in up to one third of the patients [6]. In this paper, we report a case of secondary alveolar proteinosis associated with Behçet's disease, which was diagnosed by BAL findings and lack of circulating anti-GM-CSF antibody, with spontanoeus resolution.
Case presentation
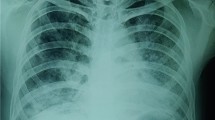
A 51-year-old man with stable Behçet's disease reported dry cough and dyspnea upon exertion which had developed three weeks earlier. Past medical history was unremarkable except for Behçet's disease diagnosed at the age of 45. There was no history of occupational or exogenous exposure. The patient commenced on topical steroid and cyclosporine treatment for one month that was completed two weeks before admission for uveitis. Physical examination revealed fever (37.4°C), papular skin lesions and fine rales at the base of both lungs. Laboratory data on admission were as follows: hemoglobin, 13.1 g/dl; hematocrit, 40.2%; leukocytes, 6400/mm3; platelets, 223,000/mm3; erythrocyte sedimentation rate, 42 mm/h; and C-reactive protein, 12.4 mg/dl. Serum biochemistry and urinanalysis were normal. Chest x-ray showed ground glass opacification and interstitial pattern particularly in the right lung. Arterial blood gases at room air were as follows: pH: 7.42, pO2: 74 mm Hg, and pCO2: 36.8 mm Hg. Pulmonary function tests were normal: forced ventilatory capacity (FVC), 3700 mL, 96% predicted; forced expiratory volume in 1 second (FEV1), 2800 mL, 91% predicted; diffusion lung capacity (DLCO), 82% predicted. CT scan of the chest revealed ground glass opacification of alveolar spaces with thickening of inter-obular and intralobular septa typical of the crazy paving pattern (Figure 1). The blood cultures were negative. Serologies for cytomegalovirus, respiratory syncytial virus, adenovirus, influenza, legionella and Mycoplasma pneumoniae were also negative. Empirical treatment with moxifloxacin was instituted. Bronchoscopy was performed because the fever persisted. The bronchial system was normal with no endobronchial pathology and no sign of infection. Bronchoalveolar lavage (BAL) fluid was cloudy in appearence. BAL differential cytology showed the composition to be 74% macrophages, 21% neutrophils, and 5% lymphocytes. Light microscopic examination of the fluid showed large amounts of amorphous, lipoproteinaceous material that was periodic acid-Schiff stain (PAS) positive (Figure 2). Smear, special stains, culture of the BAL fluid, and tissue were negative for tuberculosis, bacteria, Pneumocystis carinii, fungi or malignant cells. Histology of the transbronchial biopsy specimen demonstrated a PAS-positive intra-alveolar eosinophilic material consistent with PAP. Previous studies have reported anti-GM-CSF antibody as being present in the serum of patients with idiopathic PAP. Anti-GM-CSF antibodies were not detected in the serum and BAL fluid. Based on these findings the patient was diagnosed as secondary PAP associated with Behçet's disease. While the patient was followed as an outpatient at our department his clinical status was stable with progressive recovery of dyspnea and cough. Physical examination returned to normal. Chest CT performed four weeks later revealed complete resolution of the ground glass opacification and septal thickening (Figure 3). No recurrence was observed during the follow up period.

Chest CT demonstrating ground glass opacities with thickened interlobular septa in both lungs, the typical crazy paving pattern seen in pulmonary alveolar proteinosis.

Eosinophilic material in the alveolar spaces with scattered alveolar macrophages and lymphocytes (hematoxylyn-eosin stain ×100).

CT of the chest showing complete resolution of the ground glass pattern and interstitial thickening of the interlobular septa.
Discussion
PAP is a rare disorder characterized by abundant accumulation of phospholipids and proteinaceous material within the alveoli of the lungs. Since this disease was first described in 1958, fewer than 500 cases have been reported in the medical literature. The great majority of these cases are of the acquired variety. Advances in the cellular and molecular basis of the disease have led to a better understanding of the pathogenesis of PAP. The link between GM-CSF and human idiopathic PAP was revealed when anti-GM-CSF antibodies were detected in the serum and BAL fluid of patients with the disease. The idiopathic form accounts for more than 90% of cases and is believed to be caused by autoantibodies targeting granulocyte-macrophage-colonystimulating factor. In contrast, the role of GM-CSF in secondary PAP is unclear [4, 5]. Secondary PAP develops in the setting of a predisposing factor such as hematological malignancies, immunodeficiency syndromes, chronic infection or immunosuppressive medications [2, 7, 8].
PAP has only rarely been associated with autoimmune diseases. In a review of over 400 published cases of PAP, a diagnosis of Behçet's disease coexisted in only one patient reported by Uchiyama et al. [9]. We believe that the presence of alveolar proteinosis may be underestimated because of three factors. One, alveolar proteinosis associated with Behçet's syndrome may easily be missed as there are no specific features of alveolar consolidation to suggest the diagnosis. Second, the diagnosis can only be made with confidence on the basis of high resolution computed tomography (HRCT) appearence. Third, correct stains are needed for pathologic diagnosis. Consequently, alveolar proteinosis may be overlooked or misdiagnosed as another inflammatory disease such as pneumonia, alveolar hemorrhage or drug reaction which are common in the setting of autoimmune disease.
The absence of anti-GM-CSF antibodies strongly suggested that our patient had developed secondary PAP in the setting of Behçet's disease. The development of PAP may have been associated with infection but there was no evidence of current or earlier infection. Although few studies have described an association between secondary PAP and prolonged corticosteroid therapy, the effect of local steroid treatment may be considered negligible for immunosuppression as there are no data to suggest immunosuppression due to short course local steroid treatment. Furthermore, patients who receive corticosteroid treatment for other conditions do not develop alveolar proteinosis because only a few studies have described an association between secondary PAP and long-term corticosteroid therapy [10]. The predominant mechanism for alveolar proteinosis in our patient was cycloserine induced immunosuppression. Inhibition of CD4-T cells by cyclosporine may have produced deficiency of macrophage recruitment and phagocytosis causing impairment of surfactant clearance in this case. Autoimmune disease and vasculitis may also have contributed to the development of alveolar proteinosis to a variable extent by unknown mechanisms. Although alveolar proteinosis may run a fatal course, spontaneous recovery may also occur [1, 6]. To our knowledge, this is only the second reported case of secondary PAP associated with Behçet's disease and the first case of a spontaneous remission. Our patient draws attention to the fact that secondary alveolar proteinosis with spontaneous resolution may occur in the setting of Behçet's disease. Therefore, unnecessary diagnostic interventions or treatment modalities for secondary alveolar proteinosis associated with Behçet's disease should be reconsidered in stable patients. Further case database and multicenter trials are needed to establish the real incidence and prognosis of alveolar proteinosis in Behçet's disease.
Conflict of interests statement
None of the authors has any conflict of interest to declare in relation to the subject matter of this manuscript.
References
Rosen SH, Castleman B, Liebow AA: Pulmonary alveolar proteinosis. N Engl J Med. 1958, 258: 1123-1142. 10.1056/NEJM195806052582301.
Seymour JF, Presneill JJ: Pulmonary alveolar proteinosis: progress in the first 44 years. Am J Respir Crit Care Med. 2002, 166: 215-235. 10.1164/rccm.2109105.
Trapnell BC, Whitsett JA, Nakata K: Pulmonary alveolar proteinosis. N Engl J Med. 2003, 349: 2527-2539. 10.1056/NEJMra023226.
Kitamura T, Tanaka N, Watanabe J, Uchida K, Kanegasaki S, Yamada Y, Nakata K: Idiopathic pulmonary alveolar proteinosis as an autoimmune disease with neutralizing antibody against granulocyte/macrophage colony-stimulating factor. J Exp Med. 1999, 190: 875-880. 10.1084/jem.190.6.875.
Kitamura T, Uchida K, Tanaka N, Tsuchiya T, Watanabe J, Yamada Y, Hanaoka K, Seymour JF, Schoch OD, Doyle I, Inoue Y, Sakatani M, Kudoh S, Azuma A, Nukiwa T, Tomita T, Katagiri M, Fujita A, Kurashima A, Kanegasaki S, Nakata K: Serological diagnosis of idiopathic pulmonary alveolar proteinosis. Am J Respir Crit Care Med. 2000, 162: 658-662.
Prakash UB, Barham SS, Carpenter HA, Dines DE, Marsh HM: Pulmonary alveolar phospholipoproteinosis: experience with 34 cases and a review. Mayo Clin Proc. 1987, 62: 499-518.
Shah PL, Hansell D, Lawson PR, Reid KB, Morgan C: Pulmonary alveolar proteinosis: clinical aspects and current concepts on pathogenesis. Thorax. 2000, 55: 67-77. 10.1136/thorax.55.1.67.
Huizar I, Kavuru MS: Alveolar proteinosis syndrome: pathogenesis, diagnosis and management. Curr Opin Pulm Med. 2009, 15: 491-498. 10.1097/MCP.0b013e32832ea51c.
Uchiyama M, Nagao T, Hattori A, Fujii T, Ichiwata T, Nakata K, Tani K, Hayashi T: Pulmonary alveolar proteinosis in a patient with Behcet's disease. Respirology. 2009, 14: 305-308. 10.1111/j.1440-1843.2008.01450.x.
Samuels MP, Warner JO: Pulmonary alveolar lipoproteinosis complicating juvenile dermatomyositis. Thorax. 1988, 43: 939-940. 10.1136/thx.43.11.939.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Tetikkurt, C., Tetikkurt, S., Ozdemir, I. et al. Alveolar proteinosis in Behçet's disease. Multidiscip Respir Med 5, 264 (2010). https://doi.org/10.1186/2049-6958-5-4-264
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/2049-6958-5-4-264