
Abstract
Background
The Amazon River is by far the world’s largest in terms of volume and area, generating a fluvial export that accounts for about a fifth of riverine input into the world’s oceans. Marine microbial communities of the Western Tropical North Atlantic Ocean are strongly affected by the terrestrial materials carried by the Amazon plume, including dissolved (DOC) and particulate organic carbon (POC) and inorganic nutrients, with impacts on primary productivity and carbon sequestration.
Results
We inventoried genes and transcripts at six stations in the Amazon River plume during June 2010. At each station, internal standard-spiked metagenomes, non-selective metatranscriptomes, and poly(A)-selective metatranscriptomes were obtained in duplicate for two discrete size fractions (0.2 to 2.0 μm and 2.0 to 156 μm) using 150 × 150 paired-end Illumina sequencing. Following quality control, the dataset contained 360 million reads of approximately 200 bp average size from Bacteria, Archaea, Eukarya, and viruses. Bacterial metagenomes and metatranscriptomes were dominated by Synechococcus, Prochlorococcus, SAR11, SAR116, and SAR86, with high contributions from SAR324 and Verrucomicrobia at some stations. Diatoms, green picophytoplankton, dinoflagellates, haptophytes, and copepods dominated the eukaryotic genes and transcripts. Gene expression ratios differed by station, size fraction, and microbial group, with transcription levels varying over three orders of magnitude across taxa and environments.
Conclusions
This first comprehensive inventory of microbial genes and transcripts, benchmarked with internal standards for full quantitation, is generating novel insights into biogeochemical processes of the Amazon plume and improving prediction of climate change impacts on the marine biosphere.
Similar content being viewed by others

Background
The Amazon River runs nearly 6,500 km across the South American continent before emptying into the Western Tropical North Atlantic Ocean; in terms of both volume and watershed area it is the world’s largest riverine system [1]. The river carries a significant load of terrestrially-derived nutrients to the ocean, and this has global consequences on marine primary productivity and carbon sequestration [2, 3]. Productive phytoplankton blooms harboring cyanobacteria, coastal diatom species, and oceanic diatoms with endosymbiotic diazotrophs take advantage of the riverine nutrient supplements and enhance carbon export from the upper ocean to deeper waters via sinking particles [3, 4]. Heterotrophic bacteria also remineralize organic nutrients in the plume, further fueling primary production and increasing the flux of organic material to deep water.
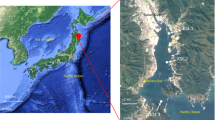
We inventoried the microbial genes and transcripts at six stations in the Amazon River plume aboard the R/V Knorr between 22 May and 25 June, 2010 (Figure 1) using Illumina sequencing with 150 × 150 bp overlapping paired-end reads. Metagenomic and metatranscriptomic data have typically been analyzed within a relative framework (that is, % of metagenome and % of metatranscriptome), but this approach is problematic for dynamic communities because a change in the abundance of one type of gene or transcript imposes a change in the percent contribution of the others. By incorporating internal standards, we are able to assess meta-omics datasets within an absolute framework that facilitates comparisons of communities sampled at different times and places in the environment. In the Amazon plume sequence libraries, known copy numbers of internal standards were added at the initiation of sample processing and consisted of genomic DNA from an exotic bacterium for the metagenomes (Thermus thermophilus HB8) and artificial mRNAs and poly(A)-tailed mRNAs for the metatranscriptomes; these standards were identified, counted, and removed from the natural sequences during quality control steps.

Location of sampling sites in the Amazon River plume in June, 2010.
For each station, metagenomes and non-selective metatranscriptomes were each obtained in duplicate for two discrete size fractions (0.2 to 2.0 μm and 2.0 to 156 μm), while poly(A)-selective metatranscriptomes were obtained in duplicate only for the 2.0 to 156 μm size fraction (to increase coverage of the eukaryotic community), resulting in a total of 60 datasets (6 stations x 5 data types × 2 replicates) (Table 1). The data collection consisted of 360 million reads following quality control (removal of poor quality reads, removal of rRNAs from metatranscriptomes, removal of internal standards, and joining of overlapping 150 bp paired ends) and provides an unprecedented view of the metabolic functions of the Bacteria, Archaea, and Eukarya mediating carbon and nutrient cycling in the Amazon River plume.
Methods
Detailed sample collection and processing methodology can be found in Additional file 1. Sample sites in the Amazon River plume were chosen to represent a range of salinity, nutrient concentrations, and microbial communities (Additional file 2). Microbial cells were collected by filtration and preserved in RNAlater (Applied Biosystems, Austin, TX, USA). During sample processing, internal standards were added to each sample prior to cell lysis. Samples collected for non-selective metatranscriptomics were processed by extracting total RNA, removing residual DNA, depleting rRNA, linearly amplifying the remaining transcripts, and making double-stranded cDNA for library preparation and sequencing. Poly(A)-selective metatranscriptome samples were processed similarly except that poly(A)-tailed mRNAs were selectively isolated, eliminating the need for rRNA depletion steps. Metagenomic samples were processed by extracting DNA and removing residual proteins and RNA. Following sample processing, cDNA or DNA was sheared and libraries were constructed for paired-end sequencing (150 × 150) using either the Genome Analyzer IIx, HiSeq 2000, MiSeq, or HiSeq 2500 platform (Illumina Inc., San Diego, CA).
From 60 samples, we obtained 8.21 × 108 raw sequences containing 1.23 × 1011 nt. Following sequence quality control, 3.59 × 108 reads with a mean length of 195 bp were obtained. Internal standards were quantified and removed, along with any remaining rRNA sequences. Remaining reads were annotated against the RefSeq Protein database or a custom marine database using RAPSearch2 [5], and abundance per liter was calculated based on internal standard recovery [6] (Additional file 2).
Biological and chemical data measured concurrently with sample collection provides environmental context for sequence data. These metadata include temperature, salinity, oxygen concentration, irradiance, chlorophyll concentration, nutrient concentrations, and bacterial abundance and production (Additional file 2). Datasets describing the phytoplankton communities and other features of the June 2010 plume ecosystem have been previously published [1, 4, 7, 8].
Quality assurance
The She-ra program [9] was used to join the paired-end Illumina reads using the default parameters and a quality metric score of 0.5. Seqtrim [10] was used to trim the joined reads using the default parameters. rRNA and internal standard sequences were identified in the metatranscriptomes using a Blastn search against a custom database containing representative rRNA sequences and internal standard sequences; sequences with a bit score ≥ 50 were identified as either rRNA or internal standards and removed from the datasets. Internal standards were identified in metagenomes by first performing a Blastn search (bit score cutoff ≥ 50) against the T. thermophilus HB8 genome. Hits were subsequently queried against the RefSeq protein database using Blastx (bit score cutoff ≥ 40) to identify and quantify all T. thermophilus HB8 protein encoding reads, and these reads were removed from the datasets.
Initial findings
Metagenomic reads from surface waters of the six Amazon River plume stations were assigned to bacterial, archaeal, eukaryotic, and viral taxa based on best hits to reference genomes. Among autotrophic bacteria, Synechococcus was the largest contributor to the metagenomes at locations closest to the river mouth (Stations 10, 3; approximately 1.5 × 1012 genes L−1) and was replaced by Prochlorococcus at more oceanic locations (Stations 25, 27) (Table 2). Among heterotrophic bacteria, SAR86 had the largest gene abundance closest to the river mouth (Station 10; approximately 8.6 × 1011 genes L−1). SAR11 clade members (HTCC7211, HIMB5) were also abundant here, and became the dominant contributor of heterotrophic bacterial genes at more oceanic stations (up to 5.7 × 1012 genes L−1) (Table 2). Genes binning to SAR324 genomes were abundant at three stations (Station 2, 3, and 23; Table 2), with the Amazon plume sequences aligning with heterotrophic members of this group [11]. Station 2 had a distinctive bacterial community relative to the other plume stations, dominated by genes from Verrucomicrobia related to Coraliomargarita akajimensis DSM 45221 and strain DG1235 and with substantial contributions from SAR116 taxa (IMCC1322, HIMB100). Coraliomargarita akajimensis DSM 45221 was also among the most abundant genome bins at Station 25 (Table 2).
Among eukaryotic taxa, diatoms and the green alga Micromonas contributed the greatest number of genes at lower salinities, while Haptophytes (binning to Phaeocystis antarctica), dinoflagellates (binning to Alexandrium tamarense CCMP1771) and relatives of the green alga Pyraminomonas obovata CCMP722 increased in importance at more saline stations (Table 2). Among Archaea, members of the ammonia-oxidizing genus Nitrosopumilus and related genera contributed the most genes at stations closest to the river mouth, although they were 100-fold lower in numbers compared to the most abundant bacterial taxa. There were very few archaeal genes at the outermost stations (Stations 25 and 27), and these binned largely to methanogen sequences. The viral sequences were dominated by cyanobacterial phages (Table 2).
Patterns of gene and transcript abundance provided insights into transcriptional activity by taxon and habitat (that is, cells that were free-living versus those that were particle-associated) for the dominant bacterial groups. Particle-associated Verrucomicrobia (Order Puniceicoccales) maintained cellular transcript inventories of up to 14 transcripts/gene for particle-associated cells and averaged 2 transcripts/gene overall (Figure 2). In contrast, members of the Flavobacteria class averaged < 0.5 transcripts/gene. Particle-associated cells in each of these major taxa typically had more transcripts per gene copy than did free-living cells (averaging 2.0 versus 0.15 transcripts/gene) (Figure 2). Abundance of transcripts originating from particle-associated versus free-living bacteria varied along the plume, with mRNAs from free-living cells contributing only 30 to 60% of the metatranscriptome in landward stations, but > 90% at outer plume stations. Environmental data (Additional file 2) indicate that Station 10 had the lowest salinity (22.6) and Station 27 the highest (36.0). Station 10 was the most strongly influenced by riverine inputs, particularly of inorganic nitrogen.

Inventories of genes and transcripts for eight bacterial taxa in surface waters of the Amazon plume. Symbols represent the mean of duplicate analyses at six stations, color-coded by taxon and size fraction (particle-associated or free-living). Lines indicate a 1:1 ratio of transcripts:genes (black) or 10:1 and 1:10 ratios (gray). The purple line indicates the ratio of transcripts:genes for exponentially growing laboratory cultures of Escherichia coli[12, 13]. Dominant bacterial groups are as follows: Oscillatoriales = Trichodesmium; Prochlorales = Prochlorococcus; Chroococcales = Synechococcus; Nostocales = Richelia; Puniceicoccales = Verrucomicrobia.
Future directions
The Amazon River plume is immense in scale and sensitive to anthropogenic forcing. This multi-omics dataset is the first of four high-throughput metagenomic and metatranscriptomic sequence collections being produced for the Amazon River Continuum as part of the ANACONDAS and ROCA projects (http://amazoncontinuum.org). These projects aim to improve predictive capabilities for climate change impacts on the marine biosphere, focusing on the Amazon ecosystem, and to better our understanding of feedbacks on the carbon cycle. Processes in the river and ocean are tightly linked from physical, biological, and biogeochemical perspectives. Thus, the complete data collection will include two datasets from the Amazon plume (June 2010 and July 2013) and two from the Amazon River (Óbidos to Macapá and Belém; June 2011 and July 2013). These high-coverage, size-discrete, and replicated datasets are all benchmarked with internal genomic and mRNA standards for comparative quantitative metagenomics and metatranscriptomics. Insights from these meta-omics datasets are enhancing predictive capabilities regarding the interplay between marine microbial communities, biogeochemical cycling, and carbon sequestration in the ocean.
Availability of supporting data
Sequences from June 2012 Amazon Continuum study are available from NCBI under accession numbers [SRP039390] (metagenomes), [SRP037995] (non-selective metatranscriptomes), and [SRP039544] (poly(A)-selected metatranscriptomes). The NCBI sequences are fastq files from which internal standard sequences and rRNA sequences (metatranscriptomes only) have been removed prior to deposition. Sequences are also available at the Community Cyberinfrastructure for Advanced Microbial Ecology Research and Analysis (CAMERA) database under project number CAM_P_0001194. The CAMERA sequences are QC’d fasta files of joined paired-end reads, also with internal standards and rRNA sequences (metatranscriptomes only) removed. Metadata accompanying the omics datasets are provided in Additional file 2. ANACONDAS and ROCA project data are also available at the BCO-DMO data repository (http://www.bco-dmo.org/project/2097).
Abbreviations
- bp:
-
base pairs
- DOC:
-
dissolved organic carbon
- nt:
-
nucleotides
- POC:
-
particulate organic carbon.
References
Coles VJ, Brooks MT, Hopkins J, Stukel MR, Yager PL, Hood RR: The pathways and properties of the Amazon River plume in the tropical north Atlantic ocean. J Geophys Res: Oceans. 2013, 118: 6894-6913. 10.1002/2013JC008981.
Richey JE, Nobre C, Deser C: Amazon River discharge and climate variability: 1903 to 1985. Science. 1989, 246: 101-103. 10.1126/science.246.4926.101.
Subramaniam A, Yager PL, Carpenter EJ, Mahaffey C, Bjorkman K, Cooley S, Kustka AB, Montoya JP, Sanudo-Wilhelmy SA, Shipe R, Capone DG: Amazon River enhances diazotrophy and carbon sequestration in the tropical north Atlantic ocean. Proc Natl Acad Sci U S A. 2008, 105: 10460-10465. 10.1073/pnas.0710279105.
Goes JI, Gomes HR, Chekalyuk AM, Carpenter EJ, Montoya JP, Coles VJ, Yager PL, Berelson WM, Capone DG, Foster RA, Steinberg DK, Subramaniam A, Hafez MA: Influence of the Amazon River discharge on the biogeography of phytoplankton communities in the Western Tropical North Atlantic. Prog Oceanogr. 2014, 120: 29-40.
Zhao Y, Tang H, Ye Y: RAPSearch2: a fast and memory-efficient protein similarity search tool for next-generation sequencing data. Bioinformatics. 2012, 28: 125-126. 10.1093/bioinformatics/btr595.
Satinsky BM, Gifford SM, Crump BC, Moran MA: Use of internal standards for quantitative metatranscriptome and metagenome analysis. Methods in Enzymology. Edited by: Edward FD. 2013, Burlington, MA: Academic, 531: 237-250. 12
Barada LP, Cutter L, Montoya JP, Webb EA, Capone DG, Sanudo-Wilhelmy SA: The distribution of thiamin and pyridoxine in the Western Tropical North Atlantic Amazon river plume. Front Microbiol. 2013, 4: 25-
Chong LS, Berelson WM, McManus J, Hammond DE, Rollins NE, Yager PL: Carbon and biogenic silica export influenced by the Amazon River Plume: patterns of remineralization in deep-sea sediments. Deep Sea Res Part I: Oceanogr Res Papers. 2014, 85: 124-137.
Rodrigue S, Materna AC, Timberlake SC, Blackburn MC, Malmstrom RR, Alm EJ, Chisholm SW: Unlocking short read sequencing for metagenomics. PLoS One. 2010, 5: e11840-10.1371/journal.pone.0011840.
Falgueras J, Lara AJ, Fernandez-Pozo N, Canton FR, Perez-Trabado G, Claros MG: SeqTrim: a high-throughput pipeline for pre-processing any type of sequence read. BMC Bioinformatics. 2010, 11: 38-10.1186/1471-2105-11-38.
Chitsaz H, Yee-Greenbaum JL, Tesler G, Lombardo MJ, Dupont CL, Badger JH, Novotny M, Rusch DB, Fraser LJ, Gormley NA, Schulz-Trieglaff O, Smith GP, Evers DJ, Pevzner PA, Lasken RS: Efficient de novo assembly of single-cell bacterial genomes from short-read data sets. Nat Biotechnol. 2011, 29: 915-921. 10.1038/nbt.1966.
Neidhardt FC, Umbarger HE: Chemical composition of Escherichia coli. Escherichia Coli and Salmonella Typhimurium: Cellular and Molecular Biology. Edited by: Neidhardt FC, Curtiss RIII, Ingraham JL, Lin ECC, Low KB, Magasanik B, Reznikoff WS, Riley M, Schaechter M, Umbarger HE. 1996, Washington, DC: ASM Press, 13-16. 2
Taniguchi Y, Choi PJ, Li GW, Chen H, Babu M, Hearn J, Emili A, Xie XS: Quantifying E. coli proteome and transcriptome with single-molecule sensitivity in single cells. Science. 2010, 329: 533-538. 10.1126/science.1188308.
Acknowledgements
We appreciate the assistance of Roger Nilsen, Camille English, and Shulei Sun, and we thank P Yager and scientists of the ROCA and ANACONDAS projects for helpful discussions. This research was funded by the Gordon and Betty Moore Foundation and NSF grant OCE-0934095. Resources and technical expertise were provided by the University of Georgia’s Georgia Advanced Computing Resource Center and CAMERA.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
BMS: conception and design of protocols, sample processing, data analysis, writing and final approval of the manuscript. BLZ: sample collection, sample processing, critical revision and final approval of the manuscript. MD: sample processing, protocol design, critical revision and final approval of the manuscript. CBS: sample processing, protocol design, critical revision and final approval of the manuscript. SS: data analysis, critical revision and final approval of the manuscript. JHP: critical revision and final approval of the manuscript. BCC: design of protocols, data analysis, critical revision and final approval of the manuscript. MAM: conception and design of protocols, data analysis, writing and final approval of the manuscript. All authors read and approved the final manuscript.
Electronic supplementary material
40168_2014_45_MOESM1_ESM.pdf
Additional file 1:Detailed methods. Description of metagenome and metatranscriptome sample processing, sequencing, and data analysis, including internal standard additions and analysis. (PDF 107 KB)
40168_2014_45_MOESM2_ESM.xlsx
Additional file 2:Metadata. Metadata accompanying the metagenomic and metatranscriptomic datasets, including sample station locations,environmental conditions and library sizes and statistics. (XLSX 34 KB)
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Satinsky, B.M., Zielinski, B.L., Doherty, M. et al. The Amazon continuum dataset: quantitative metagenomic and metatranscriptomic inventories of the Amazon River plume, June 2010. Microbiome 2, 17 (2014). https://doi.org/10.1186/2049-2618-2-17
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/2049-2618-2-17