
Abstract
Background
Several studies indicated that Erythropoietin (Epo) may provide remarkable neuroprotection in some neurological diseases. It also showed the significant decrease of Epo immunoreactivity in the cerebral cortex and hippocampus in aged rats, suggesting the role of Epo in the pathogenesis of age-related neurodegenerative diseases such as AD.
Methods
The protective effect of Epo was studied in differentiated PC12 cells treated with Abeta. The viability of the cells, the apoptosis of the cells and the level of Bax, Bcl-2, cleaved caspase-3 and cleaved PARP expression were detected by MTT, Hoechst 33258 staining and Western blotting respectively.
Results
20 μM Abeta (25-35) could induce a decreased viability and a increased apoptosis in PC12 cell in a time-dependent manner. However, 20 μM Abeta (35-25) had no effect on cell viability and apoptosis. Western blot analysis also showed that Abeta(25-35) treatment could decrease the expression of Bcl-2 (P < 0.05) and increase the expression of Bax (P < 0.05), Cleaved casapase-3 (P < 0.05), and Cleaved PARP (P < 0.05). The pretreatment of Epo could effectively reverse all the above changes induced by Abeta(25-35) (P < 0.05). Furthermore, the protective effect of Epo could be blocked by PI3K inhibitor LY294002 (P < 0.05).
Conclusions
Epo prevented cell injuries in PC12 cells exposed to the Abeta(25-35) and this effect may depend on the PI3K⁄Akt pathway. Our study provided an important evidence for the potential application of Epo in the therapy of Alzheimer's disease.
Similar content being viewed by others
Background
Apoptosis is a particular type of programmed cell death controlled by precise intrinsic genetic programme in order to regulate cell population. Among the mechanisms of cell death, apoptosis has been proposed to explain the cell loss observed in many neurodegenerative disorders including Alzheimer's disease (AD) [1–3]. AD is a neurodegenerative disorder of the central nervous system (CNS), which correlate with the appearance of neurofibrillary tangles (NFTs) and senile plaques (SPs) [4]. The major component of SPs is beta-amyloid peptide (Abeta), which is believed to be the most probable cause of AD [3, 5]. Many studies have shown that Abeta can directly induce neuronal death via apoptosis [2, 6, 7].
Erythropoietin (Epo) was originally characterized as the principal regulator of erythropoiesis [8]. Many experimental studies have shown that both Epo and its specific receptor (erythropoietin receptor, EpoR) expressing in the CNS, provide remarkable neuroprotection in many neurological diseases [9–13]. Recent research has demonstrated significant decreases in Epo immunoreactivity in the cerebral cortex and hippocampus of aged rats [14] which suggested the role of Epo in the pathogenesis of age-related neurodegenerative diseases such as AD. Therefore, we studied the possible relationship between Epo and Abeta-induced cell apoptosis. In the present study, we observed that Abeta(25-35) peptide at 20-μM concentrations could induce apoptosis in PC12 cells and Epo could reverse these changes through PI3K/Akt signaling pathway. Our results identifed a potential molecular targets for AD therapy.
Materials and methods
Cell culture and drug treatment
Abeta(25-35) (Sigma-Aldrich, St. Louis, MO) or Abeta(35-25) (Sigma-Aldrich, St. Louis, MO) was dissolved in water to obtain a 2 mM stock solution. Aliquots were stored at -20°C and thawed at 37°C for 5 ~ 7 d for use. Differentiated rat pheochromocytoma PC12 cells (provided by the Institute of Biochemistry and Cell Biology, Chinese Academy of Science, Shanghai) were plated in 100-mm culture dishes (Corning Incorporated, Corning, NY, USA) in DMEM containing 10% (v/v) heat-inactivated FBS, 5% horse serum, 1% penicillin, and 1% streptomycin. The cells were grown at 37°C in a humid 5% CO2 environment, and the medium was routinely replaced every 2 d. The media were replaced with serum-free media 12 h prior to drug treatment. The cells were then treated with Abeta(25-35) or Abeta(35-25) for 24 h. Epo (R&D systems, USA) at various concentrations were added into the cultures 1 h prior to the 24-h Abeta(25-35) exposure. 20 μM LY294002 (Sigma-Aldrich, St. Louis, MO, dissolved in DMSO) were added into the cultures 1 h prior to the Epo treatment.
Analysis of cell viability
Cell viability was assessed by MTT assay. Briefly, PC12 cells were seeded in 96-well culture plates at a density of 1 × 104 cells per well. After the treatment of Abeta(25-35), Abeta(35-25), Epo or LY294002, the cells were subjected to the assay as previously reported [15, 16].
Hoechst 33258 staining
For Hoechst 33258 staining, cells were fixed with 4% paraformaldehyde. Cell nuclei were stained with fluorescent dye Hoechst 33258 (Sigma, St. Louis, MO) at a final concentration of 5 μg/ml in PBS, for 20 min at room temperature in a dark chamber, and then observed in a fluorescence microscope (OLYMPUS 1 × 70, Japan) and photographed.
Western blotting
The Western blotting analysis procedure was conducted as previously reported [16]. After the treatment, cells were washed twice with cold phosphate buffered saline and lysed on ice with cell lysis buffer(10 mM Tris, pH 7.4, 100 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM NaF, 20 mM Na4P2O7, 2 mM Na3VO4, 0.1% SDS, 0.5% sodium deoxycholate, 1% Triton-X 100, 10% glycerol, 1 mM PMSF (made from a 0.3 M stock in DMSO), 60 μg/mL aprotinin, 10 μg/mL leupeptin, 1 μg/mL pepstatin) for 30 mininutes. The soluble fraction was obtained by centrifugation at 14000 g for 20 min at 4°C. The concentration of the protein was determined by the BCA assay (Pierce Biotechnology, Rockford, IL). Equal amounts of the protein (20 μg) were separated in an 8-10% SDS-polyacrylamide gel; the resolved proteins were electrotransferred onto PVDF or nitrocellulose membranes (Bio-Rad, Hercules, CA). The membranes were subsequently blocked with 5% nonfat milk in TBST for 1 h at room temperature and incubated with appropriate concentrations of primary antibody (1:200 for Bax and Bcl-2 (Santa Cruz Biotechnology, Inc, CA, USA), 1:5000 for beta-actin (Sigma-Aldrich, St. Louis, MO), 1:1000 for Cleaved caspase-3 and PARP (Cell Signaling Technology, Beverly, MA)) at 4°C overnight. The membranes were then washed 3 times with TBST and probed with the corresponding secondary antibodies conjugated with HRP (Cell Signaling Technology, Beverly, MA) at room temperature for 1 h. After washing, the signals were developed using the ECL Advanced Western Blotting Detection kit (Amersham, UK). Band intensities were quantified by densitometric analysis by using an AxioCam digital camera (ZEISS, Germany) and the KS400 photo analysis system (Ver. 3.0).
Statistics
Data are expressed as mean ± standard deviation (S.D.) and were analyzed using SPSS 11.0 statistical software (SPSS Inc., Chicago, IL, USA). Each procedure was performed in duplicate in 3 ~ 5 independent experiments. Statistical analyses were performed using one-way ANOVA, followed by the two-tailed Student's t test. Multiple comparison tests were applied when appropriate, and statistical significance was assumed at P < 0.05.
Results
Effects of Abeta(25-35) on cell viability and cell apoptosis determined by MTT and Hoechst 33258 staining respectively
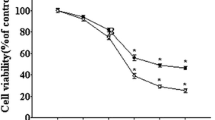
The MTT assay was used to determine the effect of 20 μM Abeta (25-35) on the viability of the PC12 cell cultures. As shown in the following graph, 20 μM Abeta (25-35) induced a decrease in PC12 cell viability in a time-dependent manner (Figure 1A). We also used the control peptide 20 μM Abeta(35-25) to determine the effect of 20 μM Abeta(35-25) on the cell viability As shown in the following graph, 20 μM Abeta (35-25) had no effect on PC12 cell viability (Figure 1B). Hoechst 33258 staining also showed 10 μM Abeta (25-35) and 20 μM Abeta(25-35) could induce PC12 cell apoptosis. However, 10 μM Abeta (35-25) and 20 μM Abeta (35-25) had no effect on PC12 cell apoptosis (Figure 2A and 2B).

Effects of Abeta (25-35) on cell viability. The MTT assay was used to determine the cell viability. As shown in the following graph, 20 μM Abeta(25-35) induced a decrease in PC12 cell viability in a time-dependent manner (P < 0.05) (A), However, 20 μM Abeta(35-25) had no effect on PC12 cell viability (P > 0.05) (B). (*P < 0.05 vs. the controls). The data shown represent 5 independent experiments

Effects of Abeta (25-35) on cell apoptosis. Hoechst33258 was performed to detect the cell apoptosis (Con, Control group), showing nuclear condensation and fragmentation (arrows) (A). 10 randomized representative fields were analyzed in one experiment. 10 μM and 20 μM Abeta(25-35) could effectively induce PC12 cell apoptosis (P < 0.05) (A and B), and 20 μM Abeta(35-25) had no effect on PC12 cell apoptosis (A and B). The data shown represent 5 independent experiments (*P < 0.05 vs. the controls) (B)
Effects of Epo on Abeta(25-35)-induced PC12 cell viability and cell apoptosis determined by MTT and Hoechst 33258 staining respectively
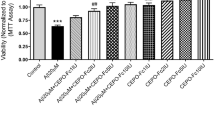
We added 3 different concentrations of Epo (5, 10, 20 u) into the serum-deprived media of PC12 cells 1 h prior to the 24-h 20 μM Abeta (25-35) exposure. As shown in the following graph, various concentrations of Epo (5, 10, 20 u) could effectively prevent a decrease of cell viability induced by 20 μM Abeta (25-35) (P < 0.05) (Figure 3). Hoechst 33258 staining also showed 3 different concentrations of Epo (5, 10, 20 u) can effectively prevent cell apoptosis induced by Abeta (25-35) (P < 0.05) (Figure 4A and 4B).

Effect of Epo on cell viability induced by 20 μM Abeta (25-35) . The MTT assay was used to determine the cell viability. As shown in the following graph, Various concentrations of Epo (5, 10, 20 u) could effectively prevent a decrease of cell viability induced by 20 μM Abeta(25-35) (P < 0.05) (B). (*P < 0.05 vs. the controls). The data shown represent 5 independent experiments

Effect of Epo on cell apoptosis induced by 20 μM Abeta (25-35) . Hoechst33258 was performed to detect the cell apoptosis (Con, Control group). We found 3 different concentrations of Epo (5, 10, 20 u) could effectively prevent cell apoptosis induced by Abeta(25-35) (P < 0.05) (A and B). The data shown represent 5 independent experiments (*P < 0.05 vs. the controls) (B)
Effects of Epo on Abeta(25-35)-induced PC12 cell apoptosis determined by Western blotting
Using Western blotting analysis, we found that the Abeta(25-35) treatment of PC12 cells could decrease the expression of Bcl-2 (P < 0.05) (Figure 5A) and increase the expression of Bax (P < 0.05) (Figure 5A), Cleaved casapase-3 (P < 0.05) (Figure 5B), and Cleaved PARP (P < 0.05) (Figure 5C). Three different Epo concentrations can prevent all the above changes induced by Abeta(25-35) (P < 0.05) (Figure 5A-C).

Effect of Epo on 20 μM Abeta (25-35) -induced cell apoptosis. Western blotting analysis indicated that the Abeta(25-35) treatment of PC12 cells could decrease the expression of Bcl-2 (P < 0.05) (A), and increase the expression of Bax (P < 0.05) (A), Cleaved casapase-3 (P < 0.05) (B), and Cleaved PARP (P < 0.05) (C). Three different Epo concentrations could prevent all the changes induced by Abeta(25-35) (P < 0.05) (A-C). Three independent experiments were performed in duplicate (*: P < 0.05 vs. the controls and #: P < 0.05 vs. 20 μM Abeta(25-35))
PI3K/Akt involvement in the effects of Epo on Abeta (25-35)-induced cell injuries
Stimulation of EpoRs by Epo has previously been shown to activate the PI3K⁄Akt signal transduction pathway [17, 18], which regulates cell survival and proliferation [19]. We treated the cells with PI3K inhibitor LY294002 and found the LY294002 treatment caused a slight increase in cell apoptosis in PC12 cells with or without Abeta(25-35) treatment (Figure 6A, B and Figure 7A-C) This suggested that the PI3K/Akt pathway was involved in Abeta(25-35)-induced cell apoptosis, When the PI3K pathway was inhibited by LY294002 in PC12 cells, we found that the effects of Epo on Abeta(25-35)-induced cell injuries were diminished (P < 0.05) (Figure 8, Figure 6A, B and Figure 7A-C).

Involvement of PI3K/Akt in the effects of Epo on Abeta (25-35) -induced cell apoptosis determined by Hoechst33258 staining. As shown in the following graph, We treated the cells with PI3K inhibitor LY294002 and found the protective effects of Epo on the Abeta(25-35)-induced cell apoptosis were diminished (P < 0.05). Three independent experiments were performed in duplicate (*: P < 0.05 vs. the controls and #: P < 0.05 vs. 20 μM Abeta(25-35))

Involvement of PI3K/Akt in the effects of Epo on Abeta (25-35) -induced cell apoptosis determined by Western blotting analysis. As shown in the graph, We treated the cells with PI3K inhibitor LY294002 and found the protective effects of Epo on Abeta(25-35)-induced cell apoptosis were diminished (P < 0.05) (A-C). Three independent experiments were performed in duplicate (*: P < 0.05 vs. the controls and #: P < 0.05 vs. 20 μM Abeta(25-35))

Involvement of PI3K/Akt in the effects of Epo on the Abeta (25-35) - decreased cell viability determined by MTT. We treated the cells with PI3K inhibitor LY294002 and found the protective effects of Epo on the Abeta(25-35)-decreased cell viability were diminished (P < 0.05). Three independent experiments were performed in duplicate (*: P < 0.05 vs. the controls and #: P < 0.05 vs. 20 μM Abeta(25-35)).
Discussion
Abeta is the major component of SPs, which are considered to play a causal role in the development and progress of AD [20, 21]. The molecular mechanisms underlying Abeta-mediated neurotoxicity remain unclear. Recently, many in vitro and vivo studies have shown that Abeta can directly induce neuronal death via the mechanism of apoptosis [2, 3, 22]. Epo is widely known for its role as a hematopoetic hormone. Epo binds to specific receptors present in the human brain can be synthesized by astrocytes as well as neurons [23]. Epo was shown to be capable of crossing the blood-CSF barrier via receptor-mediated transport [24, 25] and to act as a neurotrophic factor supporting the differentiation and regeneration of neurons [26]. Its protective effect under conditions of neuronal injury was also reported [27, 28]. Therefore, we proposed that the Epo system in the CNS can act as an endogenous system for protecting against neurodegenerative diseases such as AD. Among the fragments studied so far, the Abeta(25-35) represents the shortest fragment of Abeta, processed in vivo by brain proteases [29]. This peptide is the functional domain of Abeta required for neurotoxic effect, retaining the toxicity of the full-length peptide [30, 31]. It is highly cytotoxic to neuronal cells [32–34] and is widely used in both in vitro and in vivo experiments [35, 36]. In the present study, we used Abeta(25-35) to observe the toxic effect of Abeta and the protective effect of Epo. Abeta(35-25), a 11 amino acid with a reverse sequence of Abeta(25-35) was used as a control. We discovered that aggregated 20 μM Abeta(25-35) could decrease cell viability in a time-dependent manner (Figure 1A), However, 20 μM Abeta (35-25) had no effect on PC12 cell viability (Figure 1B.). Hoechst 33258 staining showed Abeta(25-35) can induce PC12 cell apoptosis while Abeta(35-25) had no effect on PC12 cell apoptosis (Figure 2A and 2B). Epo could attenuate the decreased cell viability (Figure 3) and increased cell apoptosis (Figure 4A and 4B) induced by Abeta(25-35).
Apoptosis is a tightly regulated process which involves changes in the expression of a distinct set of genes [37]. Bcl-2 is a key member of the anti-apoptotic Bcl-2 family, which plays a key role in regulating mitochondrial-mediated apoptotic cell death [38–40]. Over-expression of Bcl-2 can protect neuronal cells from neurotoxic insult. In contrast, Bax belongs to the pro-survival subfamily, which promotes apoptosis by translocating into the mitochondrial membrane and facilitating cytochrome c release [41]. In the present study, we found 20 μM Abeta(25-35) exposure could induce an increase of Bax expression and decrease Bcl-2 expression in serum-deprived cultured PC12 cells (Figure 5A), and Epo could effectively attenuate these changes (Figure 5A).
Caspases are a family of cysteine proteases and are critical mediators of cell apoptosis, which play an important role in the apoptotic process [42]. Caspase-3 acts as an apoptotic executor, it can activate DNA fragmentation factor, which in turn activate endonucleases to cleave nuclear DNA, and ultimately leads to cell death [43, 44]. Activation of caspase-3 appears to be a key event in execution of the apoptotic cascade in CNS diseases such as AD and Down's syndrome [45, 46]. In this study, we also found 20 μM Abeta(25-35) exposure could induce an increase of Cleaved caspase-3 expression (Figure 5B), and Epo could effectively attenuate these changes (Figure 5B).
Significant evidence indicates that caspase-3 is either partially or totally responsible for the proteolytic cleavage of many key proteins, including PARP. PARP is a nuclear DNA-binding protein of 110 kDa that is constitutively expressed in eukaryotes and that comprises up to 1% of the total nuclear proteins [47, 48]. PARP is important for cell viability, and cleavage of PARP facilitates cellular disassembly and serves as a marker of cells undergoing apoptosis [49]. In this study, we also found 20 μM Abeta(25-35) exposure could induce an increase of Cleaved PARP expression and Epo could effectively attenuate these changes (Figure 5C) with the same trend as the expression of Cleaved caspase-3 (Figure 5B).
Epo elicits its effects by binding to specific cell surface receptors. Evidence shows that Epo can induce activation of JAK-2/STAT-5 [50, 51], PI3K/Akt kinase [19], MAPK [52, 53], and PKC [54]. In the present study, we examined the effects of Epo on Abeta(25-35)-induced cell apoptosis in PC12 cells. We found Abeta(25-35)-mediated cell apoptosis could be appropriately attenuated by Epo (Figure 5A-C). Further, we found that LY294002, a PI3K inhibitor, attenuated the effect of Epo on Abeta(25-35)-induced-cell injuries (Figures 8, 6, 7), indicating that the protective effect of Epo is dependent on PI3K signaling. Our findings provide new molecular insight into the neuroprotective effect of Epo and suggest its possible therapeutic role in the management of AD.
Conclusions
In this report, we report that Epo prevented cell injuries in PC12 cells exposed to the beta-amyloid peptide and that this effect may depend on the PI3K⁄Akt pathway. The present study provides new molecular insight into the neuroprotective effect of Epo and suggests its possible therapeutic role in the management of AD.
Abbreviations
- AD:
-
Alzheimer's disease
- DMSO:
-
Dimethyl sulphoxide
- Epo:
-
Erythropoietin
- EpoR:
-
Erythropoietin receptor
- FBS:
-
Foetal bovine serum
- MAPK:
-
Migoten activated protein kinase
- PARP:
-
Poly (ADP-ribose) polymerase
- PI3K:
-
Phosphorinositide 3-kinase
- PVDF:
-
Polyvinylidene difluoride
References
Yoshioka K: Molecular mechanism of neuronal cell death in Alzheimer's disease. Nippon Ronen Igakkai Zasshi 1998, 35(4):265-267.
Cotman CW, Anderson AJ: A potential role for apoptosis in neurodegeneration and Alzheimer's disease. Mol Neurobiol 1995, 10(1):19-45.
Cotman CW, Su JH: Mechanisms of neuronal death in Alzheimer's disease. Brain Pathol 1996, 6(4):493-506.
Selkoe DJ: Translating cell biology into therapeutic advances in Alzheimer's disease. Nature 1999, 399(6738 Suppl):A23-A31.
Selkoe DJ: Alzheimer's disease: genes, proteins, and therapy. Physiol Rev 2001, 81(2):741-766.
Satou T, Cummings BJ, Cotman CW: Immunoreactivity for Bcl-2 protein within neurons in the Alzheimer's disease brain increases with disease severity. Brain Res 1995, 697(1-2):35-43.
Su JH, Deng G, Cotman CW: Bax protein expression is increased in Alzheimer's brain: correlations with DNA damage, Bcl-2 expression, and brain pathology. J Neuropathol Exp Neurol 1997, 56(1):86-93.
Lasne F, de Ceaurriz J: Recombinant erythropoietin in urine. Nature 2000, 405(6787):635.
Digicaylioglu M, Lipton SA: Erythropoietin-mediated neuroprotection involves cross-talk between Jak2 and NF-kappaB signalling cascades. Nature 2001, 412(6847):641-647.
Sinor AD, Greenberg DA: Erythropoietin protects cultured cortical neurons, but not astroglia, from hypoxia and AMPA toxicity. Neurosci Lett 2000, 290(3):213-215.
Sakanaka M, et al.: In vivo evidence that erythropoietin protects neurons from ischemic damage. Proc Natl Acad Sci USA 1998, 95(8):4635-4640.
Siren AL, et al.: Erythropoietin prevents neuronal apoptosis after cerebral ischemia and metabolic stress. Proc Natl Acad Sci USA 2001, 98(7):4044-4049.
Ehrenreich H, et al.: Exploring recombinant human erythropoietin in chronic progressive multiple sclerosis. Brain 2007, 130(Pt 10):2577-2588.
Chung YH, et al.: Age-related changes in erythropoietin immunoreactivity in the cerebral cortex and hippocampus of rats. Brain Res 2004, 1018(1):141-146.
Yang HQ, et al.: New protein kinase C activator regulates amyloid precursor protein processing in vitro by increasing alpha-secretase activity. Eur J Neurosci 2007, 26(2):381-391.
Sun ZK, et al.: Protective effects of erythropoietin on tau phosphorylation induced by beta-amyloid. J Neurosci Res 2008, 86(13):3018-3027.
Sui X, Krantz SB, Zhao ZJ: Stem cell factor and erythropoietin inhibit apoptosis of human erythroid progenitor cells through different signalling pathways. Br J Haematol 2000, 110(1):63-70.
Myklebust JH, et al.: Activation of phosphatidylinositol 3-kinase is important for erythropoietin-induced erythropoiesis from CD34(+) hematopoietic progenitor cells. Exp Hematol 2002, 30(9):990-1000.
Bao H, et al.: Protein kinase B (c-Akt), phosphatidylinositol 3-kinase, and STAT5 are activated by erythropoietin (EPO) in HCD57 erythroid cells but are constitutively active in an EPO-independent, apoptosis-resistant subclone (HCD57-SREI cells). Blood 1999, 93(11):3757-3773.
Hardy J, Selkoe DJ: The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 2002, 297(5580):353-356.
Pereira C, et al.: Cell degeneration induced by amyloid-beta peptides: implications for Alzheimer's disease. J Mol Neurosci 2004, 23(1-2):97-104.
Clementi ME, et al.: Alzheimer's amyloid beta-peptide (1-42) induces cell death in human neuroblastoma via bax/bcl-2 ratio increase: an intriguing role for methionine 35. Biochem Biophys Res Commun 2006, 342(1):206-213.
Juul SE, et al.: Erythropoietin and erythropoietin receptor in the developing human central nervous system. Pediatr Res 1998, 43(1):40-49.
Brines ML, et al.: Erythropoietin crosses the blood-brain barrier to protect against experimental brain injury. Proc Natl Acad Sci USA 2000, 97(19):10526-10531.
Martinez-Estrada OM, et al.: Erythropoietin protects the in vitro blood-brain barrier against VEGF-induced permeability. Eur J Neurosci 2003, 18(9):2538-2544.
Konishi Y, et al.: Trophic effect of erythropoietin and other hematopoietic factors on central cholinergic neurons in vitro and in vivo. Brain Res 1993, 609(1-2):29-35.
Ehrenreich H, et al.: Erythropoietin: novel approaches to neuroprotection in human brain disease. Metab Brain Dis 2004, 19(3-4):195-206.
Lewczuk P, et al.: Survival of hippocampal neurons in culture upon hypoxia: effect of erythropoietin. Neuroreport 2000, 11(16):3485-3488.
Kubo T, Nishimura S, Oda T: Amyloid beta-peptide alters the distribution of early endosomes and inhibits phosphorylation of Akt in the presence of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT). Brain Res Mol Brain Res 2002, 106(1-2):94-100.
Iversen LL, et al.: The toxicity in vitro of beta-amyloid protein. Biochem J 1995, 311(Pt 1):1-16.
Pike CJ, et al.: Structure-activity analyses of beta-amyloid peptides: contributions of the beta 25-35 region to aggregation and neurotoxicity. J Neurochem 1995, 64(1):253-265.
Yankner BA, Duffy LK, Kirschner DA: Neurotrophic and neurotoxic effects of amyloid beta protein: reversal by tachykinin neuropeptides. Science 1990, 250(4978):279-282.
D'Ursi AM, et al.: Solution structure of amyloid beta-peptide (25-35) in different media. J Med Chem 2004, 47(17):4231-4238.
Giunta S, et al.: Transferrin neutralization of amyloid beta 25-35 cytotoxicity. Clin Chim Acta 2004, 350(1-2):129-136.
Misiti F, et al.: Abeta(31-35) peptide induce apoptosis in PC 12 cells: contrast with Abeta(25-35) peptide and examination of underlying mechanisms. Neurochem Int 2005, 46(7):575-583.
Kosuge Y, et al.: Comparative study of endoplasmic reticulum stress-induced neuronal death in rat cultured hippocampal and cerebellar granule neurons. Neurochem Int 2006, 49(3):285-293.
Cummings MC, Winterford CM, Walker NI: Apoptosis. Am J Surg Pathol 1997, 21(1):88-101.
Yang J, et al.: Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science 1997, 275(5303):1129-1132.
Tsujimoto Y: Role of Bcl-2 family proteins in apoptosis: apoptosomes or mitochondria? Genes Cells 1998, 3(11):697-707.
Zhong LT, et al.: bcl-2 inhibits death of central neural cells induced by multiple agents. Proc Natl Acad Sci USA 1993, 90(10):4533-4537.
Wolter KG, et al.: Movement of Bax from the cytosol to mitochondria during apoptosis. J Cell Biol 1997, 139(5):1281-1292.
Grutter MG: Caspases: key players in programmed cell death. Curr Opin Struct Biol 2000, 10(6):649-655.
Wang XJ, Xu JX: Salvianic acid A protects human neuroblastoma SH-SY5Y cells against MPP + -induced cytotoxicity. Neurosci Res 2005, 51(2):129-138.
Lee MK, et al.: Resveratrol protects SH-SY5Y neuroblastoma cells from apoptosis induced by dopamine. Exp Mol Med 2007, 39(3):376-384.
Stadelmann C, et al.: Activation of caspase-3 in single neurons and autophagic granules of granulovacuolar degeneration in Alzheimer's disease. Evidence for apoptotic cell death. Am J Pathol 1999, 155(5):1459-1466.
Awasthi A, Matsunaga Y, Yamada T: Amyloid-beta causes apoptosis of neuronal cells via caspase cascade, which can be prevented by amyloid-beta-derived short peptides. Exp Neurol 2005, 196(2):282-289.
Alvarez-Gonzalez R, et al.: Selective loss of poly(ADP-ribose) and the 85-kDa fragment of poly(ADP-ribose) polymerase in nucleoli during alkylation-induced apoptosis of HeLa cells. J Biol Chem 1999, 274(45):32122-32126.
Lee DH, Park T, Kim HW: Induction of apoptosis by disturbing mitochondrial-membrane potential and cleaving PARP in Jurkat T cells through treatment with acetoxyscirpenol mycotoxins. Biol Pharm Bull 2006, 29(4):648-654.
Oliver FJ, et al.: Importance of poly(ADP-ribose) polymerase and its cleavage in apoptosis. Lesson from an uncleavable mutant. J Biol Chem 1998, 273(50):33533-33539.
Damen JE, et al.: Tyrosine 343 in the erythropoietin receptor positively regulates erythropoietin-induced cell proliferation and Stat5 activation. Embo J 1995, 14(22):5557-5568.
Miura O, et al.: Induction of tyrosine phosphorylation by the erythropoietin receptor correlates with mitogenesis. Mol Cell Biol 1991, 11(10):4895-4902.
Joneja B, Wojchowski DM: Mitogenic signaling and inhibition of apoptosis via the erythropoietin receptor Box-1 domain. J Biol Chem 1997, 272(17):11176-11184.
Sakamoto H, Kitamura T, Yoshimura A: Mitogen-activated protein kinase plays an essential role in the erythropoietin-dependent proliferation of CTLL-2 cells. J Biol Chem 2000, 275(46):35857-35862.
Valk P, et al.: Enhancement of erythropoietin-stimulated cell proliferation by Anandamide correlates with increased activation of the mitogen-activated protein kinases ERK1 and ERK2. Hematol J 2000, 1(4):254-263.
Acknowledgements
This study was supported by grants from the State Key Basic Research Program (No.2010CB945200), Shanghai Key Discipline Program (No.S30202), Shanghai Natural Scientific Fund (No. 09JC1416402,09ZR1419100), Program for Outstanding Medical Academic Leader (No. LJ 06003), and Shanghai Jiao Tong University Medical and Engineering Joint Key Project (No. YG2010ZD102).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
Z-KS and H-QY made substantial contributions to conception and design, acquisition of data, and analysis and involved in drafting the manuscript. Z-QW and JP participated in the design of the study and performed the statistical analysis. ZH made interpretation of data and involved in revising it critically for important intellectual content. S-DC were the general supervision of the research group, acquisition of funding, and involved in revising it critically for important intellectual content. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Zhi-Kun, S., Hong-Qi, Y., Zhi-Quan, W. et al. Erythropoietin prevents PC12 cells from beta-amyloid-induced apoptosis via PI3K⁄Akt pathway. Transl Neurodegener 1, 7 (2012). https://doi.org/10.1186/2047-9158-1-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/2047-9158-1-7