
Abstract
Background
Tumor-related methylated DNA and circulating tumor cells (CTC) in the peripheral blood might be of prognostic importance in breast cancer. Thus, the aim of our study was to examine free methylated DNA and CTC in the blood from breast cancer patients and to correlate it with clinicopathological features known to influence prognosis.
Materials and methods
We prospectively obtained serum samples from 85 patients with breast cancer and 22 healthy volunteers. Sera were analysed by methylation specific PCR (MethyLight PCR) for five genes: adenomatous polyposis coli (APC), ras association domain family protein 1A (RASSF1A), estrogen receptor 1 (ESR1), CDKN2A (p16) and glutathione s-transferase pi 1 (GSTP1). Beta actin (ACTB) served as control. In parallel matched peripheral blood of 63 patients was used to assay for circulating tumor cells in the peripheral blood by a modified immunomagnetic AdnaTest BreastCancerSelect with PCR detection for EPCAM, MUC1, MGB1 and SPDEF.
Results
We found a hypermethylation in the APC gene in 29% (25/85), in RASSF1A in 26% (22/85), in GSTP1 in 18% (14/76) and in ESR1 in 38% (32/85) of all breast cancer patients. No hypermethylation of CDKN2A was found (0/25). Blood samples of patients were defined CTC positive by detecting the EPCAM 13% (8/63), MUC1 16% (10/63), MGB 9% (5/55), SPDEF 12% (7/58) and in 27% detecting one or more genes (15/55). A significant difference was seen in methylated APC DNA between cancer patients and healthy volunteers. Moreover, methylated APC, RASSF1 and CTC were significantly different in metastatic versus non-metastatic disease. In addition, the presence of methylated APC, RASSF1A and CTC correlated significantly with AJCC-staging (p = 0.001, p = 0.031 and 0.002, respectively). High incidences of methylations were found for the genes RASSF1 and ESR1 in healthy individuals (both 23% 5/22). Methylated GSTP1 was predominantly found in the serum of patients with large primaries (p = 0.023) and was highly significantly correlated with positive Her2/neu status (p = 0.003). Elevated serum CA15.3 was strongly correlated with methylated APC and CTC detection (both p = 0.000). Methylated ESR1 failed to exhibit significant correlations with any of the above mentioned parameters. The presence of CTC in peripheral blood was significantly associated with methylated APC (p = 0.012) and methylated GSTP1 (p = 0.001).
Conclusion
The detection of methylated APC and GSTP1 DNA in serum correlated with the presence of CTC in the blood of breast cancer patients. Both methylated DNA and CTC correlated with a more aggressive tumor biology and advanced disease.
Similar content being viewed by others

Introduction
Haematogenous dissemination of tumor cells is the main mechanism for distant metastasis and the leading cause of cancer-related death in breast cancer (BC). However, early spread of tumor cells usually remains undetected even by high-resolution imaging technologies. Currently, it is still not possible to accurately identify BC patients with a high potential for a metastatic disease. Therefore, the assessment of individual risk factors remains of great importance and could help in the decision support for a more tailored treatment approach in the near future. As such, the development of new molecular staging methods enabling systemic tumor cell spreading might represent a highly desirable approach for individual tumor therapy [1]. Tumor cells entering the circulation depend on the organ microenvironment in order to be able to colonize tissues and to proliferate [2–6]. One of the promising markers for a current risk classification system is the presence of circulating tumor cells (CTC) in the blood of tumor patients [7]. Braun et al. [8] showed that the presence of micrometastasis in the bone marrow at the time of diagnosis of BC is an independent predictor of poor prognosis. Also Benoy et al. [9] described the presence of CK19+ disseminated tumor cells in the bone marrow by reverse transcriptase-PCR as an independent prognostic factor in untreated patients with BC [9–11]. In other studies the persistence of CTC after therapy showed a strong association with prognosis, particularly in women with hormone receptor-negative disease and adjuvant therapy [7].
Altered gene expression is held responsible for a transformed behaviour of tumor tissue [12–15] and may distinguish tumor from healthy cells [16, 17]. However, it remains unclear which genes allow a specific detection of malignancy. Epigenetic modifications, such as DNA methylation, are one of the most common molecular alterations in human neoplasia [18, 19]. DNA methylation refers to the addition of a methyl group to the cytosine ring of those cytosines that precede a guanosine (referred to as CpG dinucleotides) to form 5-methylcytosine. CpG dinucleotides are found at increased frequency in the promoter region of many genes, and their methylation is frequently associated with gene silencing [20]. Of clinical interest in the view of molecular diagnosis and prognosis is the fact, that several studies have shown tumor-specific epigenetic alterations in the DNA recovered from plasma or serum of patients with various malignancies [21–23]. Additionally, increased concentrations of free DNA are detected in blood of several cancer patients, whereas only small amounts of free circulating DNA are found in healthy individuals [24]. Gal et al. [25] reported about an average of approximately four times more free DNA in the serum of breast cancer patients, compared to healthy individuals. Hypermethylation of ras association domain family protein 1A (RASSF1A), adenomatous polyposis coli (APC) and/or ESR1 identified in serum DNA from breast cancer patients was found to be associated with a worse outcome [26]. Furthermore, methylated RASSF1A and neurogenic differentiationof gene promoters in serum are candidate biomarkers for monitoring the efficacy of adjuvant therapy in breast cancer patients [27, 28]. Although methylated GSTP1 DNA is predominately reported as a marker of prostate cancer, Papadopoulu et al. [29] demonstrated its predictional impact also in breast cancer. The parallel analysis of different methylated markers takes into account the inter- individual variations of gene expression and methylation.
However, the mechanism of releasing DNA into the bloodstream remains unclear. There are hypothesis of lysis of CTC, DNA leakage from cells as the result of tumor necrosis or apoptosis, or spontaneous release of DNA into the circulation from primary and metastatic tumors [30].
The aim of this study was to validate tumor-specific epigenetic alterations in the cell-free DNA found in the peripheral blood of breast cancer patients and to assess whether a correlation exists between tumor-specific methylated DNA, CTC and the clinical status of patients diagnosed with breast cancer. A combined molecular assessment of the circulating DNA as well as CTC might help to improve the evaluation of cancer stage and overall prognosis in breast cancer. Thus, we examined the tumor associated APC, RASSF1, GSTP1, ESR1 genes, while ACTB served as a control. Due to the fact that elevated levels of methylated DNA have mainly been reported in women with advanced disease, we decided to investigate these molecular markers in a patient collective with predominate progressive tumors.
Materials and methods
Patients and sample collection
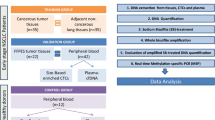
We prospectively obtained serum samples from 86 patients with breast cancer and 23 healthy volunteers and matched peripheral blood from 63 patients. All patients gave informed consent for the study and the examination of blood samples was carried out after approval from the Institutional Review Board of the University of Düsseldorf, Germany.
Blood samples from all patients were taken during the course of radiotherapy. The patient population consisted of 65 patients with primary adjuvant therapy and 21 patients with recurrency and secondary adjuvant treatment. The clinico-pathological variables are listed in Table 1. Median age of the control population was 44 (range, 24-61) years and 60 (range, 34-81) years in the breast cancer population.
Isolation of serum DNA
Blood (4 ml) from each donor was collected in serum separator tubes (Becton Dickinson) and centrifuged (2000 g, 15 min) at room temperature. Then, serum was aliquotted and cryopreserved at -80°C until use. For both, normal sera and cancer sera analysis 1 ml serum was used for DNA extraction. The genomic DNA from serum samples was extracted using the UltraSens VirusKit (QIAGEN, Hilden, Germany) according to a modified manufacturer's protocol. After the Proteinase-K digestion the DNA extract was reloaded a second time on the DNA extraction columns. For DNA elution 65 μl of AVE buffer (Qiagen, Hilden, Germany) were added and centrifuged for 1 min at 6,000 × g. A second 65 μl volume AVE was added onto the column and both eluates were pooled. DNA was quantified in triplicate by a real-time quantitative PCR assay "on demand" for the RNase-P gene and compared to RT-PCR reactions with three-fold serial dilutions of known concentrations of DNA as template (Applied Biosystems, Foster City, CA, USA).
Analysis of methylated DNA
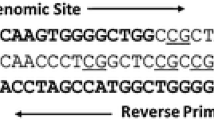
Extracted DNA samples were subjected to sodium bisulphite conversion using the CpGenome™ DNA Modification Kit (CHEMICON, USA). Sodium bisulphite-converted DNA was extracted in 100 μl TE-buffer analysed by a methylation specific real-time PCR, MethyLight (Eads 2000 Nucleic Acid Research). Per Gene one set of primers and probe, designed specifically for methylated, sodium bisulphite-converted DNA, were used. A methylation inspecific ACTB probe (lacking CpG dinucleotides) was used for control of quality and bisulphite conversion. Fluorogenic (FAM/TAMRA) probes and PCR primer sets for APC, RASSF1A, GSTP1, ESR1, CDKN2A, and ACTB were custom synthesised by Eurogentec (Köln, Germany) according to previous published sequences [26, 31]. Fluorogenic PCRs were carried out in a reaction volume of 15 μl in 384-well plates in a 7900HT Sequence Detector (Applied Biosystems). PCR was carried out in separate wells for each primer/probe set, and each sample was run in triplicate. The final reaction mixture consisted of 600 nmol of each primer, 200 nmol of probe and 7,5 μl of 2×Universal Master Mix (Applied Biosystems). 4 μl of the treated DNA solution was used in each real-time reaction. Standard thermal RT-PCR cycling started with a first denaturation step of 95°C for 10 min. The amplification profile for the PCR was 95°C for 15 s and 60°C for 1 min. Data obtained during 50 cycles of amplification were analysed. Bisulphite modified DNA isolated from normal peripheral lymphocytes (PBL-DNA) served as a negative control and a previously M.Sss1-yMethyltransferase methylated PBL-DNA (New England Biolabs, Frankfurt a.M., Germany) was used as the methylation positive control. A gene was deemed methylated if the threshold cycle number for at least one of three reactions was below 40 Ct or cycle number for at least two reactions were below 45 Ct.
Analysis of CTC in Peripheral Blood (Modified AdnaGen-Protocol)
Blood (5 ml) samples were taken using EDTA collection tubes and immediately placed on ice and further preceded within 2 hours. For each donor, a modified AdnaTest Breast Cancer Select/Detect technique was used on one blood sample, according to the manufacturer's instructions. AdnaTest Breast Cancer Select BreastSelect Beads (AdnaGen, Langenhagen, Germany) (100 μl) were added to 5 ml of blood and incubated for 120 min at room temperature (5 r.p.m.). After incubation, cells were repeatedly washed with PBS and lysed by adding a Lysis/Binding buffer (AdnaGen). The supernatant was recovered.
AdnaTest Breast Cancer Detect mRNA was subsequently separated by a magnetic unit using Oligo(dT)25 Dynabeads. The total mRNA/bead mixture (29.5 μl) was reverse transcribed using 0.5 μl of RNase inhibitor (40 U/μl; recombinant RNAsin, Promega, Mannheim, Deutschland) 4 μl of RT buffer, 4 μl of dNTPs and 2 μl of Reverse Transcriptase (Sensiscript Transcription Kit, Qiagen GmbH, Hilden, Deutschland). Reverse transcription was performed in a one-step reaction (60 min at 37°C, 5 min at 93°C). The mixture was then chilled down on ice and stored at -20°C. For the analysis of tumour-associated mRNAs, a multiplex PCR and a RT-PCR was carried out.
Multiplex PCR
The primer mixture consisted of four specific primer pairs for the amplification of three tumour markers (Muc-1, HER2 and EPCAM) and one housekeeping gene (Actin). Multiplex PCR analyses were carried out in a final volume of 50 μl PCR mixture, containing 8 μl of cDNA, 4 μl primer mixtures (PrimerMix BreastDetect; AdnaGen), 25 μl of Hot Star Taq Master Mix (Qiagen) and 13 μl of distilled water. PCR analyses were performed as described by the manufacturer. Negative controls (no template control) and a diluted mixture of PCR-Amplicons (part of AdnaGen Detect KIT) were included within each daily setup. PCR amplicons were evaluated at AdnaGen LAB on a DNA 1000 LabChip BioAnalyzer 2100 (Agilent Technologies GmbH, Böblingen, Deutschland). Samples without an internal positive Actin Signal were excluded from further analyses. One gene was considered as positively detected if the amplicon concentration was higher 0,1 ng/μl according to the manufacturer's validation. HER2 signals were excluded from analysis because of high unspecific incidence in healthy donor reactions (data not shown).
RT-PCR
Two further genes were included in analysis by separate RT-PCR reactions on the 7900 HT Sequence Detector (Applied Biosystems). The same reaction conditions were used as for methyl specific PCR described above. Instead of customer designed methylation specific probes, predesigned cDNA specific expression probe sets were used. The genes were SCGB2A2 Hs00267190_m1 (MGB1) and SPDEF Hs00171942_m1 (Assays on Demand, Applied Bio systems); 2 μl of the cDNA from the AdnaGen Detect RT-Reaction were used as template for each gene. Glucuronidase [GUS-B] specific primer probe mixture (Hs99999908_m1) was used as separate control reaction. A gene was deemed RT-PCR positive if the Ct was below 41 cycles.
Statistical analysis
Correlations were checked by Pearson's two-sided χ2-test for dichotomized values. Dichotomisation of pathological markers based on the following thresholds: > 10% cell nuclei stained at least weakly positive for ER or PR, immunhistochemical Her2/neu score > 2+ and CA15.3 > 32 U/ml. Pearson linear by linear association was used for ordinal recoded variables (AJCC stage). Mann-Whitney U-test was used to compare dichotomized values and non-parametric distributed variables (DNA-level). The level of significance was determined as p < 0.05. All statistical calculations were carried out using SPSS, version 12.0 (SPSS Inc, Chicago, IL, USA).
Results
Total DNA concentrations in plasma from healthy volunteers and breast cancer patients
In our study differences between total plasma DNA concentrations in BC patients and the controls reached no statistical significance (p = 0.79). There was also no difference in plasma DNA concentrations between primary and recurrent tumor patients (p = 0.37).
Plasma DNA concentrations failed to associate with tumor-related methylated genes, circulating tumor cells, estrogen- and progesteron receptor status, ERBB2 and clinical status (TNM, Whitney U-Test).
Tumor-Related methylated DNA in serum
In total 29% of the BC patients showed a hypermethylated APC gene in comparison to 9% in the control group (p = 0.050). This was also due when APC was compared between primary and recurrent disease (p = 0.006). Hypermethylated DNA was detected in 26% of the BC patients for the RASSF1 gene, in 18% for the GSTP1 and in 38% for the ESR1 gene with no significant differences compared to the healthy group. The CDKN2A gene was only methylated in one single volunteer in the control group, which prompted us to exclude it from further examinations.
Methylated DNA concentrations tended to be correlated with tumor progression (Table 1). Statistically significant correlations were observed between T1+T2 vs. T3+T4 for the genes RASSF1 (p = 0.029), GSTP1 (p = 0.000), and in the combination of at least one of the genes (APC, RASSF1A, GSTP1) methylated (Min-1, p = 0,010) and at least two genes (APC, RASSF1A, GSTP1) methylated (Min-2, p = 0.004).
Unexpectedly low correlations were found when methylated DNA status was compared with lymph node status, tumor grading, estrogen- and progesterone receptor status. However, methylated DNA concentrations in APC (p = 0.002) and RASSF1 (p = 0.046) significantly correlated with metastatic disease. GSTP1 (p = 0.003) and ESR1 (p = 0.024) methylation correlated strongly with the Her-2/neu-status, whereas APC methylation correlated with the Ca-15.3 (p = 0.000) tumormarker level. RASSF1 showed a tendency to a more aberrant methylation in older compared to younger participants (p = 0.089). A high concordance was observed between the methylation of APC and RASSF1 and the TNM combining AJCC-Index. Table 2 shows the correlation between the hypermetylated genes, which was strongest between APC and RASSF1A (p = 0.000), while GST1 gave a more independent information. No significant associations were found between the hypermethylation of ESR1 and GST1.
Detection of circulation tumor cells
Due to the relative low incidence of single epithelial marker gene detection in immunomagnetic enriched cell fraction [13% (8/63) for EPCAM, 16% (10/63) for MUC1, 9% (5/55) for MGB1, and 13% (8/63) for SPDEF] two indices were created. The first index (CTC-2), combines the AdnaGen marker EPCAM and/or MUC1 and another combining all four described epithelial markers (CTC-4). (The Her2/neu marker, which is a third marker in the AndaDetect multiplex PCR, was also positive in 13% (8/63 BC) cases, but was excluded from analyses because of high incidence > 50% in healthy controls. 19% (12/63) of BC were positive in the two gene set and 27% (15/55) of BC were positive in the four gene index. As was observed with methylated DNA in serum, the two marker index CTC-2 showed high correlation to the higher risk groups (high AJCC-Index p = 0.002, pM+ p = 0.009, CA15.3 positive p = 0.000 and progressive disease p = 0.050). Moreover, pN+ incidences showed increases CTC positivities (p = 0.031). The four marker index CTC-4 displayed similar correlations. The addition of SPDEF and MGB1 further improved the contribution to high risk BC detection without increasing the rate of false positives. However, a false positive background in the healthy controls was observed in CTC- as well as methylated DNA detection methods.
Association of CTC and serum methylated DNA
The detection of a methylated gene in the serum DNA was significantly associated with the detection of CTC in peripheral blood. The methylated APC and GSTP1 genes were correlated with each of the four CTC parameters, with the exception of the MGB1 marker, which was only positively detected in 5 cases. EPCAM showed the highest association to methylation. Over 70% of the patients with EPCAM gene methylation were additionally positive detected for APC or GSTP1 methylation (both p = 0.001). Furthermore, both methylated genes were highly associated with the CTC marker combining indicies. However, the methylated genes RASSF1 and ESR1 failed to display significant correlation with the CTC detection, but a correlation to EPCAM was at least marginal significant (p = 0.094, p = 0.087 respectively).
Combining of parameters
Combining the different targets of methylated DNA into one score better takes into account the tumor diversity. Thus APC, RASSF1 and GSTP1 were combined to a Min-1 score. To overcome the high inspecifity rate, a second score Min-2, requiring two hypermethylations in the APC, RASSF1 and GSTP1 set was recoded. Methylated ESR1 was left out, because of the high incidence rate in healthy donors and its independence from clinical features. The association of both scores with most clinicopathological features was significant. The Min-1 score was deemed positive in 48% of the BC patients. In healthy volunteers it was detected in 38%. The more stringent index Min-2 was positive in 20% of BC and was highly associated with tumor aggressiveness, as indicated by the high correlation with increased Her2/neu, Ca-15.3, tumor size, M-state and AJCC index. The Min-1 positivity rate was generally twice as high in the more aggressive clinic groups (82% in T3+T4 vs. 39% in T1+T2; 67% in M+ vs. 34% in M0; 73% in recurrent cases vs. 39% in first diagnosis. The more stringent Min-2 score was positive in 20% of overall BC, but positive in 42% of the AJCC-IV high risk group vs. 10% in the lower groups. The incidence was also significantly six times increased from 7% in the low Her2 group to 41% in the Her2 positive group (p = 0.001). The combined methylation scores failed to reach significance when compared with the CTC detection, with the exception of the CTC marker EPCAM (Min-1 p = 0.024, Min-2 p = 0.005). We further tested one COMBI score, which consists of the stringent methylation score Min2 and the CTC score CTC-2 (EPCAM or MUC1). This COMBI-Score was positive in 35% of all BC cases. 89% of the CA15.3 positive patients turned out to be also positive for the COMBI score, while the lower risk CA15.3 negative group was only COMBI positive in 21% of all cases (p = 0.000). The incidence of the COMBI Score was even elevated in the ER negative group (50% vs. 28%) in the ER positive group (p = 0.182) and elevated in the Her2 group > 2+ (47% vs 24%, p = 0.088). But these associations failed to reach significance. Table 3 lists the significant correlations of COMBI with most clinic parameters.
Discussion
Haematological CTC are a prerequisite to distant organ metastasis in many cancer entities [7, 32–34]. Several studies suggest that only a minority of tumor cells have the ability to invade and metastasize [35–37]. Therefore, it is crucial to investigate which genes ultimately facilitate tissue invasion and disease progression.
Circulating DNA could be observed in plasma of healthy persons but was increased in cancer patients [24]. This knowledge has attracted much attention to the potential use of circulating free DNA as a tumor marker. In our study, we measured the level of circulating DNA in the serum of healthy volunteers and patients with localised or metastatic breast cancer using real-time quantitative PCR. Interestingly, DNA concentrations in BC patients and the controls reached no statistical significant difference. There was also no difference in serum DNA concentrations between primary and recurrent tumor patients. This is opposed to the results by Auwera et al., which found a 3.25-fold difference in the median levels of circulating total DNA in plasma between breast cancer patients and healthy controls [38]. Methylated DNA markers are attractive tumor markers in blood and are frequently found in a wide range of human cancers but not (or rarely) in healthy controls 22-23, 39-40. They are not limited to patients with metastatic cancer but are also present in body fluid from patients with early or organ-confined tumors 23, 41. In general, a high concordance between epigenetic alterations in primary tumor specimens and methylated DNA in blood has been reported [42–45].
APC, RASSF1A and ESR1 are known to be frequently hypermethylated in breast cancer [19]. In addition we examined GSTP1 and CDKN2A to provide a more versatile target for detection in serum [46]. Similary to Müller et al. [26] we measured methylation levels of these genes in serum instead of plasma. Except CDKN2A, BC patients showed hypermethylation of the genes. Whereas CDKN2A has been reported earlier as one of the most methylated genes in breast cancer patients, we did not find any CDKN2A methylation in serum of our study group [47, 48, 19]. Müller et al. [26] did also include CDKN2A in an evaluation set of 10 recurrent breast cancer sera and failed positive detection in all 10 cases. ESR1 showed the highest frequency of hypermethylation with 38% and was followed by APC (29%), RASSF1A (26%) GSTP1 (18%) and CDKN2A (0%). However, methylation was also detected in a smaller proportion in the control persons. In this healthy donor group ESR1 and RASSF1A showed 23% of hypermethylation, followed by APC (9%) and GSTP1 (6%). So the frequency of methylation in serum DNA of RASSF1A, GSTP1 and ESR1 was increased among women with breast cancer as compared to controls, but this tendency reached only significance for APC 29% vs. 9% (p = 0.050). Auwera et al. [38] found very similar data for APC. ESR1 also failed to reach tumor specific significance in their study. The incidence of RASSF1A methylation in our control group seems quite untypical compared with studies of Müller [26, 31], but in our study RASFF1A displays and emphasizes its tumor related importance by a significant correlation to AJCC Index (p = 0.031), tumor size (p = 0.029) and metastasis p = 0.046. Auwera et al. [38] examined two marker expressions and found APC an RASSF1A to be the most frequent detected combination. We found identical methylation status for both markers in 71% of all cases (p = 0.000). Müller et al. [31] found a hypermethylation of RASSF1A, APC and ESR1 in a test set of extended tumor disease (24 patients) but APC and ESR1 lost univariate significance in a trainigsset (62 patients).
Another association was suspected between methylation of specific genes in serum DNA and HR status. Widschwendter et al. [19] reported significant differences in the HR status between clusters of DNA methylation profiles. In particular, the methylation of RASSF1A in breast cancer has already been associated to hormone regulation. Feng et al. [49] reported a methylation of RASSF1A with a strong correlation of estrogen, progesteron and HR expression. They suspected whether methylation of growth-suppressor genes would correlate with the expression of estrogen and progesteron. Moreover, Sunami et al.50 reported on a hypermethylation of RASSF1A and a positive estrogen status.
In contrast, we found serum DNA hypermethylation of all genes examined to be independent of the estrogen- and progesteron receptor status. This might be due to the huge proportion of progressive tumors in our collective, because Sunami et al-found the receptor dependence of epigenetic alterations predominately in early stages (T0/T1) of tumor progression [50].
In our study hypermethylation of GSTP1 and ESR1 as well as the combinations Min-1 or Min-2 showed a highly significant correlation with the Her2/neu-status. GSTP1 methylation and Her2/neu expression was previous examined by Shinozaki et al. [51]. GSTP1 hypermethylation was more frequent in the lymph node metastasis positive group than in the negative group and GSTP1 showed the highest correlation to positive Her2/neu status (p = 0.0019).
In line with these results Jhaveri and Morrow reported about hypermethylation of GSTP1 and a negative estrogen status [52]. However, this is different to the studies reported above.
The biomarker CA15.3 is a glycoprotein which can be observed in peripheral blood of breast cancer patients. Although in patients with primary BC the concentration of CA15.3 can be within normal range, increased levels of CA15.3 are often observed in patients with metastatic disease. In our study CA15.3 showed a significant correlation to hypermethylation in particular with the APC gene (p = 0.000) while the other genes failed to show correlation with Ca15.3. High correlation of CA15.3 was also found by Auwera et al. [38] (p = 0.001). Controversially to our study they reported on a similar high correlation of CA15.3 and RASSF1A (p < 0.001), but like in our study hypermethylation of ESR1 was independent from Ca15.3 marker. Nearly all patients with elevated Ca.15.3 were positive for CTC (7/8), p = 0.000). As selected methylation markers, we found positive CTC markers predominately present in patients with positive lymph nodes, metastatic tumors progressive disease and high AJCC Index. Similar to hypermethylated DNA in serum, CTC detection is not exclusively specific to breast cancer. In peripheral blood variable numbers of epithelial cells have been found to be related to benign epithelial proliferative diseases, inflammation, tissue trauma and/or surgical interventions [52–54]. Therefore cell search systems are exposed for false positive results using immuno-mediated CTC detection techniques which can occur by specific labelling of non-tumor epithelial cells or non-specific labelling of non-tumor non-epithelial cells [55]. Further, the PCR-based tumor associated gene detection used in this study might interfere with background gene expression in nearly silenced, but leaky genes.
Up to date, there is no agreement on the mechanisms responsible for the presence of tumor DNA in peripheral blood. However, patients with CTC in peripheral blood had significantly higher total DNA levels in plasma than patients with no CTC. Although plasma or serum total DNA is measured in many studies, it has never reached acceptance level as a specific tumor marker. The more tumor specific, or at least tumor associated feature of genetic or epigenetic DNA modification might display more tumor accordance.
Especially hypermethylated APC and GSTP1 were significantly correlated with the CTC detection in peripheral blood. In contrast to Auwera et al. [38] we found RASSF1 not associated with CTC. The failure to detect methylated CDKN2A in 25 BC sera might be interpreted as a circumstantial evidence, that CTC do not form the source for free methylated serum DNA, because it is methylated in a huge proportion of breast tumors and should be found to a high extend in BC sera. Schwarzenbach et al. [56] did not observe a correlation between the presence of CTC and the loss of heterozygosity in circulating DNA in the blood from breast cancer patients But these deviations might be related to a failure in methodological standardization, which is not easily realizable in rare cell detection and methylation analysis or is related to the low patient number.
Despite individual differences in the observed correlation between CTC and circulating methylated DNA, the correlation could be interpreted as follows: A) CTC are a potential source of circulating tumor-specific DNA or B) CTC as a parallel maker to free tumor DNA. But independent of causal courses, both makers secondarily correlate with a phenotypic feature of a more aggressive tumor biology. However, the prognostic importance of CTC and serum methylated DNA could not be determined at this time. Therefore, large-scale studies are necessary to validate the clinical utility of these methods.
Abbreviations
- BC:
-
breast cancer
- CTC:
-
circulating tumor cells
- APC:
-
adenomatous polyposis coli
- ESR1:
-
estrogen receptor 1
- RASSF1A:
-
ras association domain family protein 1A
- GSTP1:
-
gluthation-s-transferase pi 1
- ACTB:
-
beta actin
- MUC1:
-
Mucin 1
- CA15.3:
-
Cancer Antigen 15.3 (: MUC1)
- MGB1:
-
Mammoglobin 1 (: SCGB2A2)
- EPCAM:
-
epithelial cell adhesion molecule
- CDKN2A:
-
cyclin-dependent kinase inhibitor 2A (: p16)
- SPDEF:
-
SAM pointed domain containing ets transcription factor
- Her-2/neu :
-
ERBB2 human epidermal growth receptor 2
- FD:
-
first diagnosis
- RD:
-
recurrence disease
- HR:
-
hormone receptor status
- Min-1:
-
at least one of the gene-complex (APC, RASSF1A, GSTP1) methylated
- Min-2:
-
at least two of the gene-complex (APC, RASSF1A, GSTP1) methylated
- Ct:
-
RT-PCR Cycle threshold value.
References
Pantel K, Cote RJ, Fodstad O: Detection and clinical importance of micrometastatic disease. Journal of the National Cancer Institute 1999,91(13):1113–24. 10.1093/jnci/91.13.1113
Welch DR, Steeg PS, Rinker-Schaeffer CW: Molecular biology of breast cancer metastasis. Genetic regulation of human breast carcinoma metastasis. Breast Cancer Res 2000,2(6):408–16. 10.1186/bcr87
Cho SY, Choi HY: Causes of death and metastatic patterns in patients with mammary cancer. Ten-year autopsy study. American journal of clinical pathology 1980,73(2):232–4.
Lee YT: Breast carcinoma: pattern of metastasis at autopsy. Journal of surgical oncology 1983,23(3):175–80. 10.1002/jso.2930230311
Mochizuki S, Umemura S, Tokuda Y, Tajima T, Mitomi T, Osamura RY: A study of 46 cumulative breast cancer autopsy cases. The Tokai journal of experimental and clinical medicine 1997,22(1):19–25.
Gerber PA, Hippe A, Buhren BA, Muller A, Homey B: Chemokines in tumor-associated angiogenesis. Biological chemistry 2009,390(12):1213–23. 10.1515/BC.2009.144
Bolke E, Orth K, Gerber PA, Lammering G, Mota R, Peiper M, et al.: Gene expression of circulating tumour cells in breast cancer patients. Eur J Med Res 2009,14(10):426–32.
Braun S, Vogl FD, Naume B, Janni W, Osborne MP, Coombes RC, et al.: A pooled analysis of bone marrow micrometastasis in breast cancer. The New England journal of medicine 2005,353(8):793–802. 10.1056/NEJMoa050434
Benoy IH, Elst H, Philips M, Wuyts H, Van Dam P, Scharpe S, et al.: Prognostic significance of disseminated tumor cells as detected by quantitative real-time reverse-transcriptase polymerase chain reaction in patients with breast cancer. Clinical breast cancer 2006,7(2):146–52. 10.3816/CBC.2006.n.024
Cristofanilli M, Budd GT, Ellis MJ, Stopeck A, Matera J, Miller MC, et al.: Circulating tumor cells, disease progression, and survival in metastatic breast cancer. The New England journal of medicine 2004,351(8):781–91. 10.1056/NEJMoa040766
Cristofanilli M, Hayes DF, Budd GT, Ellis MJ, Stopeck A, Reuben JM, et al.: Circulating tumor cells: a novel prognostic factor for newly diagnosed metastatic breast cancer. J Clin Oncol 2005,23(7):1420–30. 10.1200/JCO.2005.08.140
Siegel PM, Dankort DL, Hardy WR, Muller WJ: Novel activating mutations in the neu proto-oncogene involved in induction of mammary tumors. Molecular and cellular biology 1994,14(11):7068–77.
Siegel PM, Ryan ED, Cardiff RD, Muller WJ: Elevated expression of activated forms of Neu/ErbB-2 and ErbB-3 are involved in the induction of mammary tumors in transgenic mice: implications for human breast cancer. The EMBO journal 1999,18(8):2149–64. 10.1093/emboj/18.8.2149
Oh JJ, Grosshans DR, Wong SG, Slamon DJ: Identification of differentially expressed genes associated with Her-2/ neu overexpression in human breast cancer cells. Nucleic acids research 1999,27(20):4008–17. 10.1093/nar/27.20.4008
Gendler SJ, Lancaster CA, Taylor-Papadimitriou J, Duhig T, Peat N, Burchell J, et al.: Molecular cloning and expression of human tumor-associated polymorphic epithelial mucin. The Journal of biological chemistry 1990,265(25):15286–93.
Eltahir EM, Mallinson DS, Birnie GD, Hagan C, George WD, Purushotham AD: Putative markers for the detection of breast carcinoma cells in blood. British journal of cancer 1998,77(8):1203–7. 10.1038/bjc.1998.203
Lambrechts AC, van't Veer LJ, Rodenhuis S: The detection of minimal numbers of contaminating epithelial tumor cells in blood or bone marrow: use, limitations and future of RNA-based methods. Ann Oncol 1998,9(12):1269–76. 10.1023/A:1008445604263
Egger G, Liang G, Aparicio A, Jones PA: Epigenetics in human disease and prospects for epigenetic therapy. Nature 2004,429(6990):457–63. 10.1038/nature02625
Widschwendter M, Jones PA: DNA methylation and breast carcinogenesis. Oncogene 2002,21(35):5462–82. 10.1038/sj.onc.1205606
Leonhardt H, Cardoso MC: DNA methylation, nuclear structure, gene expression and cancer. Journal of cellular biochemistry 2000, (Suppl 35):78–83.
Wong IH, Lo YM, Zhang J, Liew CT, Ng MH, Wong N, et al.: Detection of aberrant p16 methylation in the plasma and serum of liver cancer patients. Cancer research 1999,59(1):71–3.
Silva JM, Dominguez G, Villanueva MJ, Gonzalez R, Garcia JM, Corbacho C, et al.: Aberrant DNA methylation of the p16INK4a gene in plasma DNA of breast cancer patients. British journal of cancer 1999,80(8):1262–4. 10.1038/sj.bjc.6690495
Sanchez-Cespedes M, Esteller M, Wu L, Nawroz-Danish H, Yoo GH, Koch WM, et al.: Gene promoter hypermethylation in tumors and serum of head and neck cancer patients. Cancer research 2000,60(4):892–5.
Leon SA, Shapiro B, Sklaroff DM, Yaros MJ: Free DNA in the serum of cancer patients and the effect of therapy. Cancer research 1977,37(3):646–50.
Gal S, Fidler C, Lo YM, Taylor M, Han C, Moore J, et al.: Quantitation of circulating DNA in the serum of breast cancer patients by real-time PCR. British journal of cancer 2004,90(6):1211–5. 10.1038/sj.bjc.6601609
Muller HM, Widschwendter A, Fiegl H, Ivarsson L, Goebel G, Perkmann E, et al.: DNA methylation in serum of breast cancer patients: an independent prognostic marker. Cancer research 2003,63(22):7641–5.
Fiegl H, Millinger S, Mueller-Holzner E, Marth C, Ensinger C, Berger A, et al.: Circulating tumor-specific DNA: a marker for monitoring efficacy of adjuvant therapy in cancer patients. Cancer research 2005,65(4):1141–5. 10.1158/0008-5472.CAN-04-2438
Fiegl H, Jones A, Hauser-Kronberger C, Hutarew G, Reitsamer R, Jones RL, et al.: Methylated NEUROD1 promoter is a marker for chemosensitivity in breast cancer. Clin Cancer Res 2008,14(11):3494–502. 10.1158/1078-0432.CCR-07-4557
Papadopoulou E, Davilas E, Sotiriou V, Georgakopoulos E, Georgakopoulou S, Koliopanos A, et al.: Cell-free DNA and RNA in plasma as a new molecular marker for prostate and breast cancer. Annals of the New York Academy of Sciences 2006, 1075: 235–43. 10.1196/annals.1368.032
Stroun M, Maurice P, Vasioukhin V, Lyautey J, Lederrey C, Lefort F, et al.: The origin and mechanism of circulating DNA. Annals of the New York Academy of Sciences 2000, 906: 161–8.
Mueller T, Voigt W, Simon H, Fruehauf A, Bulankin A, Grothey A, et al.: Failure of activation of caspase-9 induces a higher threshold for apoptosis and cisplatin resistance in testicular cancer. Cancer research 2003, 513–21.
Matthaei H, Boelke E, Eisenberger CF, Alldinger I, Krieg A, Schmelzle M, et al.: Interdisciplinary treatment of primary hepatic angiosarcoma: emergency tumor embolization followed by elective surgery. Eur J Med Res 2007,12(12):591–4.
Matthaei H, Bolke E, Schmelzle M, Budach W, Orth K, Engers R, et al.: Modern therapy of rectal carcinoma. Eur J Med Res 2008,13(4):139–46.
Peiper M, Bolke E, Orth K, Hosch SB, Rehders A, Matthaei H, et al.: Current status of radical systematic lymphadenectomy in pancreatic cancer--a review of the literature. Eur J Med Res 2007,12(2):47–53.
Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF: Prospective identification of tumorigenic breast cancer cells. Proceedings of the National Academy of Sciences of the United States of America 2003,100(7):3983–8. 10.1073/pnas.0530291100
Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, et al.: A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 1994,367(6464):645–8. 10.1038/367645a0
Reya T, Morrison SJ, Clarke MF, Weissman IL: Stem cells, cancer, and cancer stem cells. Nature 2001,414(6859):105–11. 10.1038/35102167
Van der Auwera I, Elst HJ, Van Laere SJ, Maes H, Huget P, van Dam P, et al.: The presence of circulating total DNA and methylated genes is associated with circulating tumour cells in blood from breast cancer patients. British journal of cancer 2009,100(8):1277–86. 10.1038/sj.bjc.6605013
Silva JM, Dominguez G, Garcia JM, Gonzalez R, Villanueva MJ, Navarro F, et al.: Presence of tumor DNA in plasma of breast cancer patients: clinicopathological correlations. Cancer research 1999,59(13):3251–6.
Goessl C, Krause H, Muller M, Heicappell R, Schrader M, Sachsinger J, et al.: Fluorescent methylation-specific polymerase chain reaction for DNA-based detection of prostate cancer in bodily fluids. Cancer research 2000,60(21):5941–5.
Esteller M, Sanchez-Cespedes M, Rosell R, Sidransky D, Baylin SB, Herman JG: Detection of aberrant promoter hypermethylation of tumor suppressor genes in serum DNA from non-small cell lung cancer patients. Cancer research 1999,59(1):67–70.
Usadel H, Brabender J, Danenberg KD, Jeronimo C, Harden S, Engles J, et al.: Quanitative adenomatous polyposis coli promoter methylation analysis in tumor tissue, serum, and plasma DNA of patients with lung cancer. Cancer research 2002,62(2):371–5.
Yang HJ, Liu VW, Wang Y, Chan KY, Tsang PC, Khoo US, et al.: Detection of hypermethylated genes in tumor and plasma of cervical cancer patients. Gynecologic oncology 2004,93(2):435–40. 10.1016/j.ygyno.2004.01.039
Topaloglu O, Hoque MO, Tokumaru Y, Lee J, Ratovitski E, Sidransky D, et al.: Detection of promoter hypermethylation of multiple genes in the tumor and bronchoalveolar lavage of patients with lung cancer. Clin Cancer Res 2004,10(7):2284–8. 10.1158/1078-0432.CCR-1111-3
Hoque MO, Begum S, Topaloglu O, Jeronimo C, Mambo E, Westra WH, et al.: Quantitative detection of promoter hypermethylation of multiple genes in the tumor, urine, and serum DNA of patients with renal cancer. Cancer research 2004,64(15):5511–7. 10.1158/0008-5472.CAN-04-0799
Sharma G, Mirza S, Yang YH, Parshad R, Hazrah P, Datta Gupta S, et al.: Prognostic relevance of promoter hypermethylation of multiple genes in breast cancer patients. Cell Oncol 2009,31(6):487–500.
Dahl C, Guldberg P: A ligation assay for multiplex analysis of CpG methylation using bisulfite-treated DNA. Nucleic acids research 2007,35(21):e144. 10.1093/nar/gkm984
Wajed SA, Laird PW, DeMeester TR: DNA methylation: an alternative pathway to cancer. Annals of surgery 2001,234(1):10–20. 10.1097/00000658-200107000-00003
Feng W, Shen L, Wen S, Rosen DG, Jelinek J, Hu X, et al.: Correlation between CpG methylation profiles and hormone receptor status in breast cancers. Breast Cancer Res 2007,9(4):R57. 10.1186/bcr1762
Sunami E, Shinozaki M, Sim MS, Nguyen SL, Vu AT, Giuliano AE, et al.: Estrogen receptor and HER2/neu status affect epigenetic differences of tumor-related genes in primary breast tumors. Breast Cancer Res 2008,10(3):R46. 10.1186/bcr2098
Shinozaki M, Hoon DS, Giuliano AE, Hansen NM, Wang HJ, Turner R, et al.: Distinct hypermethylation profile of primary breast cancer is associated with sentinel lymph node metastasis. Clin Cancer Res 2005,11(6):2156–62. 10.1158/1078-0432.CCR-04-1810
Jhaveri MS, Morrow CS: Methylation-mediated regulation of the glutathione S-transferase P1 gene in human breast cancer cells. Gene 1998,210(1):1–7. 10.1016/S0378-1119(98)00021-3
Goeminne JC, Guillaume T, Symann M: Pitfalls in the detection of disseminated non-hematological tumor cells. Ann Oncol 2000,11(7):785–92. 10.1023/A:1008398228018
Fehm T, Solomayer EF, Meng S, Tucker T, Lane N, Wang J, et al.: Methods for isolating circulating epithelial cells and criteria for their classification as carcinoma cells. Cytotherapy 2005,7(2):171–85. 10.1080/14653240510027082
Paterlini-Brechot P, Benali NL: Circulating tumor cells (CTC) detection: clinical impact and future directions. Cancer letters 2007,253(2):180–204. 10.1016/j.canlet.2006.12.014
Schwarzenbach H, Muller V, Stahmann N, Pantel K: Detection and characterization of circulating microsatellite-DNA in blood of patients with breast cancer. Annals of the New York Academy of Sciences 2004, 1022: 25–32. 10.1196/annals.1318.005
Author information
Authors and Affiliations
Corresponding author
Additional information
C Matuschek, E Bölke, K Orth and H Bojar contributed equally to this work.
Rights and permissions
About this article
Cite this article
Matuschek, C., Bölke, E., Lammering, G. et al. Methylated APC and GSTP1 genes in serum DNA correlate with the presence of circulating blood tumor cells and are associated with a more aggressive and advanced breast cancer disease. Eur J Med Res 15, 277 (2010). https://doi.org/10.1186/2047-783X-15-7-277
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/2047-783X-15-7-277