
Abstract
Polarized vesicle trafficking is mediated by small GTPase proteins, such as Rabs and Arls/Arfs. These proteins have essential roles in maintaining normal cellular function, in part, through regulating intracellular trafficking. Moreover, these families of proteins have recently been implicated in the formation and function of the primary cilium. The primary cilium, which is found on almost every cell type in vertebrates, is an organelle that protrudes from the surface of the cell and functions as a signaling center. Interestingly, it has recently been linked to a variety of human diseases, collectively referred to as ciliopathies. The primary cilium has an exceptionally high density of receptors on its membrane that are important for sensing and transducing extracellular stimuli. Moreover, the primary cilium serves as a separate cellular compartment from the cytosol, providing for unique spatial and temporal regulation of signaling molecules to initiate downstream events. Thus, functional primary cilia are essential for normal signal transduction. Rabs and Arls/Arfs play critical roles in early cilia formation but are also needed for maintenance of ciliary function through their coordination with intraflagellar transport (IFT), a specialized trafficking system in primary cilia. IFT in cilia is pivotal for the proper movement of proteins into and out of this highly regulated organelle. In this review article, we explore the involvement of polarized vesicular trafficking in cilia formation and function, and discuss how defects in these processes could subsequently lead to the abnormalities observed in ciliopathies.
Similar content being viewed by others


Primary cilia are evolutionarily conserved organelles projecting from the plasma membrane in almost every vertebrate cell. In general, primary cilia serve as sensors through which cells receive signals from light, chemical, or mechanical stimuli [1]. Moreover, the involvement of primary cilia in several signaling pathways important for development and tissue homeostasis (including the Sonic hedgehog and Wnt signaling pathways) has attracted much interest and stimulated extensive studies on this ancient cellular structure [2–6]. A functional primary cilium is required to properly activate primary cilia-mediated cellular signaling. Therefore, any defects in primary cilia could lead to cellular dysfunction. Indeed, abnormalities in primary cilia have been linked to a constellation of phenotypically and genetically overlapping human diseases, which include Bardet-Biedl syndrome, Joubert syndrome, Meckel-Gruber syndrome, nephronophthisis and Sensenbrenner syndrome; all now collectively known as ciliopathies [7–10]. The clinical manifestations of these disorders can include brain malformations, skeletal abnormalities, retinal degeneration, and cystic kidney disease.
The formation and function of primary cilia are tightly regulated by polarized vesicle trafficking, not only to the primary cilium, but also in coordination with trafficking throughout the entire cell [5]. Although bioinformatic, proteomic and genetic studies have suggested that more than a thousand proteins can be localized at the primary cilium, it is still unclear why and how these proteins work together in this specialized cellular compartment [11–15]. Therefore, studying the formation and function of the primary cilium, through investigations into the function of these ciliary proteins, will help to elucidate the pathophysiological mechanisms responsible for causing the ciliopathies.
Structure and function of the primary cilium
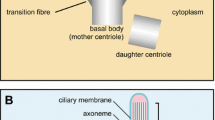
Cilia are categorized into two classes: motile and non-motile. Motile cilia, such as tracheal cilia, can be numerous on a cell surface and have the prominent function of moving mucus and fluids, but are not the focus of this review. This review focuses on non-motile cilia, also referred to more commonly as primary cilia, which are solitary and mainly serve as a sensory organelle for the cell. Importantly, most primary cilia are non-motile, except for those present in the ventral nodes of vertebrates [1, 16]. Primary cilia are polarized structures protruding from the surface of the cell into the extracellular space and are present on almost every quiescent cell in the body. The ciliary axoneme, which is composed of microtubule bundles, is the core structure of the cilium [1]. For primary cilia, the ciliary axoneme consists of a radial array of nine doublet microtubules with no central pair of singlet microtubules, and therefore is called a "9 + 0" configuration. The microtubule axoneme is nucleated at the basal body just beneath the plasma membrane (Figure 1). The basal body is a cytosolic microtubule organizing center that is derived from the mother centriole [1]. All of these structural components of the primary cilium are necessary for the proper formation and function of this signaling structure.

Structure of the primary cilium. The core structure of primary cilia is composed of microtubule bundles (ciliary axoneme) extending from the basal body, a microtubule-based structure derived from the mother centriole. The ciliary membrane is continuous with the plasma membrane, but contains a unique protein composition, such as channels and receptors. Thus, primary cilia can function as a sensory organelle for receiving and transducing extracellular stimuli into cells, such as fluid flow or via signaling molecules. The inset image is of ciliated murine inner medullary collecting duct (IMCD3) cells in which the basal body is labeled with a γ-tubulin antibody (red) and the primary cilium is marked by an Ift88 antibody (green).
Primary cilia are enriched with receptors and provide a separate highly regulated compartment in which signaling events are conveyed from the extracellular space into the cell. Sensing the extracellular environment is a major function of primary cilia. For instance, the ciliated cells of the retina (photoreceptors) and the olfactory system (olfactory sensory neurons) receive and transduce the stimuli of light and odorants to the cells, respectively [17, 18]. Primary cilia on the epithelial cells of kidney tubules act as mechanosensors for sensing fluid flow resulting in increased intracellular calcium signaling [19–21].
Recently, a vital role for primary cilia in signaling pathways important for embryonic development and tissue homeostasis has been identified. While the sonic hedgehog (Shh) pathway has been long known as a critical component of neural tube closure and organ patterning during embryonic development, it was only recently discovered that primary cilia are necessary for this signaling [2, 6, 22]. In mammalian cells when Shh is absent, the Shh receptor, Patched-1 (Ptch1), is localized at the base of the primary cilium and suppresses the activity of another transmembrane protein, Smoothened (Smo). Upon Shh stimulation, Smo is relieved from Ptch1 suppression and translocates to the primary cilium, while Ptch1 is removed from the primary cilium. This translocation of Smo to the primary cilium, after Shh stimulation, is required for activation of Shh-mediated downstream signaling [23–25].
Importantly, the integrity of the primary cilium and the proper functioning of the intraflagellar transport (IFT) system (the specialized transport system in the primary cilium) are both required for proper Shh signaling in mice [6, 26]. For example, Ift172 deficient mouse embryos display abnormal brain development resulting from defects in cilia formation, consequently leading to reduced Shh signaling [27]. In addition to Shh signaling, primary cilia are also critical for Wnt signaling [3, 28, 29], calcium signaling [30, 31], growth factor signaling [32–34], G-protein coupled receptor signaling [35, 36], and receptor tyrosine kinase signaling [37]. Interestingly, the range of different types of signaling associated with cilia is ever expanding as more receptors and proteins are identified at the primary cilium. Although it is unclear why receptors are enriched at the primary cilium as compared to the plasma membrane or why signaling mechanisms are concentrated at the primary cilium, it is clear that this unique and separate cellular compartment makes primary cilia an unusual structure for receiving and transducing a variety of signals.
Mechanisms for sorting ciliary bound proteins to the primary cilium
To date, there have been no studies which have shown that proteins can be produced in the cilium. Therefore, the unique density of proteins and receptors on the membrane around the ciliary axoneme were targeted there through a highly regulated importing process [25]. Trafficking of proteins from the cytosol and Golgi to the primary cilium, and for moving proteins along the ciliary axoneme, is regulated by polarized vesicle trafficking and intraflagellar transport, respectively. Detailed mechanisms of these two trafficking systems will be discussed later.
The transition zone (TZ), a region adjacent to the basal body, provides a selective barrier that can exclude vesicles, impede diffusion of proteins and lipids between compartments (cytosolic/plasma membrane versus ciliary), and control the entry and exit of proteins from the primary cilium [38–40]. The TZ is composed of transitional fibers (TFs), the ciliary necklace and Y-links. TFs anchor the basal body to the proximal ciliary membrane, forming a pinwheel-like structure whose protein composition is unknown. The ciliary necklace is distal to the TFs and consists of rows of protein particles around the ciliary membrane at the base of the cilium. Y-links connect axonemal microtubules to the membrane at the ciliary necklace [41]. Whereas TZ components include CC2D2A, MKS1, MKS3, MKS5, MKS6, MKS1 related-1, MKS1 related-2, NPHP1, NPHP4, Septin 2, Tctn1, and Tctn3 [40–44], the Y-links are associated with CEP290 localization [39]. Importantly, different components perform different functions at this region. For instance, while CEP20 functions as a gate keeper, controlling cilia protein composition by restricting the entrance of non-ciliary proteins, CC2D2A facilitates the entry of ciliary proteins through a role in vesicle trafficking [39, 42].
To aid in this trafficking, ciliary proteins contain a targeting sequence enabling it to efficiently localize at the primary cilium. To date, different targeting sequences have been identified: rhodopsin possesses a C-terminal targeting sequence with a VxPx motif; polycystin-2 has an N-terminal RVxP motif; and polycystin-1 harbors a C-terminal targeting sequence, KVHPSST [45, 46]. This has led to the suggestion of a VxP motif as a generic ciliary targeting sequence (CTS). However, cystin contains a N-terminal CTS with a AxEGG motif [47], whereas the G-protein coupled receptors, Sstr3, Htr6, and Mchr1, present an Ax(S/A)xQ targeting sequence in their third intracellular loop [48]. Overall, these data indicate that there is no unique consensus CTS and further suggest that there is more than one molecular mechanism involved in the recognition of such sequences.
Palmitoylation and myristoylation are post-translational modifications that provide a point of membrane association and have been identified as a requirement for some ciliary proteins in their proper targeting to the cilium. For example, the targeting sequence for rhodopsin contains two cysteine residues that are palmitoylated and which are necessary for rod outer segment targeting [49]. The same is true for fibrocystin, in which its CTS has three palmitoylated cysteine residues [50]. Lastly, cystin is myristoylated at its G2 residue and it has been shown that this acylation is required for the proper localization of cystin to the ciliary membrane [47]. Therefore, the sorting of ciliary targeted proteins to the primary cilium not only occurs through CTSs, but can also be accomplished through post-translational modifications. It is these modifications that provide the sorting information for proteins resulting in their proper trafficking to the specialized membrane microdomains of the primary cilium.
Targeting of proteins to the primary cilium also involves regulation of proteins under the control of the GDP-GTP cycle. Rab8, one such protein, interacts with the CTS of fibrocystin, as suggested by a study using a GFP fusion of the fibrocystin CTS [50]. This GFP-CTS localizes to primary cilia in cells expressing Flag-Rab8 and Flag-Rab8Q67L (a constitutively active form of Rab8a having decreased GTPase activity, thereby maintaining the protein in the GTP bound state), but not Flag-Rab8T22N (a dominant negative form of Rab8 having a higher affinity for GDP than GTP) expressing cells. Rab8T22N bound more GFP-CTS than either the wild type or the Q67L mutant, suggesting that activation of Rab8 results in the release of the GFP-CTS from Rab8. Therefore, the regulation of Rab8 activity controls the localization of fibrocystin through its CTS. Whether other ciliary localizing proteins containing CTSs interact with Rabs through their CTS, remains to be determined.
A mechanism for NPHP3 ciliary targeting has been partially described in which myristoylation of its G2 residue and regulation of GTPase activity are required. That is, NPHP3, when it is myristoylated, binds to UNC119 (HRG4). The phenylalanine residues lining the hydrophobic β-sandwich in UCN119 contribute to this NPHP3 binding. This complex is trafficked through an unknown mechanism to the primary cilium. In the cilium, ARL3-GTP binds to its effector, UNC119, releasing myristoylated NPHP3 into the cilium or ciliary membrane. RP2 then activates ARL3 GTPase activity, releasing UNC119 to reset the cycle [51]. These and other studies clearly indicate that protein sorting to the primary cilium is a tightly regulated process that may involve multiple molecular mechanisms of protein-protein recognition and protein-membrane interaction, in which the GDP-GTP state of these proteins is fundamental.
Functional primary cilia depend on polarized trafficking and intraflagellar transport
Polarized vesicle trafficking is a highly regulated delivery system in cells for transporting proteins and vesicles to their proper destinations in order to perform and maintain normal cell function (Figure 2). In most cases, the primary cilium is formed at the end of the cell cycle, after cell polarity is established. At the early stage of cilium formation (ciliogenesis), a vesicle derived from the Golgi encapsulates the distal end of the mother centriole (the origin of the basal body) as it migrates toward the apical plasma membrane. After docking of the basal body, the primary cilium elongates as the axoneme extends. Additional vesicles carry ciliary membrane proteins to the cilium, which then fuse to the plasma membrane where the cilium originates [52]. Thus, ciliogenesis requires axoneme assembly, membrane biogenesis, and a proper compartmentalization of ciliary proteins in coordination with polarized vesicle trafficking [1, 53].

Polarized vesicle trafficking mediates the formation and function of primary cilia. Enriched expression of receptors and ion channels on the ciliary membrane make the primary cilium a specialized organelle for receiving and transducing extracellular stimuli into cells. Proteins synthesized from the Golgi move to the primary cilium through polarized vesicle trafficking utilizing microtubule networks (not shown). For instance, vesicles carrying ciliary proteins leave the Golgi and move toward the basal body of the primary cilium. These vesicles can either be delivered to the surface plasma membrane and then the protein cargo moves to the ciliary membrane or they can be trafficked toward the basal body through Rab proteins, IFT20, or exocysts. Entry of protein cargo to the cilium is regulated by active forms of Rab8, a master modulator for the ciliary protein trafficking. Rab8 is recruited to the basal body of primary cilium, possibly mediated by Ahi1. The activities of Rab8 are then regulated by Rabin8, and its activity and basal body localization is modulated by the BBSome and Rab11. Once proteins are transported into the primary cilium, the IFT system continues the trafficking of these proteins or membrane receptors up and down along the ciliary axoneme. IFT-B (anterograde) and IFT-A (retrograde) are protein complexes associated with the molecular motors, kinesin-2 and cytoplasmic dynein, respectively.
Polarized vesicular trafficking in ciliogenesis and ciliary function
Polarized vesicle trafficking is a specialized cellular transport mechanism used to deliver proteins and membranes to their proper cellular compartments (Figure 2). Perturbations in this process can have significant impacts on normal cell function [54, 55]. Emerging evidence has shown an important link between polarized vesicle trafficking and the proper formation and function of the primary cilium. Not only have Golgi-derived vesicles been implicated in early ciliogenesis, but the constant trafficking of vesicles through the post-Golgi to the primary cilium has been shown to be crucial for normal ciliary structure and function [56, 57]. Polarized vesicle trafficking is mediated by Rabs along with Arf/Arl members of the Ras superfamiliy of small GTPases, which assist in the recruitment of vesicle coating complexes during vesicle budding, docking and fusion of vesicles [58, 59]. Additionally, the essential roles of these small GTPase proteins in the formation and function of primary cilia have been demonstrated in several studies (Table 1) [2, 60, 61]. The activities of Rabs and Arf/Arl proteins are regulated by their nucleotide binding status. GDP-bound Rab proteins, usually referred to as inactive forms, are switched to the GTP-bound active state through catalyzation by guanine nucleotide exchange factors (GEFs). Then, GTP-bound Rabs can modulate several cellular events through activation of their downstream effectors, including vesicle sorting proteins, tethering proteins, kinases, phosphatases and motor proteins. Conversion of active GTP-bound Rabs back to inactive GDP-bound Rabs is through GTP hydrolysis by GTPase activating proteins (GAPs). The correct membrane targeting of Rab proteins is crucial and regulated by its specific GDP dissociation inhibitors (GDIs) and membrane-bound GDI displacement factors (GDFs) [59].
Arl proteins in ciliary trafficking
Arl6 is exclusively expressed in ciliated organisms [62], and its human ortholog ARL6, encoded by BBS3, was the first identified small GTPase protein that was linked to the human ciliopathy, Bardet-Biedl syndrome (BBS) [63]. This rare inherited disorder features dysfunction in multiple organ systems, leading to retinal dystrophy, obesity, renal disease and cognitive impairments [64]. The abnormalities associated with the loss of BBS3 in BBS are thought to arise due to dysfunctional primary cilia [63]. That is, Arl6 is localized at the distal end of the basal body, near or at the TFs, a region of the ciliary compartment that controls the entry of proteins into the primary cilium [65]. Arl6 functions as a recruiter of the BBSome (a basal body localizing complex composed of seven BBS disease proteins) to the basal body and as a regulator of the function of the BBSome in ciliary protein trafficking [60, 66]. ARL13B is another small GTPase protein connected to the human ciliopathy, Joubert syndrome, an inherited neurodevelopmental disorder with midbrain-hindbrain malformations, retinal dystrophy and, occasionally, nephronophthisis [67]. Deletion of Arl13b in mice leads to the deformation of primary cilia and defective neuronal tube development due to hyperactive Shh signaling [2, 68]. Work in Caenorhabditis elegans has demonstrated that the formation of the IFT-A and IFT-B subcomplex is critical for building the ciliary structure and that the coordination of Arl13 and Arl3 is required for stabilizing this subcomplex [69–71].
Rab small GTPases
Rab GTPases are the largest family of small GTPases, and their function and distinct localization at intracellular membranes has been studied extensively [59]. However, the distribution of several Rab GTPases at the primary cilium was only recently discovered. Disruption of the ciliary localization or the activities of these Rab GTPases are associated with several ciliopathies due to impairments in cilium formation and function [72]. For example, Rab8 is a critical modulator for the formation and the function of primary cilia in addition to having an important role in vesicular trafficking between the trans-Golgi and the basolateral membrane [53, 73]. As mentioned previously, rhodopsin is present at the outer segments of photoreceptor cells. Rab8 is, in part, responsible for delivering rhodopsin-bearing post-Golgi vesicles close to the base of the photoreceptor connecting cilium, where these vesicles fuse, and rhodopsin is then transported through the connecting cilium to the outer segment [74]. Rab8 mutations that interfere with the GTP/GDP cycle of the protein have been utilized to test its role on ciliogenesis. Work in Xenopus laevis has shown that Rab8T22N (a dominant negative form of Rab8) and Rab8Q67L (a constitutively active form of Rab8) expression disrupts rhodopsin trafficking and leads to retinal degeneration, a common occurrence in human ciliopathies [75]. Expression of Rab8T22N in RPE or IMCD3 cells was shown to inhibit cilia formation, whereas expression of Rab8Q67L promoted ciliogenesis in these cell types [50, 60]. In Danio rerio, injection of Rab8T22N results in abnormalities in Kupffer's Vesicle (embryonic ciliated structure similar to the node of mammals), presumably through defects in cilia [60]. This suggests that the activity of Rab8 is critical for the biogenesis of ciliary membrane [60]. Indeed, studies of BBS and other human ciliopathies demonstrate that the ciliary localization and activity of Rab8 is critical for cilium formation and function.
The integrity of the BBSome is necessary for the regulation of Rab8 activity and only the constitutively active form of Rab8 is able to enter the primary cilium [60]. Therefore, mutations of any BBS disease protein in the BBSome would consequently affect the activity of Rab8 at the primary cilium. Moreover, it has also shown that the activity of Rab8 can also be regulated by the retinitis pigmentosa GTPase regulator (RPGR), a ciliary protein implicated in X-linked retinitis pigmentosa [76]. This suggests that there are multiple regulatory mechanisms for Rab8 activity that can be used in ciliated cells or even in different cell types.
The targeting of Rab8 to the basal body is essential for the function of Rab8 at the primary cilium. The basal body localizing protein AHI1, when mutated, can cause the human ciliopathy Joubert syndrome [77–79], and is required for recruiting Rab8 to the basal body [80, 81]. Knockdown of Ahi1 expression in IMCD3 cells impairs ciliogenesis and results in the loss of Rab8 localization to the basal body. Interestingly, these phenotypes cannot be rescued by overexpressing a constitutively activated form of Rab8, indicating the requirement for Ahi1 in the proper localization for Rab8 [80]. Also, mice with a targeted deletion of Ahi1 develop retinal degeneration with an accumulation of rhodopsin in the photoreceptor inner segments, possibly due to a decrease in the levels of photoreceptor Rab8 expression [82]. Overall, this implies that the localization of Ahi1 at the basal body is required for the ciliary targeting of Rab8.
Moreover, the central role of Rab8 in vesicle targeting to the primary cilium has been supported by many studies showing an association of Rab8 with other proteins involved in vesicular trafficking to the primary cilium [50, 83, 84]. For instance, studies in C. elegans have shown that Rab8 genetically interacts with Rabaptin5, an endocytosis regulator, and through which it forms a complex with Elipsa, an IFT particle polypeptide binding to Ift20, thereby regulating protein trafficking to the primary cilium [84]. In ciliated sensory neurons in C. elegans, coordination of Rab8 with the vesicle coating complex, AP-1, was found to be necessary for sorting and trafficking of ciliary membrane [83]. In addition to regulating proteins that participate in vesicle trafficking, Rab8 is also directly involved in targeting proteins to the primary cilium through its interaction with CTSs in these proteins. Fibrocystin, a ciliary protein which is encoded by a gene implicated in autosomal recessive polycystic kidney disease, fails to traffic to the primary cilium when Rab8 activity is inhibited in IMCD3 cells [50]. Overall, the data discussed above clearly support a fundamental role for Rab8 in modulating vesicle targeting to the primary cilium.
In addition to Rab8, several other Rab GTPases have also been identified at the primary cilium and have been shown to function in different ciliary trafficking pathways or processes. A key regulator of endosome recycling, Rab11, is enriched at the base of primary cilia, and compromising the function of Rab11 in cells abolishes ciliogenesis [85]. The mechanism by which Rab11 is involved in cilia formation is through the recruitment of Rabin8, a Rab8 specific GEF, to the centrosome, thereby stimulating centrosomal Rab8 activity during early cilium formation [86]. Rab11 also participates in ciliary targeting by recognizing conserved ciliary targeting sequences in ciliary proteins [87].
Our knowledge of the function of various Rab small GTPases in ciliary trafficking is growing through the use of ciliary fluorescence recovery after photo-bleaching (FRAP) [88]. Consistent with previous studies that have shown a direct role of Rab8 in controlling proteins entering the primary cilium, ciliary FRAP experiments have demonstrated that cells expressing a dominant negative Rab8 exhibit a slower fluorescent recovery rate for the GFP-bound ciliary proteins, Smo and Kim1 (an apical membrane receptor), indicating a disruption of ciliary anterograde transport. In addition, results of FRAP experiments in cells expressing a dominant negative Rab5 exhibited a slower fluorescent recovery rate only for Kim1 indicating that there are likely multiple pathways for delivering ciliary proteins. Conversely, inactivation of Rab23, a protein that is essential for ciliogenesis, does not block the entry of proteins into the primary cilium, but has effects on their recycling to the primary cilium. This interpretation was based on the finding that while these cells had a normal fluorescent recovery rate, they had had an increase in the fluorescence intensity recovered in the cilium supporting a role for Rab23 in protein recycling. However, expression of a dominant negative Rab23 specifically influences only Smo and not Kim1. As a result, Rab23 is necessary for maintaining the level of Smo at the primary cilium through its fundamental role in Smo recycling, providing a mechanism for Rab23 that is proposed to act as a negative regulator in Shh signaling [89]. This further supports the essential role for primary cilia in Shh signal transduction.
Exocysts
The exocyst is a conserved octameric protein complex consisting of Sec3, Sec5, Sec6, Sec8, Sec10, Sec15, Exo70, and Exo84, and it is involved in basolateral protein sorting and membrane trafficking in cells [90]. The integrity of the exocyst is essential for exocytosis in yeast [91] and for the ability of Madin-Darby canine kidney (MDCK) cells to form cysts in culture [92]. The distribution of exocysts in cells is not only at cell-cell junctions, but also at the primary cilium in polarized cells [12, 93]. Compromising the function of exocysts by knockdown of Sec10 results in the formation of shortened primary cilia, along with reduced levels of Sec8, Exo70, and Ift88 [94]. This suggests a central role for Sec10 in stabilizing the exocyst complex and possibly a role of Sec10 in trafficking Ift88 to the primary cilium. The involvement of the exocyst in early ciliogenesis has been suggested by an association of Sec8 and dishevelled (Dvl), a protein involved in the planar cell polarity (PCP) signaling pathway [95]. During basal body docking in multi-ciliated cells, Sec8 cannot co-localize with the basal body when Dvl is knocked down. The exocyst, labeled by Sec6 and Sec8, that is localized at the base of the photoreceptor-connecting cilium, co-localizes with Rab8 at the fusion sites of rhodopsin carrying vesicles, implicating a role for the exocyst as a vesicle tether at cilia [96]. In addition to Rab8, an association of exocysts with Rab10 has also been demonstrated by a direct interaction and co-localization of Sec8 and Rab10 at the basal body of a nascent primary cilium [97]. This interaction of Rab10 and Sec8 suggests that coordination of the exocyst and Rab10 is important in mediating the biogenesis of the ciliary membrane [97]. Lastly, the binding of Sec15 and Rab11 was found to regulate the function of exocysts in basolateral-to-apical transcytosis [98]. Although this study does not describe whether primary cilia were defective when Rab11 binding to Sec15 was disrupted, it does suggest another pathway through which the exocyst could be involved in regulating the primary cilium.
Microtubules
EB1 and EB3 are microtubule plus end tracking proteins that localize to the base of the primary cilium in human fibroblasts and RPE cells [99]. EB1 also localizes to the basal body in the green algae, Chlamydomonas [100]. Both proteins are important for cilia formation, likely through a role in microtubule minus end-basal body anchoring activity, since the microtubule array anchored at the basal body is disorganized in EB1 and EB3 knockdown cells having aberrant vesicle accumulation and impaired ciliogenesis. These results demonstrate the relevance of microtubules in anchoring to the basal body thereby providing a 'roadmap' for the targeting of vesicles carrying ciliary proteins to the vicinity of the basal body where they are exocytosed [99, 101].
Intraflagellar transport in ciliogenesis and ciliary function
Transport of axonemal precursors and several ciliary membrane proteins to the ciliary tip is mediated by IFT, a process which moves cargos up and down along the ciliary axoneme [102, 103]. Given that IFT is highly conserved among organisms with cilia and flagella, much of our current knowledge about IFT comes from work on Chlamydomonas. Newly synthesized ciliary proteins made in the cell body need to be transported into the cilium. This occurs through the association of these ciliary proteins with the IFT complex B (IFT-B), which then moves its cargo toward the cilium tip (anterograde transport) under the power of the molecular motor, kinesin-2 [104]. Conversely, retrograde transport delivers proteins back to the base of the primary cilium through the IFT complex A (IFT-A) using the motor protein, dynein-2 [56, 105]. At least twenty proteins have been identified to date as components of the IFT-A and IFT-B complexes in ciliated mammalian cells [22, 106]. The crucial role of IFT in the formation and function of primary cilia has been elucidated by genetic studies utilizing IFT mutants [6, 107–109]. For instance, mice with deletions of the gene encoding Ift88/Polaris, a component of IFT-B, develop polycystic kidney disease (PKD), hepatic fibrosis, and situs inversus, resulting from malformed or absent primary cilia [107, 110]. Not only is proper functioning of IFT critical for cilia formation, but it is also necessary for its maintenance. Tubulin turns over steadily on the flagella tips in Chlamydomonas, but resorption does not occur since IFT continuously delivers tubulin to the microtubule ends, thereby regulating flagella length [111]. Similarly, the outer segments of photoreceptors contain stacks of membrane discs, which receive and transduce light signals. These membrane discs undergo rapid turnover leading to the replacement of entire outer segments every two weeks [112]. Importantly, the link between the outer segment and the inner segment of the photoreceptor is through a connecting cilium, the analog to the TZ, and is critical for the proper maintenance of photoreceptor outer segments [113, 114]. Therefore, functional IFT is essential for transporting newly synthesized proteins and membrane to the outer segment through the connecting cilium [115, 116]. In support of this, mice with conditionally deleted Kif3A have an abnormal distribution of rhodopsin in the photoreceptor inner segments; an aberrant localization since rhodopsin is rapidly transported from the inner segments to the outer segments via the connecting cilium. Interestingly, it is this aberrant localization of rhodopsin in the cell body that results in the subsequent loss of photoreceptors due to increased reactive oxygen species accumulation [117]. Similar phenotypes are also observed in Ift88/Polaris mutant mice and other IFT mutant animals [110, 118, 119].
IFT is also important for cilium-dependent signaling, with the Shh pathway as the most striking example [120, 121]. Studies of IFT mutant mouse models have suggested that functional IFT is essential for trafficking of Shh signaling components in the primary cilium and for activating downstream signaling [6, 107, 122]. For instance, patterning of the neural tube and formation of the limbs are determined by a gradient of Shh signaling. Mice with deletion of Ift88 fail to form primary cilia and display phenotypes resembling mice with reduced Shh signaling, such as loss of ventral neuronal cell types and polydactyly [120, 123]. Shh phenotypes have also been observed in other anterograde IFT mutant mouse models, such as with Ift172 and Kif3A knockouts [6, 124]. Furthermore, mutant mice with defective retrograde IFT usually exhibit phenotypes reminiscent of excessive Shh signaling [22, 125, 126]. Disruption of the retrograde IFT-A complex in mice by deleting one of its components, Ift122, results in defective ciliogenesis and uncontrolled Shh signaling. More specifically, Ift122 knockout mouse embryos have a ventrolateral expansion of motoneurons and ectrosyndactyly (absence of digits), all consistent with an excess in Shh signaling [126]. However, given the diversity of phenotypes that are observed in IFT mutants and the involvement of different tissues, this would suggest that the regulatory role of IFT proteins in Shh signaling may be more complicated. Case in point, Ift88 deficient mice display an abnormal neural tube and defective limb development resulting from decreased Shh signaling, but these mutant mice also show ectopic molar tooth development due to hyperactive Shh signaling [127]. Lastly, the function of IFT in Shh signaling varies depending on the model system used. Reducing Ift88 expression in zebrafish disrupts ciliogenesis but does not affect Shh signaling, unlike the defective Shh signaling observed in Ift88 knockout mice [128, 129]. Also, in Drosophila, primary cilia are not required for proper Shh signaling and Ift Drosophila mutants do not have defective Shh signaling [62, 130, 131]. Whether the regulatory roles that IFT and primary cilia have in mediating Shh signal transduction are restricted to mammals or more generally to vertebrates still remains to be elucidated.
While IFT particles principally transport cargos along the primary cilium, they can occasionally carry vesicles containing ciliary proteins to the primary cilium from the Golgi. That is the case for Ift20, which is a component of IFT-B and is also localized at the Golgi. Like other IFT mutants, reduced expression of Ift20 in cells impairs cilia formation [132]; moreover, disrupting the Golgi localization of Ift20 through knockdown of GMAP20, a Golgi anchoring protein for IFT20, results in a loss of the membrane protein, polycystin-2, at the primary cilium [133]. These results implicate the involvement of IFT20 in the trafficking and sorting of ciliary proteins between the Golgi and the primary cilium.
Ciliopathies result from impaired ciliary trafficking
Polarized vesicle trafficking is the foundation for functional primary cilia, and disruption at any step impairs the formation and function of primary cilia. Indeed, studies of the molecular mechanisms responsible for human ciliopathies demonstrate that ciliary function is compromised when vesicle trafficking is disrupted. BBS has been extensively studied as a ciliopathy model to understand how its mutant proteins affect the primary cilium and consequently lead to the observed clinical pathologies. An individual with BBS displays malfunctions in multiple organ systems, including retinal degeneration, cognitive dysfunction, obesity, polydactyly, and cystic kidney disease. Like most ciliopathies, BBS is a genetically heterogeneous disease that can be caused by mutations in fourteen genes [134]. Seven BBS proteins (BBS1, BBS2, BBS4, BBS5, BBS7, BBS8, and BBS9) and one novel protein BBIP10 compose the BBSome. The other BBS proteins are not considered as part of the BBSome itself, but are involved in regulating either the assembly or function of the BBSome [135, 136]. Therefore, mutations in any of the BBS proteins in this network could affect ciliary function and result in similar clinical phenotypes in BBS [137]. To date, the major function for the BBSome appears to be in modulating ciliary trafficking. Recruitment of the BBSome to the basal body is regulated by Arl6, which is encoded by BBS3 as mentioned earlier [66]. The localization of the BBSome to the basal body is essential for modulating the activity and the ciliary entry of Rab8, through its association with Rabin8 via BBS1 [60]. Moreover, the BBSome also participates in the sorting and trafficking of ciliary membrane proteins. Depletion of Bbs4 in mouse neurons causes a lack of ciliary localization of the somatostatin receptor 3 (Sstr3) and the melanin-concentrating hormone receptor 1 (MchR1) [35]. This is due to a direct interaction of the BBSome and the intracellular loop 3 of Sstr3, which contains its ciliary targeting sequence [66]. Given that the composition of the BBSome resembles the structure of the canonical vesicle coat complexes and its ability to recruit lipids, it has been suggested that the BBSome may function as the ciliary membrane protein sorting machinery at the primary cilium [66].
In addition to the BBS proteins, several ciliopathy-related proteins have been linked to ciliary trafficking. CEP290 encodes for a centrosomal protein, mutations of which have been linked to several ciliopathies, including BBS [138], Joubert syndrome [79, 139], nephronophthisis, and Leber Congenital Amaurosis [140]. It has been proposed that CEP290 may function as a gatekeeper for the primary cilium to control ciliary trafficking [141]. Electron microscopic examination of flagellated Chlamydomonas has shown that cep290 is associated with the flagellar membrane and the microtubules at the transition zone of flagella, a structure providing a diffusion barrier for selective cilium transport. Also, cep290 depletion in Chlamydomonas results in malformation of the flagella along with aberrant ciliary protein composition in isolated flagella, indicative of a loss of a flagellar diffusion barrier. While there was no effect on anterograde IFT in cep290 mutants, a slight effect on retrograde IFT was observed [39]. These results indicate that the function of CEP290 in ciliary trafficking is more likely at the level of entry of proteins into the primary cilium. In further support, mutations in CEP290 are also associated with BBS [138]; however, CEP290 is not part of the BBSome. Therefore, the mechanism for how CEP290 regulates ciliary trafficking through the BBSome remains unknown.
In addition to a function of Ahi1 in modulating Wnt signaling [142–144], recent studies have now shown that Ahi1 is also linked to polarized vesicle trafficking in ciliated cells [80, 82]. Knockdown of Ahi1 expression in IMCD3 cells not only reduces cilium formation, but also affects the endocytosis of chlorea toxin A and transferrin receptors. Also, Ahi1 knockdown cells exhibit abnormal Golgi structure and location suggesting that polarized vesicle trafficking has been severely affected with loss of Ahi1 [80]. However, additional studies are needed examining whether this abnormal Golgi structure and non-apical positioning occurs in cells from individuals with AHI1 mutations. Consistent with the results from IMCD3 cells, neurons isolated from mice with a targeted deletion of Ahi1 fail to form primary cilia [80]. However, work in another Ahi1 knockout mouse has shown that primary cilia are found in normal numbers, but clearly have signaling defects [142–144]. This discrepancy in ciliogenesis, and also in the presence of cystic kidney disease [143] (unpublished observations), may be accounted for by the background strain of the mice used or whether the gene was targeted conditionally or not. Studies showing significant decreases in ciliogenesis in Ahi1 knockout mice were on a pure inbred line using a traditional knockout strategy, whereas the studies not showing differences in cilia formation were on a mixed background and were conditional knockouts. In support of the background strain hypothesis, all Ahi1 knockout mice on a pure C57BL6/J or C3H/HeJ background die within 12 hours following birth; however, the same knockout mice on a FVB/NJ or BALB/cJ background have a significant increase in survival, even into adulthood (unpublished observations). Importantly, this is not a result of maternal behavior. These observations suggest that there are likely modifying genes in the various inbred strains of mice that may account for the differences in ciliogenesis observed in different knockout mice. This raises an important consideration when comparing results from knockout mice on different genetic backgrounds and provides a difficult caveat for interpreting results demonstrating the impact of genes on cilia formation and function.
The finding of ciliopathy-disease proteins modulating ciliary protein trafficking clearly indicates the importance of these processes and how disruptions in ciliary trafficking could result in the developmental abnormalities and organ malfunction observed in ciliopathies. Moreover, given the genetic heterogeneity and phenotypic variability displayed in the ciliopathies, this would suggest that the regulatory mechanisms in primary cilia are mediated by multiple proteins, and are likely cell-type dependent. Areas of new interest in the field, with important implications for understanding the function of primary cilia, are how ciliary proteins leave and are recycled back to the primary cilium. This new avenue of research could result in therapeutic strategies for the treatment of the ciliopathies. Primary cilia, originally thought of as vestigial structures, actually have dynamic and complex vesicle trafficking regulation through which signaling can be properly performed. Through the study of the unique roles of primary cilia in cellular function, we hope to understand better and possibly treat the vast array of clinical symptoms that result when cilia are dysfunctional.
Abbreviations
- BBS:
-
Bardet-Biedl syndrome
- CTS:
-
Ciliary targeting sequence
- Dvl:
-
Disheveled
- FRAP:
-
Fluorescence recovery after photo-bleaching
- GAP:
-
GTPase activating protein
- GDI:
-
GDP dissociation inhibitor
- GDF:
-
GDP dissociation inhibitor displacement factor
- GEF:
-
Guanine nucleotide exchange factor
- IFT:
-
Intraflagellar transport
- IMCD:
-
Inner medullary collecting duct
- MDCK:
-
Madin-Darby canine kidney
- PCP:
-
Planar cell polarity
- PKD:
-
Polycystic kidney disease
- RPE:
-
Retinal pigment epithelium
- RPGR:
-
Retinitis pigmentosa GTPase regulator
- TFs:
-
Transition fibers
- TZ:
-
Transition zone.
References
Satir P, Christensen ST: Overview of structure and function of mammalian cilia. Annu Rev Physiol. 2007, 69: 377-400. 10.1146/annurev.physiol.69.040705.141236.
Caspary T, Larkins CE, Anderson KV: The graded response to Sonic Hedgehog depends on cilia architecture. Dev Cell. 2007, 12: 767-778. 10.1016/j.devcel.2007.03.004.
Otto EA, Schermer B, Obara T, O'Toole JF, Hiller KS, Mueller AM, Ruf RG, Hoefele J, Beekmann F, Landau D, Foreman JW, Goodship JA, Strachan T, Kispert A, Wolf MT, Gagnadoux MF, Nivet H, Antignac C, Walz G, Drummond IA, Benzing T, Hildebrandt F: Mutations in INVS encoding inversin cause nephronophthisis type 2, linking renal cystic disease to the function of primary cilia and left-right axis determination. Nat Genet. 2003, 34: 413-420. 10.1038/ng1217.
Singla V, Reiter JF: The primary cilium as the cell's antenna: signaling at a sensory organelle. Science. 2006, 313: 629-633. 10.1126/science.1124534.
Gerdes JM, Davis EE, Katsanis N: The vertebrate primary cilium in development, homeostasis, and disease. Cell. 2009, 137: 32-45. 10.1016/j.cell.2009.03.023.
Huangfu D, Liu A, Rakeman AS, Murcia NS, Niswander L, Anderson KV: Hedgehog signalling in the mouse requires intraflagellar transport proteins. Nature. 2003, 426: 83-87. 10.1038/nature02061.
Badano JL, Mitsuma N, Beales PL, Katsanis N: The ciliopathies: an emerging class of human genetic disorders. Annu Rev Genomics Hum Genet. 2006, 7: 125-148. 10.1146/annurev.genom.7.080505.115610.
Fliegauf M, Benzing T, Omran H: When cilia go bad: cilia defects and ciliopathies. Nat Rev Mol Cell Biol. 2007, 8: 880-893. 10.1038/nrm2278.
Bredrup C, Saunier S, Oud MM, Fiskerstrand T, Hoischen A, Brackman D, Leh SM, Midtbo M, Filhol E, Bole-Feysot C, Nitschké P, Gilissen C, Haugen OH, Sanders JS, Stolte-Dijkstra I, Mans DA, Steenbergen EJ, Hamel BC, Matignon M, Pfundt R, Jeanpierre C, Boman H, Rødahl E, Veltman JA, Knappskog PM, Knoers NV, Roepman R, Arts HH: Ciliopathies with skeletal anomalies and renal insufficiency due to mutations in the IFT-A gene WDR19. Am J Hum Genet. 2011, 89: 634-643. 10.1016/j.ajhg.2011.10.001.
Hildebrandt F, Benzing T, Katsanis N: Ciliopathies. N Engl J Med. 2011, 364: 1533-1543. 10.1056/NEJMra1010172.
Inglis PN, Boroevich KA, Leroux MR: Piecing together a ciliome. Trends Genet. 2006, 22: 491-500. 10.1016/j.tig.2006.07.006.
Liu Q, Tan G, Levenkova N, Li T, Pugh EN, Rux JJ, Speicher DW, Pierce EA: The proteome of the mouse photoreceptor sensory cilium complex. Mol Cell Proteomics. 2007, 6: 1299-1317. 10.1074/mcp.M700054-MCP200.
Mitchell KA, Szabo G, Otero Ade S: Methods for the isolation of sensory and primary cilia--an overview. Methods Cell Biol. 2009, 94: 87-101.
Ostrowski LE, Blackburn K, Radde KM, Moyer MB, Schlatzer DM, Moseley A, Boucher RC: A proteomic analysis of human cilia: identification of novel components. Mol Cell Proteomics. 2002, 1: 451-465. 10.1074/mcp.M200037-MCP200.
Pazour GJ, Agrin N, Leszyk J, Witman GB: Proteomic analysis of a eukaryotic cilium. J Cell Biol. 2005, 170: 103-113. 10.1083/jcb.200504008.
Okada Y, Takeda S, Tanaka Y, Izpisua Belmonte JC, Hirokawa N: Mechanism of nodal flow: a conserved symmetry breaking event in left-right axis determination. Cell. 2005, 121: 633-644. 10.1016/j.cell.2005.04.008.
Glees P, Spoerri PE: Surface processes of olfactory receptors. Cell Tissue Res. 1978, 188: 149-152.
Richardson TM: Cytoplasmic and ciliary connections between the inner and outer segments of mammalian visual receptors. Vision Res. 1969, 9: 727-731. 10.1016/0042-6989(69)90010-8.
Low SH, Vasanth S, Larson CH, Mukherjee S, Sharma N, Kinter MT, Kane ME, Obara T, Weimbs T: Polycystin-1, STAT6, and P100 function in a pathway that transduces ciliary mechanosensation and is activated in polycystic kidney disease. Dev Cell. 2006, 10: 57-69. 10.1016/j.devcel.2005.12.005.
Chauvet V, Tian X, Husson H, Grimm DH, Wang T, Hiesberger T, Igarashi P, Bennett AM, Ibraghimov-Beskrovnaya O, Somlo S, Caplan MJ: Mechanical stimuli induce cleavage and nuclear translocation of the polycystin-1 C terminus. J Clin Invest. 2004, 114: 1433-1443.
Hanaoka K, Qian F, Boletta A, Bhunia AK, Piontek K, Tsiokas L, Sukhatme VP, Guggino WB, Germino GG: Co-assembly of polycystin-1 and -2 produces unique cation-permeable currents. Nature. 2000, 408: 990-994. 10.1038/35050128.
Goetz SC, Anderson KV: The primary cilium: a signalling centre during vertebrate development. Nat Rev Genet. 2010, 11: 331-344. 10.1038/nrg2774.
Corbit KC, Aanstad P, Singla V, Norman AR, Stainier DY, Reiter JF: Vertebrate Smoothened functions at the primary cilium. Nature. 2005, 437: 1018-1021. 10.1038/nature04117.
Rohatgi R, Milenkovic L, Scott MP: Patched1 regulates hedgehog signaling at the primary cilium. Science. 2007, 317: 372-376. 10.1126/science.1139740.
Berbari NF, O'Connor AK, Haycraft CJ, Yoder BK: The primary cilium as a complex signaling center. Curr Biol. 2009, 19: R526-R535. 10.1016/j.cub.2009.05.025.
Haycraft CJ, Banizs B, Aydin-Son Y, Zhang Q, Michaud EJ, Yoder BK: Gli2 and Gli3 localize to cilia and require the intraflagellar transport protein polaris for processing and function. PLoS Genet. 2005, 1: e53-10.1371/journal.pgen.0010053.
Gorivodsky M, Mukhopadhyay M, Wilsch-Braeuninger M, Phillips M, Teufel A, Kim C, Malik N, Huttner W, Westphal H: Intraflagellar transport protein 172 is essential for primary cilia formation and plays a vital role in patterning the mammalian brain. Dev Biol. 2009, 325: 24-32. 10.1016/j.ydbio.2008.09.019.
Cano DA, Murcia NS, Pazour GJ, Hebrok M: Orpk mouse model of polycystic kidney disease reveals essential role of primary cilia in pancreatic tissue organization. Development. 2004, 131: 3457-3467. 10.1242/dev.01189.
Oishi I, Kawakami Y, Raya A, Callol-Massot C, Izpisua Belmonte JC: Regulation of primary cilia formation and left-right patterning in zebrafish by a noncanonical Wnt signaling mediator, duboraya. Nat Genet. 2006, 38: 1316-1322. 10.1038/ng1892.
Nauli SM, Alenghat FJ, Luo Y, Williams E, Vassilev P, Li X, Elia AE, Lu W, Brown EM, Quinn SJ, Ingber DE, Zhou J: Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Genet. 2003, 33: 129-137. 10.1038/ng1076.
Shiba D, Takamatsu T, Yokoyama T: Primary cilia of inv/inv mouse renal epithelial cells sense physiological fluid flow: bending of primary cilia and Ca2+ influx. Cell Struct Funct. 2005, 30: 93-100. 10.1247/csf.30.93.
Schneider L, Clement CA, Teilmann SC, Pazour GJ, Hoffmann EK, Satir P, Christensen ST: PDGFRalphaalpha signaling is regulated through the primary cilium in fibroblasts. Curr Biol. 2005, 15: 1861-1866. 10.1016/j.cub.2005.09.012.
Schneider L, Stock CM, Dieterich P, Jensen BH, Pedersen LB, Satir P, Schwab A, Christensen ST, Pedersen SF: The Na+/H + exchanger NHE1 is required for directional migration stimulated via PDGFR-alpha in the primary cilium. J Cell Biol. 2009, 185: 163-176. 10.1083/jcb.200806019.
Chakravarthy B, Gaudet C, Menard M, Atkinson T, Chiarini A, Dal Pra I, Whitfield J: The p75 neurotrophin receptor is localized to primary cilia in adult murine hippocampal dentate gyrus granule cells. Biochem Biophys Res Commun. 2010, 401: 458-462. 10.1016/j.bbrc.2010.09.081.
Berbari NF, Lewis JS, Bishop GA, Askwith CC, Mykytyn K: Bardet-Biedl syndrome proteins are required for the localization of G protein-coupled receptors to primary cilia. Proc Natl Acad Sci USA. 2008, 105: 4242-4246. 10.1073/pnas.0711027105.
Zhang X, Cote RH: cGMP signaling in vertebrate retinal photoreceptor cells. Front Biosci. 2005, 10: 1191-1204. 10.2741/1612.
Christensen ST, Clement CA, Satir P, Pedersen LB: Primary cilia and coordination of receptor tyrosine kinase (RTK) signalling. J Pathol. 2012, 226: 172-184. 10.1002/path.3004.
Deane JA, Cole DG, Seeley ES, Diener DR, Rosenbaum JL: Localization of intraflagellar transport protein IFT52 identifies basal body transitional fibers as the docking site for IFT particles. Curr Biol. 2001, 11: 1586-1590. 10.1016/S0960-9822(01)00484-5.
Craige B, Tsao CC, Diener DR, Hou Y, Lechtreck KF, Rosenbaum JL, Witman GB: CEP290 tethers flagellar transition zone microtubules to the membrane and regulates flagellar protein content. J Cell Biol. 2010, 190: 927-940. 10.1083/jcb.201006105.
Gilula NB, Satir P: The ciliary necklace. A ciliary membrane specialization. J Cell Biol. 1972, 53: 494-509. 10.1083/jcb.53.2.494.
Williams CL, Li C, Kida K, Inglis PN, Mohan S, Semenec L, Bialas NJ, Stupay RM, Chen N, Blacque OE, Yoder BK, Leroux MR: MKS and NPHP modules cooperate to establish basal body/transition zone membrane associations and ciliary gate function during ciliogenesis. J Cell Biol. 2011, 192: 1023-1041. 10.1083/jcb.201012116.
Bachmann-Gagescu R, Phelps IG, Stearns G, Link BA, Brockerhoff SE, Moens CB, Doherty D: The ciliopathy gene cc2d2a controls zebrafish photoreceptor outer segment development through a role in Rab8-dependent vesicle trafficking. Hum Mol Genet. 2011, 20: 4041-4055. 10.1093/hmg/ddr332.
Garcia-Gonzalo FR, Corbit KC, Sirerol-Piquer MS, Ramaswami G, Otto EA, Noriega TR, Seol AD, Robinson JF, Bennett CL, Josifova DJ, García-Verdugo JM, Katsanis N, Hildebrandt F, Reiter JF: A transition zone complex regulates mammalian ciliogenesis and ciliary membrane composition. Nat Genet. 2011, 43: 776-784. 10.1038/ng.891.
Hu Q, Milenkovic L, Jin H, Scott MP, Nachury MV, Spiliotis ET, Nelson WJ: A septin diffusion barrier at the base of the primary cilium maintains ciliary membrane protein distribution. Science. 2010, 329: 436-439. 10.1126/science.1191054.
Pazour GJ, Bloodgood RA: Targeting proteins to the ciliary membrane. Curr Top Dev Biol. 2008, 85: 115-149.
Ward HH, Brown-Glaberman U, Wang J, Morita Y, Alper SL, Bedrick EJ, Gattone VH, Deretic D, Wandinger-Ness A: A conserved signal and GTPase complex are required for the ciliary transport of polycystin-1. Mol Biol Cell. 2011, 22: 3289-3305. 10.1091/mbc.E11-01-0082.
Tao B, Bu S, Yang Z, Siroky B, Kappes JC, Kispert A, Guay-Woodford LM: Cystin localizes to primary cilia via membrane microdomains and a targeting motif. J Am Soc Nephrol. 2009, 20: 2570-2580. 10.1681/ASN.2009020188.
Berbari NF, Johnson AD, Lewis JS, Askwith CC, Mykytyn K: Identification of ciliary localization sequences within the third intracellular loop of G protein-coupled receptors. Mol Biol Cell. 2008, 19: 1540-1547. 10.1091/mbc.E07-09-0942.
Tam BM, Moritz OL, Hurd LB, Papermaster DS: Identification of an outer segment targeting signal in the COOH terminus of rhodopsin using transgenic Xenopus laevis. J Cell Biol. 2000, 151: 1369-1380. 10.1083/jcb.151.7.1369.
Follit JA, Li L, Vucica Y, Pazour GJ: The cytoplasmic tail of fibrocystin contains a ciliary targeting sequence. J Cell Biol. 2010, 188: 21-28. 10.1083/jcb.200910096.
Wright KJ, Baye LM, Olivier-Mason A, Mukhopadhyay S, Sang L, Kwong M, Wang W, Pretorius PR, Sheffield VC, Sengupta P, Slusarski DC, Jackson PK: An ARL3-UNC119-RP2 GTPase cycle targets myristoylated NPHP3 to the primary cilium. Genes Dev. 2011, 25: 2347-2360. 10.1101/gad.173443.111.
Sorokin S: Centrioles and the formation of rudimentary cilia by fibroblasts and smooth muscle cells. J Cell Biol. 1962, 15: 363-377. 10.1083/jcb.15.2.363.
Leroux MR: Taking vesicular transport to the cilium. Cell. 2007, 129: 1041-1043. 10.1016/j.cell.2007.05.049.
Schwartz SL, Cao C, Pylypenko O, Rak A, Wandinger-Ness A: Rab GTPases at a glance. J Cell Sci. 2007, 120: 3905-3910. 10.1242/jcs.015909.
Zerial M, McBride H: Rab proteins as membrane organizers. Nat Rev Mol Cell Biol. 2001, 2: 107-117. 10.1038/35052055.
Pedersen LB, Veland IR, Schroder JM, Christensen ST: Assembly of primary cilia. Dev Dyn. 2008, 237: 1993-2006. 10.1002/dvdy.21521.
Sorokin SP: Reconstructions of centriole formation and ciliogenesis in mammalian lungs. J Cell Sci. 1968, 3: 207-230.
Barr FA: Rab GTPase function in Golgi trafficking. Semin Cell Dev Biol. 2009, 20: 780-783. 10.1016/j.semcdb.2009.03.007.
Stenmark H: Rab GTPases as coordinators of vesicle traffic. Nat Rev Mol Cell Biol. 2009, 10: 513-525.
Nachury MV, Loktev AV, Zhang Q, Westlake CJ, Peranen J, Merdes A, Slusarski DC, Scheller RH, Bazan JF, Sheffield VC, Jackson PK: A core complex of BBS proteins cooperates with the GTPase Rab8 to promote ciliary membrane biogenesis. Cell. 2007, 129: 1201-1213. 10.1016/j.cell.2007.03.053.
Mukhopadhyay S, Lu Y, Shaham S, Sengupta P: Sensory signaling-dependent remodeling of olfactory cilia architecture in C. elegans. Dev Cell. 2008, 14: 762-774. 10.1016/j.devcel.2008.03.002.
Avidor-Reiss T, Maer AM, Koundakjian E, Polyanovsky A, Keil T, Subramaniam S, Zuker CS: Decoding cilia function: defining specialized genes required for compartmentalized cilia biogenesis. Cell. 2004, 117: 527-539.
Fan Y, Esmail MA, Ansley SJ, Blacque OE, Boroevich K, Ross AJ, Moore SJ, Badano JL, May-Simera H, Compton DS, Green JS, Lewis RA, van Haelst MM, Parfrey PS, Baillie DL, Beales PL, Katsanis N, Davidson WS, Leroux MR: Mutations in a member of the Ras superfamily of small GTP-binding proteins causes Bardet-Biedl syndrome. Nat Genet. 2004, 36: 989-993. 10.1038/ng1414.
Tobin JL, Beales PL: Bardet-Biedl syndrome: beyond the cilium. Pediatr Nephrol. 2007, 22: 926-936. 10.1007/s00467-007-0435-0.
Wiens CJ, Tong Y, Esmail MA, Oh E, Gerdes JM, Wang J, Tempel W, Rattner JB, Katsanis N, Park HW, Leroux MR: Bardet-Biedl syndrome-associated small GTPase ARL6 (BBS3) functions at or near the ciliary gate and modulates Wnt signaling. J Biol Chem. 2010, 285: 16218-16230. 10.1074/jbc.M109.070953.
Jin H, White SR, Shida T, Schulz S, Aguiar M, Gygi SP, Bazan JF, Nachury MV: The conserved Bardet-Biedl syndrome proteins assemble a coat that traffics membrane proteins to cilia. Cell. 2010, 141: 1208-1219. 10.1016/j.cell.2010.05.015.
Cantagrel V, Silhavy JL, Bielas SL, Swistun D, Marsh SE, Bertrand JY, Audollent S, Attie-Bitach T, Holden KR, Dobyns WB, Traver D, Al-Gazali L, Ali BR, Lindner TH, Caspary T, Otto EA, Hildebrandt F, Glass IA, Logan CV, Johnson CA, Bennett C, Brancati F, International Joubert Syndrome Related Disorders Study Group, Valente EM, Woods CG, Gleeson JG: Mutations in the cilia gene ARL13B lead to the classical form of Joubert syndrome. Am J Hum Genet. 2008, 83: 170-179. 10.1016/j.ajhg.2008.06.023.
Horner VL, Caspary T: Disrupted dorsal neural tube BMP signaling in the cilia mutant Arl13b(hnn) stems from abnormal Shh signaling. Dev Biol. 2011, 355: 43-54. 10.1016/j.ydbio.2011.04.019.
Scholey JM: Intraflagellar transport motors in cilia: moving along the cell's antenna. J Cell Biol. 2008, 180: 23-29. 10.1083/jcb.200709133.
Cevik S, Hori Y, Kaplan OI, Kida K, Toivenon T, Foley-Fisher C, Cottell D, Katada T, Kontani K, Blacque OE: Joubert syndrome Arl13b functions at ciliary membranes and stabilizes protein transport in Caenorhabditis elegans. J Cell Biol. 2010, 188: 953-969. 10.1083/jcb.200908133.
Li Y, Wei Q, Zhang Y, Ling K, Hu J: The small GTPases ARL-13 and ARL-3 coordinate intraflagellar transport and ciliogenesis. J Cell Biol. 2010, 189: 1039-1051. 10.1083/jcb.200912001.
Yoshimura S, Egerer J, Fuchs E, Haas AK, Barr FA: Functional dissection of Rab GTPases involved in primary cilium formation. J Cell Biol. 2007, 178: 363-369. 10.1083/jcb.200703047.
Huber LA, Pimplikar S, Parton RG, Virta H, Zerial M, Simons K: Rab8, a small GTPase involved in vesicular traffic between the TGN and the basolateral plasma membrane. J Cell Biol. 1993, 123: 35-45. 10.1083/jcb.123.1.35.
Deretic D, Huber LA, Ransom N, Mancini M, Simons K, Papermaster DS: rab8 in retinal photoreceptors may participate in rhodopsin transport and in rod outer segment disk morphogenesis. J Cell Sci. 1995, 108 (Pt 1): 215-224.
Moritz OL, Tam BM, Hurd LL, Peranen J, Deretic D, Papermaster DS: Mutant rab8 impairs docking and fusion of rhodopsin-bearing post-Golgi membranes and causes cell death of transgenic Xenopus rods. Mol Biol Cell. 2001, 12: 2341-2351.
Murga-Zamalloa CA, Atkins SJ, Peranen J, Swaroop A, Khanna H: Interaction of retinitis pigmentosa GTPase regulator (RPGR) with RAB8A GTPase: implications for cilia dysfunction and photoreceptor degeneration. Hum Mol Genet. 2010, 19: 3591-3598. 10.1093/hmg/ddq275.
Dixon-Salazar T, Silhavy JL, Marsh SE, Louie CM, Scott LC, Gururaj A, Al-Gazali L, Al-Tawari AA, Kayserili H, Sztriha L, Gleeson JG: Mutations in the AHI1 gene, encoding jouberin, cause Joubert syndrome with cortical polymicrogyria. Am J Hum Genet. 2004, 75: 979-987. 10.1086/425985.
Ferland RJ, Eyaid W, Collura RV, Tully LD, Hill RS, Al-Nouri D, Al-Rumayyan A, Topcu M, Gascon G, Bodell A, Shugart YY, Ruvolo M, Walsh CA: Abnormal cerebellar development and axonal decussation due to mutations in AHI1 in Joubert syndrome. Nat Genet. 2004, 36: 1008-1013. 10.1038/ng1419.
Sayer JA, Otto EA, O'Toole JF, Nurnberg G, Kennedy MA, Becker C, Hennies HC, Helou J, Attanasio M, Fausett BV, Utsch B, Khanna H, Liu Y, Drummond I, Kawakami I, Kusakabe T, Tsuda M, Ma L, Lee H, Larson RG, Allen SJ, Wilkinson CJ, Nigg EA, Shou C, Lillo C, Williams DS, Hoppe B, Kemper MJ, Neuhaus T, Parisi MA et al: The centrosomal protein nephrocystin-6 is mutated in Joubert syndrome and activates transcription factor ATF4. Nat Genet. 2006, 38: 674-681. 10.1038/ng1786.
Hsiao YC, Tong ZJ, Westfall JE, Ault JG, Page-McCaw PS, Ferland RJ: Ahi1, whose human ortholog is mutated in Joubert syndrome, is required for Rab8a localization, ciliogenesis and vesicle trafficking. Hum Mol Genet. 2009, 18: 3926-3941. 10.1093/hmg/ddp335.
Kim J, Krishnaswami SR, Gleeson JG: CEP290 interacts with the centriolar satellite component PCM-1 and is required for Rab8 localization to the primary cilium. Hum Mol Genet. 2008, 17: 3796-3805. 10.1093/hmg/ddn277.
Westfall JE, Hoyt C, Liu Q, Hsiao YC, Pierce EA, Page-McCaw PS, Ferland RJ: Retinal degeneration and failure of photoreceptor outer segment formation in mice with targeted deletion of the Joubert syndrome gene, Ahi1. J Neurosci. 2010, 30: 8759-8768. 10.1523/JNEUROSCI.5229-09.2010.
Kaplan OI, Molla-Herman A, Cevik S, Ghossoub R, Kida K, Kimura Y, Jenkins P, Martens JR, Setou M, Benmerah A, Blacque OE: The AP-1 clathrin adaptor facilitates cilium formation and functions with RAB-8 in C. elegans ciliary membrane transport. J Cell Sci. 2010, 123: 3966-3977. 10.1242/jcs.073908.
Omori Y, Zhao C, Saras A, Mukhopadhyay S, Kim W, Furukawa T, Sengupta P, Veraksa A, Malicki J: Elipsa is an early determinant of ciliogenesis that links the IFT particle to membrane-associated small GTPase Rab8. Nat Cell Biol. 2008, 10: 437-444. 10.1038/ncb1706.
Knodler A, Feng S, Zhang J, Zhang X, Das A, Peranen J, Guo W: Coordination of Rab8 and Rab11 in primary ciliogenesis. Proc Natl Acad Sci USA. 2010, 107: 6346-6351. 10.1073/pnas.1002401107.
Westlake CJ, Baye LM, Nachury MV, Wright KJ, Ervin KE, Phu L, Chalouni C, Beck JS, Kirkpatrick DS, Slusarski DC, Sheffield VC, Scheller RH, Jackson PK: Primary cilia membrane assembly is initiated by Rab11 and transport protein particle II (TRAPPII) complex-dependent trafficking of Rabin8 to the centrosome. Proc Natl Acad Sci USA. 2011, 108: 2759-2764. 10.1073/pnas.1018823108.
Mazelova J, Astuto-Gribble L, Inoue H, Tam BM, Schonteich E, Prekeris R, Moritz OL, Randazzo PA, Deretic D: Ciliary targeting motif VxPx directs assembly of a trafficking module through Arf4. EMBO J. 2009, 28: 183-192. 10.1038/emboj.2008.267.
Boehlke C, Bashkurov M, Buescher A, Krick T, John AK, Nitschke R, Walz G, Kuehn EW: Differential role of Rab proteins in ciliary trafficking: Rab23 regulates smoothened levels. J Cell Sci. 2010, 123: 1460-1467. 10.1242/jcs.058883.
Eggenschwiler JT, Espinoza E, Anderson KV: Rab23 is an essential negative regulator of the mouse Sonic hedgehog signalling pathway. Nature. 2001, 412: 194-198. 10.1038/35084089.
Kang RS, Folsch H: An old dog learns new tricks: novel functions of the exocyst complex in polarized epithelia in animals. F1000 Biol Rep. 2009, 1: 83-
Novick P, Field C, Schekman R: Identification of 23 complementation groups required for post-translational events in the yeast secretory pathway. Cell. 1980, 21: 205-215. 10.1016/0092-8674(80)90128-2.
Lipschutz JH, Guo W, O'Brien LE, Nguyen YH, Novick P, Mostov KE: Exocyst is involved in cystogenesis and tubulogenesis and acts by modulating synthesis and delivery of basolateral plasma membrane and secretory proteins. Mol Biol Cell. 2000, 11: 4259-4275.
Rogers KK, Wilson PD, Snyder RW, Zhang X, Guo W, Burrow CR, Lipschutz JH: The exocyst localizes to the primary cilium in MDCK cells. Biochem Biophys Res Commun. 2004, 319: 138-143. 10.1016/j.bbrc.2004.04.165.
Zuo X, Guo W, Lipschutz JH: The exocyst protein Sec10 is necessary for primary ciliogenesis and cystogenesis in vitro. Mol Biol Cell. 2009, 20: 2522-2529. 10.1091/mbc.E08-07-0772.
Park TJ, Mitchell BJ, Abitua PB, Kintner C, Wallingford JB: Dishevelled controls apical docking and planar polarization of basal bodies in ciliated epithelial cells. Nat Genet. 2008, 40: 871-879. 10.1038/ng.104.
Mazelova J, Ransom N, Astuto-Gribble L, Wilson MC, Deretic D: Syntaxin 3 and SNAP-25 pairing, regulated by omega-3 docosahexaenoic acid, controls the delivery of rhodopsin for the biogenesis of cilia-derived sensory organelles, the rod outer segments. J Cell Sci. 2009, 122: 2003-2013. 10.1242/jcs.039982.
Babbey CM, Bacallao RL, Dunn KW: Rab10 associates with primary cilia and the exocyst complex in renal epithelial cells. Am J Physiol Renal Physiol. 2010, 299: F495-F506. 10.1152/ajprenal.00198.2010.
Oztan A, Silvis M, Weisz OA, Bradbury NA, Hsu SC, Goldenring JR, Yeaman C, Apodaca G: Exocyst requirement for endocytic traffic directed toward the apical and basolateral poles of polarized MDCK cells. Mol Biol Cell. 2007, 18: 3978-3992. 10.1091/mbc.E07-02-0097.
Schroder JM, Larsen J, Komarova Y, Akhmanova A, Thorsteinsson RI, Grigoriev I, Manguso R, Christensen ST, Pedersen SF, Geimer S, Pedersen LB: EB1 and EB3 promote cilia biogenesis by several centrosome-related mechanisms. J Cell Sci. 2011, 124: 2539-2551. 10.1242/jcs.085852.
Pedersen LB, Geimer S, Sloboda RD, Rosenbaum JL: The Microtubule plus end-tracking protein EB1 is localized to the flagellar tip and basal bodies in Chlamydomonas reinhardtii. Curr Biol. 2003, 13: 1969-1974. 10.1016/j.cub.2003.10.058.
Schroder JM, Schneider L, Christensen ST, Pedersen LB: EB1 is required for primary cilia assembly in fibroblasts. Curr Biol. 2007, 17: 1134-1139. 10.1016/j.cub.2007.05.055.
Rosenbaum JL, Witman GB: Intraflagellar transport. Nat Rev Mol Cell Biol. 2002, 3: 813-825. 10.1038/nrm952.
Pedersen LB, Rosenbaum JL: Intraflagellar transport (IFT) role in ciliary assembly, resorption and signalling. Curr Top Dev Biol. 2008, 85: 23-61.
Cole DG, Diener DR, Himelblau AL, Beech PL, Fuster JC, Rosenbaum JL: Chlamydomonas kinesin-II-dependent intraflagellar transport (IFT): IFT particles contain proteins required for ciliary assembly in Caenorhabditis elegans sensory neurons. J Cell Biol. 1998, 141: 993-1008. 10.1083/jcb.141.4.993.
Pazour GJ, Wilkerson CG, Witman GB: A dynein light chain is essential for the retrograde particle movement of intraflagellar transport (IFT). J Cell Biol. 1998, 141: 979-992. 10.1083/jcb.141.4.979.
Ishikawa H, Marshall WF: Ciliogenesis: building the cell's antenna. Nat Rev Mol Cell Biol. 2011, 12: 222-234. 10.1038/nrm3085.
Murcia NS, Richards WG, Yoder BK, Mucenski ML, Dunlap JR, Woychik RP: The Oak Ridge Polycystic Kidney (orpk) disease gene is required for left-right axis determination. Development. 2000, 127: 2347-2355.
Nonaka S, Tanaka Y, Okada Y, Takeda S, Harada A, Kanai Y, Kido M, Hirokawa N: Randomization of left-right asymmetry due to loss of nodal cilia generating leftward flow of extraembryonic fluid in mice lacking KIF3B motor protein. Cell. 1998, 95: 829-837. 10.1016/S0092-8674(00)81705-5.
Takeda S, Yonekawa Y, Tanaka Y, Okada Y, Nonaka S, Hirokawa N: Left-right asymmetry and kinesin superfamily protein KIF3A: new insights in determination of laterality and mesoderm induction by kif3A-/- mice analysis. J Cell Biol. 1999, 145: 825-836. 10.1083/jcb.145.4.825.
Pazour GJ, Dickert BL, Vucica Y, Seeley ES, Rosenbaum JL, Witman GB, Cole DG: Chlamydomonas IFT88 and its mouse homologue, polycystic kidney disease gene tg737, are required for assembly of cilia and flagella. J Cell Biol. 2000, 151: 709-718. 10.1083/jcb.151.3.709.
Marshall WF, Rosenbaum JL: Intraflagellar transport balances continuous turnover of outer doublet microtubules: implications for flagellar length control. J Cell Biol. 2001, 155: 405-414. 10.1083/jcb.200106141.
Deretic D: A role for rhodopsin in a signal transduction cascade that regulates membrane trafficking and photoreceptor polarity. Vision Res. 2006, 46: 4427-4433. 10.1016/j.visres.2006.07.028.
Young RW: Passage of newly formed protein through the connecting cilium of retina rods in the frog. J Ultrastruct Res. 1968, 23: 462-473. 10.1016/S0022-5320(68)80111-X.
Young RW, Droz B: The renewal of protein in retinal rods and cones. J Cell Biol. 1968, 39: 169-184. 10.1083/jcb.39.1.169.
Pazour GJ, Baker SA, Deane JA, Cole DG, Dickert BL, Rosenbaum JL, Witman GB, Besharse JC: The intraflagellar transport protein, IFT88, is essential for vertebrate photoreceptor assembly and maintenance. J Cell Biol. 2002, 157: 103-113. 10.1083/jcb.200107108.
Jimeno D, Feiner L, Lillo C, Teofilo K, Goldstein LS, Pierce EA, Williams DS: Analysis of kinesin-2 function in photoreceptor cells using synchronous Cre-loxP knockout of Kif3a with RHO-Cre. Invest Ophthalmol Vis Sci. 2006, 47: 5039-5046. 10.1167/iovs.06-0032.
Marszalek JR, Liu X, Roberts EA, Chui D, Marth JD, Williams DS, Goldstein LS: Genetic evidence for selective transport of opsin and arrestin by kinesin-II in mammalian photoreceptors. Cell. 2000, 102: 175-187. 10.1016/S0092-8674(00)00023-4.
Krock BL, Perkins BD: The intraflagellar transport protein IFT57 is required for cilia maintenance and regulates IFT-particle-kinesin-II dissociation in vertebrate photoreceptors. J Cell Sci. 2008, 121: 1907-1915. 10.1242/jcs.029397.
Sukumaran S, Perkins BD: Early defects in photoreceptor outer segment morphogenesis in zebrafish ift57, ift88 and ift172 Intraflagellar Transport mutants. Vision Res. 2009, 49: 479-489. 10.1016/j.visres.2008.12.009.
Haycraft CJ, Zhang Q, Song B, Jackson WS, Detloff PJ, Serra R, Yoder BK: Intraflagellar transport is essential for endochondral bone formation. Development. 2007, 134: 307-316. 10.1242/dev.02732.
Huangfu D, Anderson KV: Cilia and Hedgehog responsiveness in the mouse. Proc Natl Acad Sci USA. 2005, 102: 11325-11330. 10.1073/pnas.0505328102.
Zhang Q, Murcia NS, Chittenden LR, Richards WG, Michaud EJ, Woychik RP, Yoder BK: Loss of the Tg737 protein results in skeletal patterning defects. Dev Dyn. 2003, 227: 78-90. 10.1002/dvdy.10289.
Liu A, Wang B, Niswander LA: Mouse intraflagellar transport proteins regulate both the activator and repressor functions of Gli transcription factors. Development. 2005, 132: 3103-3111. 10.1242/dev.01894.
Marszalek JR, Ruiz-Lozano P, Roberts E, Chien KR, Goldstein LS: Situs inversus and embryonic ciliary morphogenesis defects in mouse mutants lacking the KIF3A subunit of kinesin-II. Proc Natl Acad Sci USA. 1999, 96: 5043-5048. 10.1073/pnas.96.9.5043.
Tran PV, Haycraft CJ, Besschetnova TY, Turbe-Doan A, Stottmann RW, Herron BJ, Chesebro AL, Qiu H, Scherz PJ, Shah JV, Yoder BK, Beier DR: THM1 negatively modulates mouse sonic hedgehog signal transduction and affects retrograde intraflagellar transport in cilia. Nat Genet. 2008, 40: 403-410. 10.1038/ng.105.
Cortellino S, Wang C, Wang B, Bassi MR, Caretti E, Champeval D, Calmont A, Jarnik M, Burch J, Zaret KS, Larue L, Bellacosa A: Defective ciliogenesis, embryonic lethality and severe impairment of the Sonic Hedgehog pathway caused by inactivation of the mouse complex A intraflagellar transport genes Ift122/Wdr10, partially overlapping with the DNA repair gene Med1/Mbd4. Dev Biol. 2009, 325: 225-237. 10.1016/j.ydbio.2008.10.020.
Ohazama A, Haycraft CJ, Seppala M, Blackburn J, Ghafoor S, Cobourne M, Martinelli DC, Fan CM, Peterkova R, Lesot H, Yoder BK, Sharpe PT: Primary cilia regulate Shh activity in the control of molar tooth number. Development. 2009, 136: 897-903. 10.1242/dev.027979.
Lunt SC, Haynes T, Perkins BD: Zebrafish ift57, ift88, and ift172 intraflagellar transport mutants disrupt cilia but do not affect hedgehog signaling. Dev Dyn. 2009, 238: 1744-1759. 10.1002/dvdy.21999.
Huang P, Schier AF: Dampened Hedgehog signaling but normal Wnt signaling in zebrafish without cilia. Development. 2009, 136: 3089-3098. 10.1242/dev.041343.
Han YG, Kwok BH, Kernan MJ: Intraflagellar transport is required in Drosophila to differentiate sensory cilia but not sperm. Curr Biol. 2003, 13: 1679-1686. 10.1016/j.cub.2003.08.034.
Ray K, Perez SE, Yang Z, Xu J, Ritchings BW, Steller H, Goldstein LS: Kinesin-II is required for axonal transport of choline acetyltransferase in Drosophila. J Cell Biol. 1999, 147: 507-518. 10.1083/jcb.147.3.507.
Follit JA, Tuft RA, Fogarty KE, Pazour GJ: The intraflagellar transport protein IFT20 is associated with the Golgi complex and is required for cilia assembly. Mol Biol Cell. 2006, 17: 3781-3792. 10.1091/mbc.E06-02-0133.
Follit JA, San Agustin JT, Xu F, Jonassen JA, Samtani R, Lo CW, Pazour GJ: The Golgin GMAP210/TRIP11 anchors IFT20 to the Golgi complex. PLoS Genet. 2008, 4: e1000315-10.1371/journal.pgen.1000315.
Tobin JL, Beales PL: The nonmotile ciliopathies. Genet Med. 2009, 11: 386-402. 10.1097/GIM.0b013e3181a02882.
Jin H, Nachury MV: The BBSome. Curr Biol. 2009, 19: R472-R473. 10.1016/j.cub.2009.04.015.
Seo S, Baye LM, Schulz NP, Beck JS, Zhang Q, Slusarski DC, Sheffield VC: BBS6, BBS10, and BBS12 form a complex with CCT/TRiC family chaperonins and mediate BBSome assembly. Proc Natl Acad Sci USA. 2010, 107: 1488-1493. 10.1073/pnas.0910268107.
Zaghloul NA, Katsanis N: Mechanistic insights into Bardet-Biedl syndrome, a model ciliopathy. J Clin Invest. 2009, 119: 428-437. 10.1172/JCI37041.
Leitch CC, Zaghloul NA, Davis EE, Stoetzel C, Diaz-Font A, Rix S, Alfadhel M, Lewis RA, Eyaid W, Banin E, Dollfus H, Beales PL, Badano JL, Katsanis N: Hypomorphic mutations in syndromic encephalocele genes are associated with Bardet-Biedl syndrome. Nat Genet. 2008, 40: 443-448. 10.1038/ng.97.
Valente EM, Silhavy JL, Brancati F, Barrano G, Krishnaswami SR, Castori M, Lancaster MA, Boltshauser E, Boccone L, Al-Gazali L, Fazzi E, Signorini S, Louie CM, Bellacchio E, International Joubert Syndrome Related Disorders Study Group, Bertini E, Dallapiccola B, Gleeson JG: Mutations in CEP290, which encodes a centrosomal protein, cause pleiotropic forms of Joubert syndrome. Nat Genet. 2006, 38: 623-625. 10.1038/ng1805.
den Hollander AI, Koenekoop RK, Yzer S, Lopez I, Arends ML, Voesenek KE, Zonneveld MN, Strom TM, Meitinger T, Brunner HG, Hoyng CB, van den Born LI, Rohrschneider K, Cremers FP: Mutations in the CEP290 (NPHP6) gene are a frequent cause of Leber congenital amaurosis. Am J Hum Genet. 2006, 79: 556-561. 10.1086/507318.
Betleja E, Cole DG: Ciliary trafficking: CEP290 guards a gated community. Curr Biol. 2010, 20: R928-R931. 10.1016/j.cub.2010.09.058.
Lancaster MA, Gopal DJ, Kim J, Saleem SN, Silhavy JL, Louie CM, Thacker BE, Williams Y, Zaki MS, Gleeson JG: Defective Wnt-dependent cerebellar midline fusion in a mouse model of Joubert syndrome. Nat Med. 2011, 17: 726-731. 10.1038/nm.2380.
Lancaster MA, Louie CM, Silhavy JL, Sintasath L, Decambre M, Nigam SK, Willert K, Gleeson JG: Impaired Wnt-beta-catenin signaling disrupts adult renal homeostasis and leads to cystic kidney ciliopathy. Nat Med. 2009, 15: 1046-1054. 10.1038/nm.2010.
Lancaster MA, Schroth J, Gleeson JG: Subcellular spatial regulation of canonical Wnt signalling at the primary cilium. Nat Cell Biol. 2011, 13: 700-707.
Acknowledgements
This work was supported in part by the National Institutes of Health [MH71801 to R.J.F.] and the March of Dimes Foundation [5-FY09-29 to R.J.F.]. This work was also supported in part by the Empire State Stem Cell Fund through the New York State Department of Health Contract #C024324 to R.J.F. The opinions expressed here are solely those of the author and do not necessarily reflect those of the Empire State Stem Cell Board, the New York State Department of Health, or the State of New York. The authors wish to thank Linda Crane Bonin for critically reading our manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
YH wrote the first draft. KT made substantial changes to the manuscript. RJF commented, rewrote, and edited the draft, in addition to commenting on and editing the drafts. All authors read and approved the manuscript.
Yi-Chun Hsiao, Karina Tuz contributed equally to this work.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Hsiao, YC., Tuz, K. & Ferland, R.J. Trafficking in and to the primary cilium. Cilia 1, 4 (2012). https://doi.org/10.1186/2046-2530-1-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/2046-2530-1-4