
Abstract
Inflammaging is characterized by the upregulation of the inflammatory response that occurs with advancing age; its roots are strongly embedded in evolutionary theory.
Inflammaging is believed to be a consequence of a remodelling of the innate and acquired immune system, resulting in chronic inflammatory cytokine production.
Complex interrelated genetic, environmental and age-related factors determine an individual’s vulnerability or resilience to inflammaging. These factors include polymorphisms to the promoter regions of cytokines, cytokine receptors and antagonists, age-related decreases in autophagy and increased adiposity. Anti-inflammaging describes the upregulation of the hypothalamic-pituitary axis in response to inflammaging, leading to higher levels of cortisol, which in turn may be detrimental, contributing to less successful ageing and frailty. This may be countered by the adrenal steroid dehydroepiandrosterone, which itself declines with age, leaving certain individuals more vulnerable. Inflammaging and anti-inflammaging have both been linked with a number of age-related outcomes, including chronic morbidity, functional decline and mortality. This important area of research offers unique insights into the ageing process and the potential for screening and targeted interventions.
Similar content being viewed by others


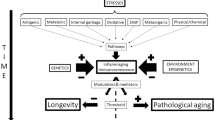
Review
Introduction
The immune system protects an organism from disease. The first reference to immunity dates back to the plague of Athens in 430 BC when it was noticed that people who recovered from the disease could then nurse others without contracting it a second time. It is recognized that the immune system, comprising both innate (nonspecific) and acquired (specific) components, is an intricate defence system that is highly conserved across vertebrate species, and has, from an evolutionary perspective, undergone strong pressures to maximize survival to allow procreation. The significant improvements in human survival and lifespan to well beyond childbearing ages have been totally ‘unpredicted’ by evolution. As a consequence, human immune systems are exposed to considerable additional antigenic exposure outside the forces of natural selection. It is in this situation that immunity begins to exert negative effects on human ageing (antagonistic pleiotropy), leading to gradual systemic failures [1, 2].
The immune system of older people declines in reliability and efficiency with age, resulting in greater susceptibility to pathology as a consequence of inflammation, for example, cardiovascular disease, Alzheimer's disease, autoreactivity and vaccine failure, as well as an increased vulnerability to infectious disease [3–5]. These changes are further compounded by reduced responsiveness and impaired communication between all cells of the immune system. The overall change to the immune system with age is termed immunosenescence and has a multifactorial aetiology; a consequence of the complexity of the immune system as well as of multiple genetic and environmental influences [3]. Immunosenescence of the innate immune system is primarily characterized by reduced cellular superoxide production and capability for phagocytosis. Involution of the thymus and reduced responsiveness to new antigen load, owing to reduced naïve: memory cell ratio and expansion of mature cell clones, characterizes immunosenescence of the acquired immune system (Table 1).
Research into age-related changes of the immune system is gathering pace as its importance within the context of multiple pathologies in ageing populations is realized. As part of this advance, Franceschi and colleagues [6] described the phenomenon of ‘inflammaging’ at the turn of the millennium as part of the spectrum of immunosenescence. Inflammaging denotes an upregulation of the inflammatory response that occurs with age, resulting in a low-grade chronic systemic proinflammatory state. It is characterized by raised levels of proinflammatory cytokines interleukin-1 (IL-1), interleukin-6 (IL-6) and tumour necrosis factor (TNF); all of which have been shown to rise with age [7] and be involved in the pathogenesis of most age-associated diseases [8]. C-reactive protein (CRP), an acute phase protein produced by the liver in response to IL-6, is also a useful marker of inflammaging, more commonly used in clinical practice, as well as a robust predictor of risk for cardiovascular and other diseases [9, 10].
Inflammaging
Inflammaging is believed to be a consequence of a cumulative lifetime exposure to antigenic load caused by both clinical and subclinical infections as well as exposure to noninfective antigens [1]. The consequent inflammatory response, tissue damage and production of reactive oxygen species that cause oxidative damage also elicits the release of additional cytokines, principally from cells of the innate immune system [11] but also from the acquired immune response. This results in a vicious cycle, driving immune system remodelling and favouring a chronic proinflammatory state where pathophysiological changes, tissue injury and healing proceed simultaneously. Irreversible cellular and molecular damage that is not clinically evident slowly accumulates over decades (Figure 1).

Cycle of inflammaging.
Immunosenescence of the acquired system has received a great deal of attention in recent years and is by a cellular ‘exhaustion’, which can contribute to inflammaging. Thymic output is reduced with age resulting in reduced T-cell repertoire and increased oligoclonal expansion of memory and effector-memory cells [12]. This imbalance results in a reduced ability to clear novel pathogens (prolonging infection duration) as well as an increase in functionally distinct T-cell populations, which have an amplified proinflammatory phenotype [13]. The CD8+ T cell population is altered to a greater extent than the CD4+ population and is associated with specificity to single antigens, particularly latent viral infections, such as cytomegalovirus, Epstein-Barr and varicella zoster virus. Increases in numbers of antigen-specific cells with age are associated with an increase in the number of terminally differentiated ‘senescent’ cells, which occupy a large proportion of immune space. These cells, particularly CD8+, are extremely potent producers of inflammatory cytokines and have been heavily associated with reduced antiviral immunity and inflammatory related pathologies [14]. With age, these antigen-specific cells do not proliferate as well, and on mitogenic and viral stimulation produce more IL-6 and TNF than their younger counterparts [15]. Subsequently antigenic load is a known driver of immunosenescence and has been associated with increased IL-6 and mortality [16]. Although it was recently shown that cytomegalovirus infection did not drive inflammaging (IL-6, TNF and CRP) in healthy older people over a 10-year period, the study could not account for other latent infections, suggesting a combined role of other antigenic stimulus in systemic inflammaging [17]. Therefore, T-cell immunosenescence is characterized by an increased proinflammatory phenotype and most probably contributes to inflammaging in a manner dependent on previous exposure to and reactivation of antigenic challenges and exhaustion of T-cell repertoire.
Of the innate immune system, monocytes and macrophages are suggested to contribute to inflammaging more than any other cell type. Monocyte changes with age contribute to inflammaging by a functional shift towards a proinflammatory phenotype and reduced function [18]. Monocytes consist of three distinct subtypes, differentiated by expression of CD14 and CD16; CD14++/CD16− (classical), CD14++/CD16+ (intermediate) and CD14+/CD16++ (nonclassical), with varying degrees of functional capabilities [19]. The CD16+ positive monocytes constitutively produce more IL-6, IL-1β and TNF basally and with stimulation, with older people having a significantly larger proportion of CD16+ cells than younger people [20, 21]. Furthermore, the CD16+ population have increased adherence and migrate towards endothelial lesions via CX3CR1, contributing to increased atheroma plaque formation [22]. Subsequently, monocytes can contribute to inflammaging by chronically increased production of inflammatory cytokines and prolongation of the immune response.
A two-hit hypothesis states that the extent to which an individual is vulnerable to inflammaging and its physiological consequences is dependent first on the degree of increased proinflammatory environment and then on the resilience of that individual to withstand it. This is reflected in part by the balance of increasing levels of proinflammatory cytokines with anti-inflammatory cytokines, and also concentrations of soluble cytokine receptors and cytokine receptor antagonists. These are believed to affect the degree to which an individual is susceptible to an antigenic load, the rate at which age-related pathologies progress and individual vulnerability to frailty [2, 7] (Figure 2). Indeed, centenarians, a population deemed to have aged successfully, have strong and effective anti-inflammatory responses with a reduced proinflammatory capacity [23]. This degree of susceptibility is multifactorial and a consequence of complex genetic and environmental interactions as well as of the ageing process itself.

Relationship between pro- and anti-inflammation and ageing.
Factors influencing susceptibility to inflammaging
There is now convincing evidence that susceptibility to inflammaging is, in part, genetically determined through polymorphisms of promoter regions of genes that either affect cytokine production rates or alter their protein structure to produce a functional variant. For example, polymorphisms in the promoter C/G 174 on the IL-6 gene have been shown to affect serum IL-6 concentrations [24]. Similar associations have been found in alleles for IL-10 [25] and polymorphisms of the toll-like receptor 4 residing on the membrane of dendritic cells. Macrophages positively affect inflammatory responses and are associated with an increased risk of acute myocardial infarction [26].
Telomeres may also be implicated; these are protein complexes made from several thousand repetitive DNA sequences located at the end of chromosomes to protect them from deterioration or fusion. Telomere repetitive sequences shorten with each cell division, eventually reaching a critical number leading to cellular senescence and death. There is evidence that this process is associated with inflammation, although this is controversial, and the direction of this relationship is unclear and could be bi-directional. For example, raised IL-6 and TNF was associated with shorter telomere length in 1,962 individuals of the Health, Ageing and Body Composition Study [27]. Shorter telomere length has also been found in cohorts of patients with chronic inflammatory diseases of liver, kidney and lung [28]. Negative relationships with telomere length have also been demonstrated with CRP [29] and IL-6 [30].
Ageing is associated with decreases in autophagy mechanisms, further contributing to proinflammatory environments [31]. Autophagy is a cellular housekeeping mechanism that is responsible for the removal of dysfunctional intracellular protein (for example, dead organelles, damaged scaffold proteins) via lysomal degradation. This prevents the activation of inflammasomes; intracellular multi-protein sensors that stimulate the inflammatory response after recognizing danger signals emanating from proteins, such as the intracellular danger-associated molecular patterns that occur as a consequence of either tissue injury or necrosis. The consequence of a decline of autophagy with age is therefore an increased activation of the inflammasome and greater proinflammatory responses [32].
Ageing, though often visually characterized by a decrease in subcutaneous tissue and sarcopenia [33], is associated with a linear accumulation of adipose tissue, both around the viscera and via the fatty infiltration of several organs, including liver, bone and muscle. This adipose tissue, once thought of as inert, is now considered a major endocrine and paracrine organ [34]; to date, approximately one hundred adipokines have been identified, falling into several functional groups [35]; many are proinflammatory. For example, the classic adipokine, leptin, not only has a primary role in energy homeostasis but also causes the stimulation and differentiation of monocytes into macrophages, activates NK-lymphocytes and induces the production of a number of proinflammatory cytokines, including TNF and IL-6 [36]. Consequently, greater degrees of adiposity are directly asso ciated with greater proinflammatory environments, partly via proinflammatory adipokines and also from immune cells residing within adipose tissue.
Anti-inflammaging
As with all complex organisms, single biological systems rarely work in isolation. There is extensive cross-talk between the immune and endocrine axes, which, together with the neural system, form the major communication network in the body [37].
Cortisol is a glucocorticoid hormone secreted by the adrenal gland in response to pituitary secretion of adrenal corticotrophic hormone, which itself is stimulated by corticotrophin releasing hormone from the hypothalamus; together these form the hypothalamic-pituitary-adrenal (HPA) axis. Cortisol plays key roles in the stress response and is also immunosuppressive [38]. Neuronal cells within the HPA axis contain multiple cytokine receptors, particularly for IL-1, IL-6 and TNF [39], and it has been demonstrated in human beings in vivo that injection of IL-6 or TNF induces a marked change in HPA axis [40]. Therefore, an inevitable physiological response to inflammaging is an increase in circulating cortisol levels [41].
This mechanism has been termed anti-inflammaging and although it represents an appropriate attempt to counter the inflammaging process it may also have negative implications. These include the paradox of both inflammaging and the global immunosuppression seen with increasing age, as well as associations with frailty via catabolic effects on several tissue types, such as liver (gluconeogenesis), muscle (protein catabolism) and bone (resorption) [41].
Dehydroepiandrosterone (DHEA) and its sulphated precursor, DHEA sulphate (DHEAS), have opposing actions to cortisol and may protect individuals from the negative effects of anti-inflammaging. They are secreted from the adrenal cortex and, in smaller amounts from the testis and ovary, although subsequently converted to sex steroids in the latter; they account for 30% to 50% of testosterone in men and 100% of oestrogen in post-menopausal women. Like cortisol, DHEAS is also secreted in response to adrenal corticotrophic hormone but serum levels are more stable as a consequence of stronger binding to albumin [42]. Circulating DHEAS concentrations reach a maximum in early adulthood and subsequently decline with age to approximately 10% to 20% of maximal levels by 70 years [43]. There is large interindividual variability in circulating DHEAS concentrations; some post-menopausal women have barely detectable serum concentrations whilst others have normal values. The fall in DHEAS is accompanied by a parallel fall in androgens, resulting in women with low DHEAS levels experiencing a deficit of sex steroids for their remaining lifetime and negative implications for the ageing process [43].
More recently, an immunomodulating role for DHEAS has been realized [38]; low levels are associated with chronic inflammatory conditions and the molecule counteracts the effects of cortisol via antagonism of glucocorticoid receptors, either directly or via its downstream metabolites. Further, DHEAS may directly inhibit glucocorticoid receptor production. This importance as a counterbalance to cortisol has been demonstrated in a number of in-vitro studies where the immunosuppression of neutrophil function that occurs with cortisol has been successfully overcome by co-incubation with DHEAS [44]. Additionally, a number of studies have consistently demonstrated the steroid to be an enhancer of IL-2 secretion from T-helper 1 (Th1) cells whilst negatively affecting secretion from T-helper 2 (Th2) cells. Therefore, it has been suggested that observed declines in DHEAS levels with age, at least in part, explains the cytokine dysregulation seen with age and specifically the shift towards Th2 cytokine profiles.
From this discussion it is apparent that both cortisol and DHEAS have opposing effects relating to the immune system. Cortisol causes immune-suppression and its concentration increases with age, whilst DHEA(S) antagonizes the effects of cortisol and is immune modulating, and its concentration falls with age. Therefore, when considering the effects of the HPA axis on inflammaging, the ratio of cortisol to DHEAS (Figure 3) may more accurately reflect the HPA axis than either cortisol or DHEAS alone. An in-vivo study of cortisol and DHEAS in patients with septic shock after a hip fracture showed that higher cortisol levels, lower DHEAS levels, and a higher cortisol: DHEAS ratio correlated with poorer clinical outcomes [45]. This ratio has also been positively associated with metabolic syndrome and all-cause, cancer and other medical cause mortality in Vietnam veterans [38, 46].

Representation of changes in adrenocorticoid hormones with age.
Cortisol: DHEAS ratios have the potential for manipulation via DHEAS supplementation. To date, DHEAS supplementation has been trialled in a number of chronic inflammatory conditions, usually with nonbeneficial outcomes. However, doses used have always been low. The only true beneficial effect of DHEAS supplementation has been seen in patients with systemic lupus erythematosus, where the drug successfully reduces disease activity and the number of exacerbations [47–49].
Associations with chronic disease and ageing
Inflammaging, in particular elevations in levels of TNF, IL-6, IL-1 and CRP, are strong independent risk factors for morbidity and mortality in older people [50]. Epidemiological and mechanistic studies suggest that many age-related diseases are initiated or worsened by systemic inflammation, including neurodegeneration and atherosclerosis [51, 52]. For example, proinflammatory cytokines have been shown to interact with the processing and production of Aβ peptide, the pathological hallmark feature of Alzheimer’s disease, and autopsy studies have shown that the brains of patients who had Alzheimer’s disease have dramatically higher levels of proinflammatory markers than high-pathology controls who have pathological features of Alzheimer’s disease without its symptoms [53].
Inflammaging also affects the anabolic-catabolic balance within myocytes, causing a shift towards catabolism, atrophy and progression of sarcopenia; this is a major contributor to functional decline and frailty. As with other age-related disease processes, it is believed that the degree of shift towards catabolism affects the rate of sarcopenic decline, and cachexia in older people may represent this process in extremis. Proinflammatory cytokines appear to play a particularly important role through activation of NF-κB and the ubiquitin system which is implicated in myofibre proteolysis [54]. It may be that interindividual variations in vulnerability to inflammaging in part explain the different rates at which individuals become sarcopenic. A number of community-based observational studies have demonstrated associations between proinflammatory cytokine profiles and sarcopenia. For example, cross-sectional analysis from the InCHIANTI study (1,020 men and women over 65 years) demonstrated a significant association between inflammation (IL-6, IL-1R, CRP) and both poor physical performance and reduced muscle strength [55]. Longitudinal associations also exist. Schaap used data from 2,177 men and women, (mean age 73), to show significant associations between TNF and 5-year change in muscle mass [56]. Associations with sarcopenia have also been demonstrated in the Framingham Heart Study (IL-6) [57] and the Longitudinal Aging Study Amsterdam (IL-6, CRP) [58] and have recently undergone systematic review [59]. Inflammaging is also implicated in the progression of osteoporosis [60] and dementia [51]; both strongly implicated in the frailty syndrome.
Higher cortisol levels have been associated with increased mortality in patients with stroke [61], sepsis [62], heart failure [63] and sarcopenia [64]. Low levels of DHEA have been demonstrated in patients with chronic inflammatory diseases, including inflammatory bowel disease, rheumatoid arthritis, systemic lupus erythematosus and pemphigus [65]. Moreover, associations have also been established between low DHEAS concentrations and cardiovascular disease, sarcopenia, osteoporosis and all-cause mortality [66–68].
The cumulative consequence of having a greater degree of inflammaging and anti-inflammaging is increased susceptibility to, and a faster progression of, all age-related diseases. This results in an increased vulnerability to stressors and reduced functional ability and is associated with the development of the frailty syndrome [50]. Interestingly, a 10-year study of 254 healthy, community-dwelling older people demonstrated that those individuals with baseline markers of greater inflammaging (white cell count) and anti-inflammaging (cortisol to DHEAS ratio) were significantly more likely to be frail at 10-year follow-up. Intriguingly, the authors speculate that this might offer a tool to screen for people who are ‘less robust’ in late middle age to allow early intervention before functional decline and the development of frailty [50].
Conclusions
Inflammaging is a pathological phenomenon and a central concept that brings together our understanding of age-related chronic disease, functional decline and frailty across the lifecourse. It is a consequence of lifelong exposure of the immune system to antigenic stimuli and complex genetic, environmental and age-related mechanisms that expose varying degrees of vulnerability or resilience. An important consequence of inflammaging is activation of the HPA axis, anti-inflammaging and a widening of the gap between molar concentrations of cortisol and DHEA(S). This ‘double-edged’ sword provides an appropriate counter to inflammaging but also has multi-system consequences in terms of disease progression and immunosuppression.
As populations age, a better understanding of these processes is essential to identify older people who are at risk of ageing-related chronic diseases, in order to facilitate successful, early, targeted interventions.
Authors’ information
DB is supported by a National Institute of Health Research (NIHR) Doctoral Research Fellowship. DBB is supported by a Biotechnology and Biological Sciences Research Council grant. HPP is supported by the University of Southampton National Institute of Health Clinical Lectureship Scheme. HCR is a Senior Clinical Lecturer in Geriatric Medicine at the University of Southampton.
Abbreviations
- CRP C:
-
Reactive protein
- DHEAS:
-
DHEA sulphate
- HPA:
-
Hypothalamic-pituitary-adrenal
- IL:
-
Interleukin
- MHC:
-
Major histocompatibility complex
- NIHR:
-
National Institute of Health Research
- Th:
-
T-helper
- TNF:
-
Tumour necrosis factor
References
De Martinis M, Franceschi C, Monti D, Ginaldi L: Inflamm-ageing and lifelong antigenic load as major determinants of ageing rate and longevity. FEBS Lett. 2005, 579 (10): 2035-2039. 10.1016/j.febslet.2005.02.055.
Franceschi C, Capri M, Monti D, Giunta S, Olivieri F, Sevini F, Panourgia MP, Invidia L, Celani L, Scurti M, Cevenini E, Castellani GC, Salvioli S: Inflammaging and anti-inflammaging: a systemic perspective on aging and longevity emerged from studies in humans. Mech Ageing Dev. 2007, 128 (1): 92-105. 10.1016/j.mad.2006.11.016.
Weiskopf D, Weinberger B, Grubeck-Loebenstein B: The aging of the immune system. Transpl Int. 2009, 22 (11): 1041-1050. 10.1111/j.1432-2277.2009.00927.x.
Targonski PV, Jacobson RM, Poland GA: Immunosenescence: role and measurement in influenza vaccine response among the elderly. Vaccine. 2007, 25 (16): 3066-3069. 10.1016/j.vaccine.2007.01.025.
Pawelec G: Immunosenescence: impact in the young as well as the old?. Mech Ageing Dev. 1999, 108 (1): 1-7.
Franceschi C, Bonafe M, Valensin S, Olivieri F, De LM, Ottaviani E, De Benedictis G: Inflamm-aging. An evolutionary perspective on immunosenescence. Ann NY Acad Sci. 2000, 908: 244-254.
Vasto S, Candore G, Balistreri CR, Caruso M, Colonna-Romano G, Grimaldi MP, Listi F, Nuzzo D, Lio D, Caruso C: Inflammatory networks in ageing, age-related diseases and longevity. Mech Ageing Dev. 2007, 128 (1): 83-91. 10.1016/j.mad.2006.11.015.
De Martinis M, Franceschi C, Monti D, Ginaldi L: Inflammation markers predicting frailty and mortality in the elderly. Exp Mol Pathol. 2006, 80 (3): 219-227. 10.1016/j.yexmp.2005.11.004.
Ansar W, Ghosh S: C-reactive protein and the biology of disease. Immunol Res. 2013, 56 (1): 131-142. 10.1007/s12026-013-8384-0.
Buckley DI, Fu R, Freeman M, Rogers K, Helfand M: C-reactive protein as a risk factor for coronary heart disease: a systematic review and meta-analyses for the U.S. Preventive Services Task Force. Ann Intern Med. 2009, 151 (7): 483-495. 10.7326/0003-4819-151-7-200910060-00009.
Cannizzo ES, Clement CC, Sahu R, Follo C, Santambrogio L: Oxidative stress, inflamm-aging and immunosenescence. J Proteomics. 2011, 74 (11): 2313-2323. 10.1016/j.jprot.2011.06.005.
Pawelec G: Hallmarks of human “immunosenescence”: adaptation or dysregulation?. Immun Ageing. 2012, 9 (1): 15-10.1186/1742-4933-9-15.
Fagiolo U, Cossarizza A, Scala E, Fanales-Belasio E, Ortolani C, Cozzi E, Monti D, Franceschi C, Paganelli R: Increased cytokine production in mononuclear cells of healthy elderly people. Eur J Immunol. 1993, 23 (9): 2375-2378. 10.1002/eji.1830230950.
Effros RB, Dagarag M, Spaulding C, Man J: The role of CD8+ T-cell replicative senescence in human aging. Immunol Rev. 2005, 205: 147-157. 10.1111/j.0105-2896.2005.00259.x.
Vescovini R, Biasini C, Fagnoni FF, Telera AR, Zanlari L, Pedrazzoni M, Bucci L, Monti D, Medici MC, Chezzi C, Franceschi C, Sansoni P: Massive load of functional effector CD4+ and CD8+ T cells against cytomegalovirus in very old subjects. J Immunol. 2007, 179 (6): 4283-4291.
Wikby A, Nilsson BO, Forsey R, Thompson J, Strindhall J, Löfgren S, Ernerudh J, Pawelec G, Ferguson F, Johansson B: The immune risk phenotype is associated with IL-6 in the terminal decline stage: findings from the Swedish NONA immune longitudinal study of very late life functioning. Mech Ageing Dev. 2006, 127 (8): 695-704. 10.1016/j.mad.2006.04.003.
Bartlett DB, Firth CM, Phillips AC, Moss P, Baylis D, Syddall H, Sayer AA, Cooper C, Lord JM: The age-related increase in low-grade systemic inflammation (Inflammaging) is not driven by cytomegalovirus infection. Aging Cell. 2012, 11 (5): 912-915. 10.1111/j.1474-9726.2012.00849.x.
Shaw AC, Joshi S, Greenwood H, Panda A, Lord JM: Aging of the innate immune system. Curr Opin Immunol. 2010, 22 (4): 507-513. 10.1016/j.coi.2010.05.003.
Ziegler-Heitbrock L, Ancuta P, Crowe S, Dalod M, Grau V, Hart DN, Leenen PJ, Liu YJ, MacPherson G, Randolph GJ, Scherberich J, Schmitz J, Shortman K, Sozzani S, Strobl H, Zembala M, Austyn JM, Lutz MB: Nomenclature of monocytes and dendritic cells in blood. Blood. 2010, 116 (16): e74-e80. 10.1182/blood-2010-02-258558.
Belge KU, Dayyani F, Horelt A, Siedlar M, Frankenberger M, Frankenberger B, Espevik T, Ziegler-Heitbrock L: The proinflammatory CD14+CD16+DR++ monocytes are a major source of TNF. J Immunol. 2002, 168 (7): 3536-3542.
Sadeghi HM, Schnelle JF, Thoma JK, Nishanian P, Fahey JL: Phenotypic and functional characteristics of circulating monocytes of elderly persons. Exp Gerontol. 1999, 34 (8): 959-970. 10.1016/S0531-5565(99)00065-0.
Stolla M, Pelisek J, von Brühl ML, Schäfer A, Barocke V, Heider P, Lorenz M, Tirniceriu A, Steinhart A, Bauersachs J, Bray PF, Massberg S, Schulz C: Fractalkine is expressed in early and advanced atherosclerotic lesions and supports monocyte recruitment via CX3CR1. PLoS One. 2012, 7 (8): e43572-10.1371/journal.pone.0043572.
Bruunsgaard H, Andersen-Ranberg K, Hjelmborg JB, Pedersen BK, Jeune B: Elevated levels of tumor necrosis factor alpha and mortality in centenarians. Am J Med. 2003, 115 (4): 278-283. 10.1016/S0002-9343(03)00329-2.
Rea IM, Ross OA, Armstrong M, McNerlan S, Alexander DH, Curran MD, Middleton D: Interleukin-6-gene C/G 174 polymorphism in nonagenarian and octogenarian subjects in the BELFAST study. Reciprocal effects on IL-6, soluble IL-6 receptor and for IL-10 in serum and monocyte supernatants. Mech Ageing Dev. 2003, 124 (4): 555-561. 10.1016/S0047-6374(03)00036-8.
Lio D, Candore G, Crivello A, Scola L, Colonna-Romano G, Cavallone L, Hoffmann E, Caruso M, Licastro F, Caldarera CM, Branzi A, Franceschi C, Caruso C: Opposite effects of interleukin 10 common gene polymorphisms in cardiovascular diseases and in successful ageing: genetic background of male centenarians is protective against coronary heart disease. J Med Genet. 2004, 41 (10): 790-794. 10.1136/jmg.2004.019885.
Balistreri CR, Candore G, Accardi G, Bova M, Buffa S, Bulati M, Forte GI, Listì F, Martorana A, Palmeri M, Pellicanò M, Vaccarino L, Scola L, Lio D, Colonna-Romano G: Genetics of longevity. Data from the studies on Sicilian centenarians. Immun Ageing. 2012, 9 (1): 8-10.1186/1742-4933-9-8.
O’Donovan A, Pantell MS, Puterman E, Dhabhar FS, Blackburn EH, Yaffe K, Cawthon RM, Opresko PL, Hsueh WC, Satterfield S, Newman AB, Ayonayon HN, Rubin SM, Harris TB, Epel ES, Health Aging and Body Composition Study: Cumulative inflammatory load is associated with short leukocyte telomere length in the Health, Aging and Body Composition Study. PLoS One. 2011, 6 (5): e19687-10.1371/journal.pone.0019687.
Houben JM, Moonen HJ, van Schooten FJ, Hageman GJ: Telomere length assessment: biomarker of chronic oxidative stress?. Free Radic Biol Med. 2008, 44 (3): 235-246. 10.1016/j.freeradbiomed.2007.10.001.
Aviv A, Valdes A, Gardner JP, Swaminathan R, Kimura M, Spector TD: Menopause modifies the association of leukocyte telomere length with insulin resistance and inflammation. J Clin Endocrinol Metab. 2006, 91 (2): 635-640.
Shiels PG S, McGlynn LM, MacIntyre A, Johnson PC, Batty GD, Burns H, Cavanagh J, Deans KA, Ford I, McConnachie A, McGinty A, McLean JS, Millar K, Sattar N, Tannahill C, Velupillai YN, Packard CJ: Accelerated telomere attrition is associated with relative household income, diet and inflammation in the pSoBid cohort. PLoS One. 2011, 6 (7): e22521-10.1371/journal.pone.0022521.
Pallauf K, Rimbach G: Autophagy, polyphenols and healthy ageing. Ageing Res Rev. 2012, 12 (1): 237-252.
Salminen A, Kaarniranta K, Kauppinen A: Inflammaging: disturbed interplay between autophagy and inflammasomes. Aging (Albany NY). 2012, 4 (3): 166-175.
Rudin E, Barzilai N: Inflammatory peptides derived from adipose tissue. Immun Ageing. 2005, 2 (1): 1-10.1186/1742-4933-2-1.
Trayhurn P, Drevon CA, Eckel J: Secreted proteins from adipose tissue and skeletal muscle - adipokines, myokines and adipose/muscle cross-talk. Arch Physiol Biochem. 2011, 117 (2): 47-56. 10.3109/13813455.2010.535835.
Trayhurn P, Wood IS: Adipokines: inflammation and the pleiotropic role of white adipose tissue. Br J Nutr. 2004, 92 (3): 347-355. 10.1079/BJN20041213.
de Heredia FP, Sonia G-M, Ascension M: Chronic and degenerative diseases: obesity, inflammation and the immune system. Proc Nutr Soc. 2012, 71: 332-338. 10.1017/S0029665112000092.
Straub RH, Cutolo M, Zietz B, Scholmerich J: The process of aging changes the interplay of the immune, endocrine and nervous systems. Mech Ageing Dev. 2001, 122 (14): 1591-1611. 10.1016/S0047-6374(01)00289-5.
Phillips AC, Carroll D, Gale CR, Lord JM, Arlt W, Batty GD: Cortisol, DHEA sulphate, their ratio, and all-cause and cause-specific mortality in the Vietnam Experience Study. Eur J Endocrinol. 2010, 163 (2): 285-292. 10.1530/EJE-10-0299.
Turnbull AV, Rivier CL: Regulation of the hypothalamic-pituitary-adrenal axis by cytokines: actions and mechanisms of action. Physiol Rev. 1999, 79 (1): 1-71.
Straub RH, Miller LE, Scholmerich J, Zietz B: Cytokines and hormones as possible links between endocrinosenescence and immunosenescence. J Neuroimmunol. 2000, 109 (1): 10-15. 10.1016/S0165-5728(00)00296-4.
Sergio G: Exploring the complex relations between inflammation and aging (inflamm-aging): anti-inflamm-aging remodelling of inflamm-aging, from robustness to frailty. Inflamm Res. 2008, 57 (12): 558-563. 10.1007/s00011-008-7243-2.
Rosenfeld RS, Rosenberg BJ, Fukushima DK, Hellman L: 24-hour secretory pattern of dehydroisoandrosterone and dehydroisoandrosterone sulfate. J Clin Endocrinol Metab. 1975, 40 (5): 850-855. 10.1210/jcem-40-5-850.
Labrie F: DHEA, important source of sex steroids in men and even more in women. Prog Brain Res. 2010, 182: 97-148.
Butcher SK, Killampalli V, Lascelles D, Wang K, Alpar EK, Lord JM: Raised cortisol:DHEAS ratios in the elderly after injury: potential impact upon neutrophil function and immunity. Aging Cell. 2005, 4 (6): 319-324. 10.1111/j.1474-9726.2005.00178.x.
Arlt W, Hammer F, Sanning P, Butcher SK, Lord JM, Allolio B, Annane D, Stewart PM: Dissociation of serum dehydroepiandrosterone and dehydroepiandrosterone sulfate in septic shock. J Clin Endocrinol Metab. 2006, 91 (7): 2548-2554. 10.1210/jc.2005-2258.
Phillips AC, Carroll D, Gale CR, Lord JM, Arlt W, Batty GD: Cortisol, DHEAS, their ratio and the metabolic syndrome: evidence from the Vietnam experience study. Eur J Endocrinol. 2010, 162 (5): 919-923. 10.1530/EJE-09-1078.
Petri MA, Mease PJ, Merrill JT, Lahita RG, Iannini MJ, Yocum DE, Ginzler EM, Katz RS, Gluck OS, Genovese MC, Van Vollenhoven R, Kalunian KC, Manzi S, Greenwald MW, Buyon JP, Olsen NJ, Schiff MH, Kavanaugh AF, Caldwell JR, Ramsey-Goldman R, St Clair EW, Goldman AL, Egan RM, Polisson RP, Moder KG, Rothfield NF, Spencer RT, Hobbs K, Fessler BJ, Calabrese LH: Effects of prasterone on disease activity and symptoms in women with active systemic lupus erythematosus. Arthritis Rheum. 2004, 50 (9): 2858-2868. 10.1002/art.20427.
Chang DM, Lan JL, Lin HY, Luo SF: Dehydroepiandrosterone treatment of women with mild-to-moderate systemic lupus erythematosus: a multicenter randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 2002, 46 (11): 2924-2927. 10.1002/art.10615.
Sawalha AH, Kovats S: Dehydroepiandrosterone in systemic lupus erythematosus. Curr Rheumatol Rep. 2008, 10 (4): 286-291. 10.1007/s11926-008-0046-1.
Baylis D, Bartlett DB, Syddall HE, Ntani G, Gale CR, Cooper C, Lord JM, Sayer AA: Immune-endocrine biomarkers as predictors of frailty and mortality: a 10-year longitudinal study in community-dwelling older people. Age (Dordr). 2012, 10.1007/s11357-012-9396-8.
Gao HM, Hong JS: Why neurodegenerative diseases are progressive: uncontrolled inflammation drives disease progression. Trends Immunol. 2008, 29 (8): 357-365. 10.1016/j.it.2008.05.002.
Libby P: Inflammation in atherosclerosis. Arterioscler Thromb Vasc Biol. 2012, 32 (9): 2045-2051. 10.1161/ATVBAHA.108.179705.
Giunta B, Fernandez F, Nikolic WV, Obregon D, Rrapo E, Town T, Tan J: Inflammaging as a prodrome to Alzheimer’s disease. J Neuroinflammation. 2008, 5: 51-10.1186/1742-2094-5-51.
Saini A, Faulkner S, Al-Shanti N, Stewart C: Powerful signals for weak muscles. Ageing Res Rev. 2009, 8 (4): 251-267. 10.1016/j.arr.2009.02.001.
Cesari M, Penninx BW, Pahor M, Lauretani F, Corsi AM, Rhys Williams G, Guralnik JM, Ferrucci : Inflammatory markers and physical performance in older persons: the InCHIANTI study. J Gerontol A Biol Sci Med Sci. 2004, 59 (3): 242-248. 10.1093/gerona/59.3.M242.
Schaap LA, Pluijm SM, Deeg DJ, Harris TB, Kritchevsky SB, Newman AB, Colbert LH, Pahor M, Rubin SM, Tylavsky FA, Visser M, Health ABC Study: Higher inflammatory marker levels in older persons: associations with 5-year change in muscle mass and muscle strength. J Gerontol A Biol Sci Med Sci. 2009, 64 (11): 1183-1189.
Payette H, Roubenoff R, Jacques PF, Dinarello CA, Wilson PW, Abad LW, Harris T: Insulin-like growth factor-1 and interleukin 6 predict sarcopenia in very old community-living men and women: the Framingham heart study. J Am Geriatr Soc. 2003, 51 (9): 1237-1243. 10.1046/j.1532-5415.2003.51407.x.
Rohleder N, Kudielka BM, Hellhammer DH, Wolf JM, Kirschbaum C: Age and sex steroid-related changes in glucocorticoid sensitivity of pro-inflammatory cytokine production after psychosocial stress. J Neuroimmunol. 2002, 126 (1–2): 69-77.
Beyer I, Mets T, Bautmans I: Chronic low-grade inflammation and age-related sarcopenia. Curr Opin Clin Nutr Metab Care. 2012, 15 (1): 12-22. 10.1097/MCO.0b013e32834dd297.
Lencel P, Magne D: Inflammaging: the driving force in osteoporosis?. Med Hypotheses. 2011, 76 (3): 317-321. 10.1016/j.mehy.2010.09.023.
Christensen H, Boysen G, Johannesen HH: Serum-cortisol reflects severity and mortality in acute stroke. J Neurol Sci. 2004, 217 (2): 175-180. 10.1016/j.jns.2003.09.013.
Sam S, Corbridge TC, Mokhlesi B, Comellas AP, Molitch ME: Cortisol levels and mortality in severe sepsis. Clin Endocrinol (Oxf). 2004, 60 (1): 29-35. 10.1111/j.1365-2265.2004.01923.x.
Güder G, Bauersachs J, Frantz S, Weismann D, Allolio B, Ertl G, Angermann CE, Störk S: Complementary and incremental mortality risk prediction by cortisol and aldosterone in chronic heart failure. Circulation. 2007, 115 (13): 1754-1761. 10.1161/CIRCULATIONAHA.106.653964.
Waters DL, Qualls CR, Dorin RI, Veldhuis JD, Baumgartner RN: Altered growth hormone, cortisol, and leptin secretion in healthy elderly persons with sarcopenia and mixed body composition phenotypes. J Gerontol A Biol Sci Med Sci. 2008, 63 (5): 536-541. 10.1093/gerona/63.5.536.
Straub RH, Lehle K, Herfarth H, Weber M, Falk W, Preuner J, Scholmerich J: Dehydroepiandrosterone in relation to other adrenal hormones during an acute inflammatory stressful disease state compared with chronic inflammatory disease: role of interleukin-6 and tumour necrosis factor. Eur J Endocrinol. 2002, 146 (3): 365-374. 10.1530/eje.0.1460365.
Trivedi DP, Khaw KT: Dehydroepiandrosterone sulfate and mortality in elderly men and women. J Clin Endocrinol Metab. 2001, 86 (9): 4171-4177. 10.1210/jc.86.9.4171.
Mazat L, Lafont S, Berr C, Debuire B, Tessier JF, Dartigues JF, Baulieu EE: Prospective measurements of dehydroepiandrosterone sulfate in a cohort of elderly subjects: relationship to gender, subjective health, smoking habits, and 10-year mortality. Proc Natl Acad Sci USA. 2001, 98 (14): 8145-8150. 10.1073/pnas.121177998.
Valenti G, Denti L, Maggio M, Ceda G, Volpato S, Bandinelli S, Ceresini G, Cappola A, Guralnik JM, Ferrucci L: Effect of DHEAS on skeletal muscle over the life span: the InCHIANTI study. J Gerontol A Biol Sci Med Sci. 2004, 59 (5): 466-472. 10.1093/gerona/59.5.M466.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors’ declared that they have no competing interest.
Authors’ contributions
DB and HPP prepared the first draft and DBB added immunological input. All authors read and approved the final draft.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Baylis, D., Bartlett, D.B., Patel, H.P. et al. Understanding how we age: insights into inflammaging. Longev Healthspan 2, 8 (2013). https://doi.org/10.1186/2046-2395-2-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/2046-2395-2-8