
Abstract
Nitric oxide (NO), carbon monoxide (CO) and hydrogen sulfide (H2S) are lipid-soluble, endogenously produced gaseous messenger molecules collectively known as gasotransmitters. Over the last several decades, gasotransmitters have emerged as potent cytoprotective mediators in various models of tissue and cellular injury. Specifically, when used at physiological levels, the exogenous and endogenous manipulation of these three gases has been shown to modulate ischemia/reperfusion injury by inducing a number of cytoprotective mechanisms including: induction of vasodilatation, inhibition of apoptosis, modulation of mitochondrial respiration, induction of antioxidants, and inhibition of inflammation. However, while the actions are similar, there are some differences in the mechanisms by which these gasotransmitters induce these effects and the regulatory actions of the enzyme systems can vary depending upon the gas being investigated. Furthermore, there does appear to be some crosstalk between the gases, which can provide synergistic effects and additional regulatory effects. This review article will discuss several models and mechanisms of gas-mediated cytoprotection, as well as provide a brief discussion on the complex interactions between the gasotransmitter systems.
Similar content being viewed by others


Introduction
Nitric oxide (NO), carbon monoxide (CO) and hydrogen sulfide (H2S) are lipid-soluble, endogenously-produced gaseous messenger molecules [1]. Together, they make up the family of labile biological mediators termed gasotransmitters. Historically, these gases were considered to be highly toxic and hazardous to the environment. However, it was found that under normal physiological conditions in mammals these molecules are enzymatically regulated and endogenously produced. Because of this discovery, the biological and physiological role of these gases has been re-evaluated. As such, an extensive amount of work has been conducted over the last several decades (last three centuries for NO) and has led to the discovery that each gasotransmitter possess a number of physiological actions. The gasotransmitters have also been extensively studied in several models of cellular and tissue injury. This work has led to the discovery that gasotransmitters and the enzymes that generate them share similar features and overlap in a variety of biological functions. Specifically, studies have found that deficiencies in the enzymes (through genetic manipulation or use of inhibitors) exacerbate ischemia-reperfusion (I/R) injury, whereas genetic overexpression of the enzymes induces cytoprotection. Furthermore, treatment with pharmacological donors or inhaled gas therapy has also been shown to provide cytoprotection. This review article will discuss the physiological significance and the fundamental mechanisms by which these gaseous molecules exert cytoprotection in several models of tissue and cellular injury, as well as provide a brief discussion on the complex interactions between the gasotransmitter systems.
Physiological and Biological Roles of Gasotransmitters
NO was the first gasotransmitter to be identified by studies dating back to the late 1700's, which investigated its pharmacological efficacy [2]. However, it was not until 1867 that evidence emerged to suggest that NO induced vasodilatory effects in patients suffering from angina pectoris [3]. Unfortunately, it's true potential and physiological significance in the field of medicine was not discovered until the 1980's, when scientists Furchgott and Zawadzki identified NO as an endogenous modulator of vascular tone [4]. NO levels are controlled at the level of synthesis, initiated by the interaction of nitric oxide synthases (NOSs) and calcium-calmodulin stimulation. There are three isoforms of NOS that have been characterized, purified, and cloned: the endothelial isoform (eNOS), the neuronal isoform (nNOS), and the inducible isoform (iNOS). These enzymes generate NO from the guanidine nitrogen of the amino acid L-arginine in the presence of oxygen and NADPH, while forming L-citrulline as a byproduct (Figure 1). NO released from the endothelium enters the target cell and initiates cGMP-dependent protein kinase phosphorylation of myosin, by activating the cytosolic enzyme soluble guanylyl cylase causing a subsequent increase in the intracellular concentration of cyclic GMP (cGMP), which then goes on to regulate smooth muscle relaxation and vasodilatation.

Enzymatic Synthesis of Nitric Oxide. Nitric oxide (NO) is produced from amino acid L-arginine by the enzymatic action of nitric oxide synthase (NOS). There are there forms of NOS: endothelial NOS (eNOS), neuronal NOS (nNOS), and inducible NOS (iNOS). NO activates guanylate cyclase (GC), which leads to increased production of 3',5'-monophosphate (cGMP).
The endogenous source of CO was first identified in 1969 when it was determined that it is derived from the breakdown of heme by the enzyme heme oxygenase (HO) (Figure 2) [5]. In 1993, a study investigating nonadrenergic/noncholinergic (NANC) neurotransmission in the enteric nervous system identified CO as a vasorelaxant and later confirmed it as the second gasotransmitter [6, 7]. Molecular cloning has revealed three known isoforms of heme oxygenase: inducible HO-1 a ubiquitously expressed transcription factor activator that is crucial in oxidative stress response; HO-2 which is constitutively active and controlled by posttranslational modification [8]; and HO-3 which is similar to HO-2 but considered a less efficient heme catalyst [9]. Similar to NO, CO has been shown to modulate vasorelaxation, vascular smooth muscle cell growth, and tissue injury through elevation of cGMP levels, it is also suggested that local effects of CO may directly influence NO release, and furthermore stimulate adaptive responses and augment gene expression [10, 11].

Enzymatic Synthesis of Carbon Monoxide. Heme is catabolised by heme oxygenases (HO), to form biliverdin, carbon monoxide, and iron. Carbon monoxide can activate soluble guanylyl cyclase, which causes an increase in cyclic guanosine monophosphate levels (cGMP). Biliverdin is subsequently converted to bilirubin by biliverdin reductase.
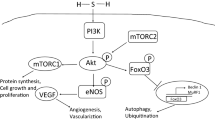
H2S was the third endogenously produced gasotransmitter to be identified. The production of H2S in mammalian systems has been attributed to three principal enzymes (Figure 3): cystathionine β-synthase (CBS), cystathionine γ-lyase (CSE or CGL) and 3-metacaptopyruvate sulfur transferase (3MST). The endogenous production of H2S was initially described in the brain and attributed to CBS activity [12]. However, recent studies have found that ~90% of total H2S production in the brain is attributed to 3MST [13]. CBS and CGL are found in all tissues; however CBS is the predominant source of H2S in the central nervous system (CNS), whereas CGL is the predominant source in the cardiovascular system. Perhaps the most characterized physiological action of H2S is its participation in memory formation as a central component of the process of long-term-potentiation of neuronal circuitry [12]. Additionally, like NO and CO, H2S also mediates smooth muscle relaxation and vasodilation. However, it does so in a guanylyl cyclase/cGMP independent manner [14, 15].

Enzymatic Synthesis of Hydrogen Sulfide. There are three enzymatic pathways involved in the synthesis of hydrogen sulfide (H2S) in mammalian systems. Cystathionine β-synthase (CBS) produces H2S through a reaction involving the generation of cystathionine from homocysteine and L-cysteine from cystathione. Cystathionine γ-lyase (CGL or CSE) produces H2S through a reaction involving the generation of L-cysteine from cystathionine. 3-mercaptopyruvate sulfur transferase (3MST) produces H2S through a reaction involving the generation of 3-mercaptopyruvate (3MP) from α-ketoglutarate (α-KG) by cysteine aminotransferase (CAT).
Cytoprotective Effects of Gasotransmitters
A unique characteristic of gasotransmitters is that they lack conventional regulatory mechanisms, as they have the ability to pass messages directly to an intracellular target without the need for receptor or plasma membrane interactions [16]. This makes the gasotransmitters particularly attractive candidates for the treatment of pathological disorders, such as I/R injury. Over the past several decades, studies using animal models and clinical investigations have defined these gaseous molecules as physiological participants in a wide range of profound biochemical and biological functions, and have defined them as potent cytoprotective mediators in various models of tissue and cellular injury. In the physiological range, the exogenous and endogenous manipulation of these three gases has been shown to modulate ischemia/reperfusion injury, vascular damage, vasodilation, oxidative stress, inflammation, and apoptosis.
Myocardial Ischemia-Reperfusion Injury
In terms of cytoprotective effects, NO has been the most investigated gasotransmitter. Specifically, much of this work has focused on the role of endogenously and exogenously derived NO in mediating the effects of myocardial I/R injury [17–21]. The role of endogenously derived NO has been studied using pharmacological inhibitors against NOS and by genetically targeting each NOS. The role of exogenously derived NO has been studied through the administration of NO in the form of authentic NO gas, NO donors, and more recently nitrite and nitrate. Perhaps the most clear-cut evidence for a protective role of endogenously derived NO in the setting of myocardial injury comes from studies aimed at investigating eNOS [22]. Studies that have employed the use of mice deficient in eNOS (eNOS-/-) have overwhelmingly shown that these mice experience exacerbated infarct sizes and increased myocardial dysfunction in response to myocardial ischemia [23–26]. In contrast, the overexpression of eNOS has been shown to reduce the size of myocardial infarction and increase myocardial function in the same experimental models of injury [27–29]. Early studies reported that a deficiency of nNOS or iNOS did not affect infarct size in response to acute myocardial ischemia [24, 28, 30–32]. However, more recent evidence suggests that nNOS plays a crucial role in preventing adverse left ventricular remodeling and ventricular arrhythmias and maintaining myocardial β-adrenergic reserve after myocardial infarction [33, 34]. Likewise, new evidence has emerged to suggest that gene transfer of iNOS affords cardioprotection against myocardial I/R injury [35, 36]. Taken together, these studies clearly demonstrate that endogenously produced NO has the ability to protect the heart from I/R injury.
Extensive work has also investigated the use of NO as a viable pharmacological approach for the treatment of I/R injury. Inhaled NO gas therapy initiated just before or during coronary artery reperfusion has been shown to be an effective means to rapidly increase the accumulation of NO metabolites in blood and tissues and to provide protection against myocardial I/R injury [37, 38]. Additionally, the class of drugs known as NONOates, which release NO in a pH-dependent, first order process have repeatedly been reported to provide cardioprotection in experimental models of myocardial I/R injury [39, 40]. NONOates are not the only pharmacological agents that can provide protection by increasing the bioavailability of NO, as it has clearly been shown that statins, metformin, adiponectin, and estrogen provide cardioprotection by increasing the production of NO from eNOS [28, 29, 41–44]. The use of NO as a therapeutic agent in the treatment of myocardial I/R injury has not been without some controversy, as there have been some studies to report negative effects. In 2001 a comprehensive review investigating the role of NO in modulating myocardial injury spanning from 1991-2001 found that 73% of the studies reported that NO (endogenous or exogenous) was cardioprotective, whereas 12% reported that NO was detrimental [17]. Further investigation of NO efficacy in myocardial I/R have suggested the cause for discrepancies between the opposing findings can be explained by dosing inconsistencies, as it is suggested that physiological levels (i.e., nanomolar) of NO promote cytoprotection, while suprapharmacological levels (i.e. high micromolar and milimolar) mediate cellular necrosis and apoptosis [17, 21].
Enhanced expression of HO-1 and its degradation products have been shown to augment multiple intracellular cytoprotective pathways. In particular, HO- 1 protein expression is significantly up-regulated in myocardial infarction [45], and hypoxia-induced upregulation of HO-1 in the heart has been shown to significantly increase CO production [46]. Predictably, studies investigating myocardial damage in HO-1 knockout mice following MI have reported [32] exacerbated myocardial injury, increased ROS production, and decreased endogenous CO production. However, at low levels exogenous CO has been shown to stimulate cardioprotection in HO-1 knockout mice, and rat hearts during I/R [47]. The role of endogenous CO in cardioprotection has also been demonstrated using carbon monoxide-releasing molecules (CO-RMs) to elicit pharmacological activities in myocardial cells against I/R injury [48]. Taken together, these studies suggest the use HO-1 induced CO production and direct administration of CO provide potential therapeutic alternatives for the pharmacological regulation of myocardial I/R injury [9, 49, 50].
An increasing number of studies also provide evidence that both exogenous and endogenous H2S exert cytoprotective effects [51], especially against myocardial I/R injury [52] Studies have found that, targeted deletion and genetic manipulation of CGL leads to modification of H2S expression in the aorta, heart, and serum [53]. Johansen first investigated exogenous pre-treatment of H2S using a Lagendorff hanging heart model, and found that H2S administration caused a reduction in infarct size and suppressed myocardial I/R injury [54]. Similarly, in vitro studies have found that pretreatment with H2S reduces myocardial necrosis, decreases cardiomyocyte death, improves mitochondrial function [55] and increases myocyte contractility [56, 57]. In vivo models of myocardial I/R have provided further support suggesting the cardioprotective effects of H2S. Studies using murine models I/R injury have shown that of treatment with H2S prior to myocardial ischemia significantly reduces infarct size, and H2S administered at the time of reperfusion has been shown to reduce infarct size and exert dose dependent cardioprotection [43, 58]. However, when the production of H2S is reduced by pharmalogical inhibition prior to myocardial ischemia, mice experience exacerbated myocardial injury [58]. Further evidence that H2S confers cardioprotection has been shown by genetically altering CGL expression. Mice deficient in CGL (CGL-/-) have been reported to experience decreased myocardial function, reduced serum H2S levels, pronounced hypertension, diminished endothelium-dependent vasodilation, and significantly larger areas of myocardial infarction compared to wild-type control animals [58–60]. However, a recent study investigating the hemodynamic effects of H2S reported that CGL-/- mice did not display a significant difference in blood pressure when compared to wild-type mice [61]. The discrepancy between these two studies might be partly due to the genetic background of the mice used, which indicates that more research is needed to confirm the effects of CGL inhibition on blood pressure. Furthermore, specific overexpression of CGL has been shown to increase H2S production in the heart, and reduce the degree of injury following myocardial I/R [58]. These findings suggest that therapy targeting endogenous and exogenous H2S may offer cytoprotection against myocardial I/R injury.
Other Models of Ischemia-Reperfusion Injury
The cytoprotective effects of gasotransmitter therapy are not limited to myocardial I/R injury, as NO and H2S have been shown to confer protection in other organ systems, such as the liver, kidney, and brain. Hepatic I/R injury is oftentimes associated with liver surgery, hepatic transplantation, and hepatic resection. NO modulates hepatocellular/tissue injury through its participation in neutrophil adhesion, platelet aggregation and maintenance of normal vascular permeability [62]. Kuroki et al investigated the role of nitroprusside in the pathogenesis of hepatic I/R injury using a rat model, and reported that it enhances hepatic microcirculation, decreases LDH serum levels and reduces hepatocyte damage [63]. H2S therapy has also been shown to reduce serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels following hepatic ischemia-reperfusion [41], and to inhibit lipid peroxidation as well as decrease inflammation [64, 65]. In 2007, Tripatara and colleagues used a rat model of renal I/R injury to demonstrate attenuation of renal dysfunction and injury in response to topical treatment with sodium nitrite [66]. In addition, Unal and colleagues [67] have investigated the effects of nitroprusside and antioxidant vitamins C and E, using rat kidney I/R models and found that nitroprusside inhibited xanthine oxidase and provided a preventive influence in renal I/R injury than the antioxidant vitamins C+E. Tripatara and colleagues also investigated the endogenous and exogenous effects of H2S in renal I/R injury, and found that CGL inhibition causes a significant decrease in renal function and that topical H2S therapy applied to the kidney prior to ischemia improves renal function and attenuates renal I/R injury [68]. More evidence regarding the efficacy of NO and H2S therapy has been demonstrated in models of cerebral ischemia. Chen examined the effect of eNOS production in cerebral ischemia using eNOS-/- mice [69]. Predictably, the eNOS-/- mice displayed a significant decrease in neurological function, attenuation of angiogenesis, and decreased cell proliferation. Other studies have reported the benefits of intravenous sodium nitrite infusion at the time of reperfusion as means to restore cerebral blood flow, and decrease infarct volume [70, 71]. Furthermore, administration of the exogenous NO donor ZJM-289 has been shown to increase eNOS expression, cGMP, and NO after cerebral ischemia. Moreover, administration of H2S following cerebral ischemia has been shown to reduce infarct size, increase H2S levels in the brain and provides neuroprotection by inducing hypothermia (30.8 ± 0.7°C) [72]. However, contrasting studies have shown H2S administration significantly increases cerebral infarct volume in rats following middle cerebral artery occlusion [73]. A recent study has reported that the neuroprotective effects of H2S are concentration dependent [74], and that administration of H2S increases fetal GSH levels in the brain, decreases cerebral I/R injury and protects against oxidative stress in utero [75]. Additionally, H2S has been shown to reduce neuronal cell death in a murine model of cardiac arrest/cardiopulmonary resuscitation [76]. Thus, additional studies are certainly needed to address the reported discrepancies in models of cerebral injury.
Exposure to CO has been shown to promote cell survival, decrease necrosis, prevent graft rejection and promote tissue protection during organ transplantation [40, 77]. Exposure of the graft donor as well as the graft (during ischemia) to exogenous CO and HO-1-derived CO has been shown to restore graft function, reduce generation of ROS and thus prevent cytotoxic tissue injury. Overexpression of HO-1 has been shown to reduce intragraft apoptosis [78] and suppress vascular injury. Akamatsu and colleagues used HO-1 preconditioning to demonstrate retention of functional viability in cardiomyocyte cellular grafts after implantation [79]. Yoshida and colleagues, exposed isolated rat hearts to CO at high pressure, and reported organ preservation, attenuation of intracellular decomposition and prevention of necrosis [80]. Other studies have demonstrated HO-1 increases survival after cardiac transplant and HO-induced CO protects tissue in mouse-to-rat cardiac transplantation [81]. Additionally, at physiological levels CO inhalation was found to exert tissue protection in lung transplantation [82], and HO-1 overexpression has been shown to regulate a cascade of cytoprotective effects in immune response to organ transplantation [83]. Currently the US food and Drug administration has granted an orphan drug safety and tolerability study for CO inhalation therapy in the reduction of delayed graft function, and solid organ transplant preservation.
In summary, extensive research performed in recent years has clearly demonstrated that the efficacy of gasotransmitter therapy in ameliorating in vitro or in vivo I/R injury. Most importantly, these studies have provided important information regarding the doses of each gas that provide cytoprotection and suggest that the use of these gases at or near the levels considered to be produced under physiological conditions in vivo is optimal to protect a number of organs including the heart, liver, kidney, and brain.
Summary of Cytoprotective Mechanisms and Evidence for Gasotransmitter Crosstalk
So far, this review has provided evidence supporting the multifaceted role of gasotransmitters in cytoprotection and as such has highlighted the similarities between all three gases. For instance, all are naturally produced in the body and are constantly participating in biological responses within target tissues and organs [84]. The rate of NO/CO/H2S production, cytoprotection and clearance vary with time, dose concentration, and enzymatic mediators. Even the enzymes responsible for biosynthesis of gastrotransmitters show parallel similarities and in the case of NO and CO can be classified as constitutive (eNOS, nNOS, and HO-2) or inducible (iNOS, and HO1). Importantly, all three gasotransmitters possess similar physiological actions that could account for the observed cytoprotective effects (Figure 4). For example, all three can: (1) induce vasodilatation by activating the sGC/cGMP pathway (NO and CO) [85] or by activating ATP-sensitive K+ (KATP) channels (H2S) [15]; (2) inhibit apoptosis by directly interacting with the apoptotic machinery [86] or by increasing the expression of anti-apoptogens, such as HSP90, HSP70, and Bcl-2 [43]; (3) modulate mitochondrial respiration [17, 58, 87]; (4) induce antioxidants [58, 81] and (5) inhibit inflammation [88–91]. However, while the actions are similar, there are some differences in the mechanisms by which these gasotransmitters induce these effects and the regulatory actions of the enzyme systems can vary depending upon the gas being investigated. Furthermore, there does appear to be some crosstalk between the gases, which can provide synergistic effects and additional regulatory effects. The rest of this article will provide a brief discussion on the complex interactions between the gasotransmitter systems.

Summary of Mechanisms by which Gasotransmitters can Induce Cytoprotection. The gasotransmitters share unique and similar pathways by which they protect against tissue and cellular injury. Both CO and NO have been shown to regulate smooth muscle relaxation through the sGC/cGMP pathway. NO and H2S have been shown to regulate cell proliferation and vascular smooth muscle relaxation balance through mitogen-activated protein kinases (MAPK), and ATP-sensitive potassium channels (KATP ). In addition, H2S and CO regulate oxidant/antioxidant balance through the transcription factor NF-E2-related factor (Nrf2).
We will begin with the interaction between NO and H2S. Like NO, H2S is produced in the endothelium as well as SMCs [92], and mediates acute regulation by vasorelaxative hormones through calmodulin and IP3 dependent pathways [93]. There appears to be a close interaction between H2S and NO, with NO amplifying the inhibitory effect of H2S and H2S tissue specific activation of eNOS [94]. In particular, NO and H2S have been suggested to collaborate in regulating vascular homeostasis and vasodilation [14]. Additional evidence suggests that NO can increase CGL activity acutely, and that chronic exposure to NO up-regulates CGL expression. Moreover, at low concentrations H2S has been shown to enhance the release of NO from vascular endothelium and increase the vasorelaxant effect of the NO donor sodium nitroprusside [15].
CO and NO share apparent similarities in structure, molecular weight and solubility [95]. Both NO and CO interact with iron (Fe) to form 5 or 6 coordinated haem complexes, which result in conformational changes and activation of the sGC/cGMP pathway [85]. Thus, many of the biological effects of CO are similar to NO, including its anti-apoptotic, anti-proliferative and anti-inflammatory mechanisms. Other studies have confirmed the participation of both NO and CO-mediated signaling cascades in immune suppression of platelet aggregation and neurotransmission [96]. In addition to regulating vascular cell growth, CO influences cell survival by blocking cytokine-mediated mitochondrial release of cytochrome C [97] and has been shown to influence hepatoprotection through the transcriptional upregulation of iNOS in the liver. Both exogenously administered or endogenously released NO stimulates HO-1 gene expression and CO production [32, 97]. Furthermore, CO and NO have been shown to participate in vasoactive cross talk, influencing: growth factors, anti-inflammatory mediators, angiogenesis and vascular remodeling [98, 99].
The crosstalk between H2S and CO has been the least studied, as there are only a few studies which have addressed this interactions. Zhang and colleagues [100] were one of the first to investigate the physiological and pathological interactions of CO and H2S and found that exogenous H2S can upregulate the CO/HO pathway during hypoxic pulmonary hypertension. Additionally, in much the same manner to that described for NO, H2S increases the expression of HO-1 in a Nrf-2 dependent manner [43].
Conclusion
The studies mentioned in this review have identified the therapeutic potential and translational opportunities of gasotransmitters as potent cytoprotective molecules. So far, the exogenous administration, endogenous manipulation and use of genetically modified animals has been successful in demonstrating gasotransmitter-mediated cytoprotection in models of I/R injury and other forms of disease. In addition, the gasotransmitters have been shown to play a pivotal role in the regulation of cell functions and in the reduction of tissue injury by activation of a number of prosurvival pathways. However, there are still a number of questions that remain to be answered, especially in relation to the interactions between the gases. For instance, the exact correlation between these gases in the various pathways of cytoprotection has yet to be fully investigated [101]. It is also not known if using some variation of NO/CO/H2S, as a combination therapy will provide synergistic effects in the treatment of ischemic disorders. Therefore, additional studies designed to examine NO/CO/H2S cross talk will provide better comprehension concerning this issue, as well as new insights into their interactions. In addition, it is important to recognize a need for the development of consistent dosing and measurement techniques for the advancement of gasotransmitters in pharmalogical research. Because the regulation, expression and function of these gaseous molecules are so complex, optimal alterations in synthesis and activity will possibly provide novel therapeutic opportunities for the treatment of a number of pathophysiological conditions.
Abbreviations
- 3MST:
-
3-metacaptopyruvate sulfur transferase
- ALT:
-
alanine aminotransferase
- AST:
-
aspartate aminotransferase
- Bcl-2:
-
B-cell lymphoma 2
- CBS:
-
cystathionine β-synthase
- CGL:
-
cystathionine γ-lyase
- cGMP:
-
cyclic guanosine monophosphate
- CNS:
-
central nervous system
- CO:
-
carbon monoxide
- CO-RM:
-
carbon monoxide -releasing molecules
- eNOS:
-
endothelial nitric oxide synthase
- Fe:
-
iron
- GC:
-
guanylyl cylase
- H2S:
-
hydrogen sulfide
- HO:
-
heme oxygenase
- HSP:
-
heat shock protein
- I/R:
-
ischemia reperfusion
- iNOS:
-
inducible nitric oxide synthase
- IP3:
-
inositol trisphosphate
- KATP:
-
ATP-sensitive K+ channels
- MI/R:
-
myocardial ischemia reperfusion
- NADPH:
-
nicotinamide adenine dinucleotide phosphate-oxidase
- NANC:
-
nonadrenergic/noncholinergic
- nNOS:
-
neuronal nitric oxide synthase
- NO:
-
nitric oxide
- NOS:
-
nitric oxide synthase
- Nrf2:
-
nuclear factor-E2-related factor-2
- SMC:
-
smooth muscle cell.
References
Nicholson CK, Calvert JW: Hydrogen sulfide and ischemia-reperfusion injury. Pharmacol Res. 2010, 62: 289-297. 10.1016/j.phrs.2010.06.002.
Szabo C, Ischiropoulos H, Radi R: Peroxynitrite: biochemistry, pathophysiology and development of therapeutics. Nat Rev Drug Discov. 2007, 6: 662-680. 10.1038/nrd2222.
Brunton TL: On the use of nitrite of amyl in angina pectoris. 1867, 2:
Furchgott RF, Zawadzki JV: The obligatory role dof endothelial cells in the relazation of arterial smooth muscle by acetylcholine. 1980, 288: 288-373.
Choi AM, Otterbein LE: Emerging role of carbon monoxide in physiologic and pathophysiologic states. Antioxid Redox Signal. 2002, 4: 227-228. 10.1089/152308602753666271.
Wang R: Resurgence of carbon monoxide: an endogenous gaseous vasorelaxing factor. Can J Physiol Pharmacol. 1998, 76: 1-15. 10.1139/y97-187.
Verma A, Hirsch D, Glatt C, Ronett G, Snyder S: Carbon monoxide: A putative neural messenger. Science. 1993, 259: 381-384. 10.1126/science.7678352.
Ryter SW, Choi AM: Heme oxygenase-1: molecular mechanisms of gene expression in oxygen-related stress. Antioxid Redox Signal. 2002, 4: 625-632. 10.1089/15230860260220120.
Perrella MA, Yet SF: Role of heme oxygenase-1 in cardiovascular function. Curr Pharm Des. 2003, 9: 2479-2487. 10.2174/1381612033453776.
Marshall HE, Stamler JS: Exhaled nitric oxide (NO), NO synthase activity, and regulation of nuclear factor (NF)-kappaB. Am J Respir Cell Mol Biol. 1999, 21: 296-297.
Foresti R, Motterlini R: The heme oxygenase pathway and its interaction with nitric oxide in the control of cellular homeostasis. Free Radic Res. 1999, 31: 459-475. 10.1080/10715769900301031.
Kimura H: Hydrogen sulfide: its production, release and functions. J Neurosci. 1996, 16: 1066-1071.
Shibuya N, Tanaka M, Yoshida M, Ogasawara Y, Togawa T, Ishii K, Kimura H: 3-Mercaptopyruvate sulfurtransferase produces hydrogen sulfide and bound sulfane sulfur in the brain. Antioxid Redox Signal. 2009, 11: 703-714. 10.1089/ars.2008.2253.
Hosoki R, Matsuki N, Kimura H: The possible role of hydrogen sulfide as an endogenous smooth muscle relaxant in synergy with nitric oxide. Biochem Biophys Res Commun. 1997, 237: 527-531. 10.1006/bbrc.1997.6878.
Zhao W, Zhang J, Lu Y, Wang R: The vasorelaxant effect of H(2)S as a novel endogenous gaseous K(ATP) channel opener. EMBO J. 2001, 20: 6008-6016. 10.1093/emboj/20.21.6008.
Wang R: The gasotransmitter role of hydrogen sulfide. 2003, 5: 493-501.
Bolli R: Cardioprotective function of inducible nitric oxide synthase and role of nitric oxide in myocardial ischemia and preconditioning: an overview of a decade of research. J Mol Cell Cardiol. 2001, 33: 1897-1918. 10.1006/jmcc.2001.1462.
Jugdutt B: Nitric oxide and cardiovascular protection. Heart Fail Rev. 2003, 8: 29-34. 10.1023/A:1022190820131.
Ferdinandy P, Schulz R: Nitric oxide, superoxide and peroxynitrite inmyocardial ischaemia-reperfusion injury and preconditioning. Br J Pharmacol. 2003, 138: 532-543. 10.1038/sj.bjp.0705080.
Schulz R, Kelm M, Heusch G: Nitric oxide in myocardial ischemia/reperfusion injury. Cardiovasc Res. 2004, 61: 402-413. 10.1016/j.cardiores.2003.09.019.
Jones SP, Bolli R: The ubiquitous role of nitric oxide in cardioprotection. 2006, 40: 16-23.
Bannenberg G, Vieira H: Therapeutic applications of the gaseous mediators carbon monoxide and hydrogen sulfide. Expert Opin Ther Pat. 2009, 19: 663-682. 10.1517/13543770902858824.
Hannan RL, John MC, Kouretas PC, Hack BD, Matherne GP, Laubach VE: Deletion of endothelial nitric oxide synthase exacerbates myocardial stunning in an isolated mouse heart model. J Surg Res. 2000, 93: 127-132. 10.1006/jsre.2000.5953.
Sumeray MS, Rees DD, Yellon DM: Infarct size and nitric oxide synthase in murine myocardium. J Mol Cell Cardiol. 2000, 32: 35-42. 10.1006/jmcc.1999.1050.
Sharp BR, Jones SP, Rimmer DM, Lefer DJ: Differential response to myocardial reperfusion injury in eNOS-deficient mice. Am J Physiol Heart CIrc Physiol. 2002, 282: H2422-2426.
Jones SP, Girod WG, Palazzo AJ, Granger DN, Grisham MB, Jourd'Heuil D, Huang PL, Lefer DJ: Myocardial ischemia-reperfusion injury is exacerbated in absence of endothelial cell nitric oxide synthase. Am J Physiol. 1999, 276: H1567-1573.
Jones AM, Wilkerson DP, Koppo K, Wilmshurst S, Campbell IT: Inhibition of nitric oxide synthase by L-NAME speeds phase II pulmonary. VO2 kinetics in the transition to moderate-intensity exercise in man. J Physiol. 2003, 552: 265-272. 10.1113/jphysiol.2003.045799.
Jones SP, Lefer DJ: Myocardial Reperfusion Injury: Insights Gained from Gene-Targeted Mice. News Physiol Sci. 2000, 15: 303-308.
Elrod JW, Greer JJ, Bryan NS, Langston W, Szot JF, Gebregzlabher H, Janssens S, Feelisch M, Lefer DJ: Cardiomyocyte-specific overexpression of NO synthase-3 protects against myocardial ischemia-reperfusion injury. Arterioscler Thromb Vasc Biol. 2006, 26: 1517-1523. 10.1161/01.ATV.0000224324.52466.e6.
Jones S, Greer J, Kakkar A, Ware P, Turnage R, Hicks M, vanHaperen R, de Crom R, Kawashima S, Yokoyama M, Lefer D: Endothelial nitric oxide synthatse overexpression attenuates myocardial reperfusion injury. Am J Physiol Heart Cir Physiol. 2004, 286: H276-282.
Xi L: Nitric oxide-dependent mechanism of anti-ischemic myocardial protection induced by monophosphoryl lipid A. Zhongguo Yao Li Xue Bao. 1999, 20: 865-871.
Liu X, Chapman G, Peyton K, Schafer A, Durante W: Carbon monoxide inhibits apoptosis in vascular smooth muscle cells. Cardiovasc Res. 2002, 55: 396-405. 10.1016/S0008-6363(02)00410-8.
Burger DE, Xiang FL, Hammoud L, Jones DL, Feng Q: Erythropoietin protects the heart from ventricular arrhythmia during ischemia and reperfusion via neuronal nitric-oxide synthase. J Pharmacol Exp Ther. 2009, 329: 900-907. 10.1124/jpet.109.150896.
Dawson D, Lygate CA, Zhang MH, Hulbert K, Neubauer S, Casadei B: nNOS gene deletion exacerbates pathological left ventricular remodeling and functional deterioration after myocardial infarction. Circulation. 2005, 112: 3729-3737. 10.1161/CIRCULATIONAHA.105.539437.
Li T, Li J, Liu J, Zhang P, Wu W, Zhou R, Li G, Zhang W, Yi M, Huang H: Polymerized placenta hemoglobin attenuates ischemia/reperfusion injury and restores the nitroso-redox balance in isolated rat heart. Free Radic Biol Med. 2009, 46: 397-405. 10.1016/j.freeradbiomed.2008.10.042.
Li Q, Guo Y, Tan W, Ou Q, Wu WJ, Sturza D, Dawn B, Hunt G, Cui C, Bolli R: Cardioprotection afforded by inducible nitric oxide synthase gene therapy is mediated by cyclooxygenase-2 via a nuclear factor-kappaB dependent pathway. Circulation. 2007, 116: 1577-1584. 10.1161/CIRCULATIONAHA.107.689810.
Yamashita H, Akamin S, Sumida Y, Inoue M, Sawada T, Nagayasu T, Oka T: Inhaled nitric oxide attenuates apoptosis in ishemia-reperfusion injury of rabbit lungs. The annals of thoracic surgery. 78: 292-297. 204
Liu X, Huang Y, Pokreisz P, Vermeersch P, Marsboom G, Swinnen M, Verbeken E, Santos J, Pellens M, Gillijns H, et al: Nitric oxide inhalation improves microvascular flow and decreases infarction size after myocardial ischemia and reperfusion. J Am Coll Cardiol. 2007, 50: 808-817. 10.1016/j.jacc.2007.04.069.
Takano H, Manchikalapudi S, Tang XL, Qiu Y, Rizvi A, Jadoon AK, Zhang Q, Bolli R: Nitric oxide synthase is the mediator of late preconditioning against myocardial infarction in conscious rabbits. Circulation. 1998, 98: 441-449.
Wang G, Liem DA, Vondriska TM, Honda HM, Korge P, Pantaleon DM, Qiao X, Wang Y, Weiss JN, Ping P: Nitric oxide donors protect murine myocardium against infarction via modulation of mitochondrial permeability transition. Am J Physiol Heart CIrc Physiol. 2005, 288: H1290-1295.
Jha S, Calvert J, Duranski M, Ramachandran A, Lefer D: Hydrogen sulfide attenuates hepatic ischemia-reperfusion injury: role of antioxidant and antiapoptotic signaling. Am J Physiol Heart Circ Physiol. 2008, 295: H801-806. 10.1152/ajpheart.00377.2008.
Li Q, Sun B, Wang X, Jin Z, Zhou Y, Dong L, Jiang LH, Rong W: A crucial role for hydrogen sulfide in oxygen sensing via modulating large conductance calcium-activated potassium channels. Antioxid Redox Signal. 2010, 12: 1179-1189. 10.1089/ars.2009.2926.
Calvert JW, Jha S, Gundewar S, Elrod JW, Ramachandran A, Pattillo CB, Kevil CG, Lefer DJ: Hydrogen sulfide mediates cardioprotection through Nrf2 signaling. Circ Res. 2009, 105: 365-374. 10.1161/CIRCRESAHA.109.199919.
Jones NC, Constantin D, Gibson CL, Prior MJ, Morris PG, Marsden CA, Murphy S: A detrimental role for nitric oxide synthase-2 in the pathology resulting from acute cerebral injury. J Neuropathol Exp Neurol. 2004, 63: 708-720.
Lakkisto P, Palojoki E, Backlund T, Saraste A, Tikkanen I, Voipio-Pulkki LM, Pulkki K: Expression of heme oxygenase-1 in response to myocardial infarction in rats. J Mol Cell Cardiol. 2002, 34: 1357-1365. 10.1006/jmcc.2002.2094.
Grilli A, De Lutiis MA, Patruno A, Speranza L, Gizzi F, Taccardi AA, Di Napoli P, De Caterina R, Conti P, Felaco M: Inducible nitric oxide synthase and heme oxygenase-1 in rat heart: direct effect of chronic exposure to hypoxia. Ann Clin Lab Sci. 2003, 33: 208-215.
Mei DS, Du YA, Wang Y: Cardioprotection and mechanisms of exogenous carbon monoxide releaser CORM-2 against ischemia/reperfusion injury in isolated rat hearts. Zhejiang Da Xue Xue Bao Yi Xue Ban. 2007, 36: 291-297.
Clark JE, Kottam A, Motterlini R, Marber MS: Measuring left ventricular function in the normal, infarcted and CORM-3-preconditioned mouse heart using complex admittance-derived pressure volume loops. J Pharmacol Toxicol Methods. 2009, 59: 94-99. 10.1016/j.vascn.2008.10.007.
Yoshida J, Ozaki K, Nalesnik M, Ueki S, Castillo-Rama M, Faleo G, Ezzelarab M, Nakao A, Ekser B, Echeverri G, et al: Ex vivo application of carbon monoxide in UW solutionprevents transplant-induced renal ischemia/reperfusion injury in pigs. Am J Transplant. 2010, 10: 763-772. 10.1111/j.1600-6143.2010.03040.x.
Guo Y, Stein A, Wu W, Tan W, Zhu X, Li Q, Dawn B, Motterlini R, Bolli R: Administration of a Co-releasing molecule at the time of reperfusion reduces infarct size in vivo. Am J Physiol Heart Circ Physiol. 2004, 286: H1649-1653. 10.1152/ajpheart.00971.2003.
Kimura Y, Kimura H: Hydrogen sulfide protects neurons from oxidative stress. FASEB J. 2004, 18: 1165-1167.
Mancardi D, Penna C, Merlino A, Del Soldato P, Wink DA, Pagliaro P: Physiological and pharmacological features of the novel gasotransmitter: hydrogen sulfide. Biochim Biophys Acta. 2009, 1787: 864-872. 10.1016/j.bbabio.2009.03.005.
Mustafa AK, Gadalla MM, Snyder SH: Signaling by gasotransmitters. Sci Signal. 2009, 2: re2-10.1126/scisignal.268re2.
Johansen D, Ytrehus K, Baxter GF: Exogenous hydrogen sulfide (H2S) protects against regional myocardial ischemia-reperfusion injury--Evidence for a role of K ATP channels. Basic Res Cardiol. 2006, 101: 53-60. 10.1007/s00395-005-0569-9.
Elsey DJ, Fowkes RC, Baxter GF: Regulation of cardiovascular cell function by hydrogen sulfide (H(2)S). Cell Biochem Funct. 2010, 28: 95-106. 10.1002/cbf.1618.
Hu Y, Chen X, Pan T, Neo K, Lee S, Khin E, Moore P, Bian J: Cardioprotection induced by hydrogen sulfide preconditioning involves activation of ERK and PI3K/Akt pathways. Pflugers Arch. 2008, 455: 607-616.
Bian JS, Yong Q, Pan T, Feng Z, Ali M, Zhou S, Moore P: Role of hydrogen sulfide in the cardioprotection caused by ischemic preconditioning in the rat heart and cardiac myocytes. J Pharmacol Exp Ther. 2006, 316: 670-678.
Elrod JW, Calvert JW, Morrison J, Doeller JE, Kraus DW, Tao L, Jiao X, Scalia R, Kiss L, Szabo C, et al: Hydrogen sulfide attenuates myocardial ischemia-reperfusion injury by preservation of mitochondrial function. Proc Natl Acad Sci USA. 2007, 104: 15560-15565. 10.1073/pnas.0705891104.
Sivarajah A, Collino M, Yasin M, Benetti E, Gallicchio M, Mazzon E, Cuzzocrea S, Fantozzi R, Thiemermann C: Anti-apoptotic and anti-inflammatory effects of hydrogen sulfide in a rat model of regional myocardial I/R. Shock. 2009, 31: 267-274. 10.1097/SHK.0b013e318180ff89.
Yang G, Wu L, Jiang B, Yang W, Qi J, Cao K, Meng Q, Mustafa AK, Mu W, Zhang S, et al: H2S as a physiologic vasorelaxant: hypertension in mice with deletion of cystathionine gamma-lyase. Science. 2008, 322: 587-590. 10.1126/science.1162667.
Ishii I, Akahoshi N, Yamada H, Nakano S, Izumi T, Suematsu M: Cystathionine gamma-Lyase-deficient mice require dietary cysteine to protect against acute lethal myopathy and oxidative injury. J Biol Chem. 2010, 285: 26358-26368. 10.1074/jbc.M110.147439.
Phillips L, Lopez-Nebllna F, Toledo A, Anaya-Prado R, Toledo-Pereyra p: Nitric Oxide Mechanism of protection in ischemia reperfusion injury. Invs Surgery. 2009, 22: 46-55. 10.1080/08941930802709470.
Kuroki I, Miyazaki T, Mizukami I, Matsumoto N, Matsumoto I: Effect of sodium nitroprusside on ischemia-reperfusion injuries of the rat liver. Hepatogastroenterology. 2004, 51: 1404-1407.
Kang K, Zhao M, Jiang H, Tan G, Pan S, Sun X: Role of hydrogen sulfide in hepatic ischemia-reperfusion-induced injury in rats. Liver Transpl. 2009, 15: 1306-1314. 10.1002/lt.21810.
Xu GY, Winston JH, Shenoy M, Zhou S, Chen JD, Pasricha PJ: The endogenous hydrogen sulfide producing enzyme cystathionine-beta synthase contributes to visceral hypersensitivity in a rat model of irritable bowel syndrome. Mol Pain. 2009, 5: 44-10.1186/1744-8069-5-44.
Tripatara P, Patel NS, Webb A, Rathod K, Lecomte FM, Mazzon E, Cuzzocrea S, Yaqoob MM, Ahluwalia A, Thiemermann C: Nitrite-derived nitric oxide protects the rat kidney against ischemia/reperfusion injury in vivo: role for xanthine oxidoreductase. J Am Soc Nephrol. 2007, 18: 570-580. 10.1681/ASN.2006050450.
Unal D, Yeni E, Erel O, Bitiren M, Vural H: Antioxidative effects of exogenous nitric oxide versus antioxidant vitamins on renal ischemia reperfusion injury. Urol Res. 2002, 30: 190-194. 10.1007/s00240-002-0254-5.
Tripatara P, Patel NS, Collino M, Gallicchio M, Kieswich J, Castiglia S, Benetti E, Stewart KN, Brown PA, Yaqoob MM, et al: Generation of endogenous hydrogen sulfide by cystathionine gamma-lyase limits renal ischemia/reperfusion injury and dysfunction. Lab Invest. 2008, 88: 1038-1048. 10.1038/labinvest.2008.73.
Chen SH, Cheung RT: Neuropeptide Y and its receptor analogs differentially modulate the immunoreactivity for neuronal or endothelial nitric oxide synthase in the rat brain following focal ischemia with reperfusion. J Biomed Sci. 2005, 12: 267-278. 10.1007/s11373-005-1359-y.
Xing Y, Xu ZX, Mang J, Qian JL: Vascular endothelial growth factor expression in focal cerebral ischemia/reperfusion in injury in rats. Zhongguo Wei Zhong Bing Ji Jiu Yi Xue. 2005, 17: 174-176.
Calvert JW, Lefer DJ: Myocardial protection by nitrite. Cardiovasc Res. 2009, 83: 195-203. 10.1093/cvr/cvp079.
Florian B, Vintilescu R, Balseanu AT, Buga AM, Grisk O, Walker LC, Kessler C, Popa-Wagner A: Long-term hypothermia reduces infarct volume in aged rats after focal ischemia. Neurosci Lett. 2008, 438: 180-185. 10.1016/j.neulet.2008.04.020.
Qu K, Chen CP, Halliwell B, Moore PK, Wong PT: Hydrogen sulfide is a mediator of cerebral ischemic damage. Stroke. 2006, 37: 889-893. 10.1161/01.STR.0000204184.34946.41.
Ren G, Bardwell J: Engineered pathways for correct disulfide bond oxidation. Antioxid Redox Signal.
Kimura Y, Goto Y, Kimura H: Hydrogen sulfide increases glutathione production and suppresses oxidative stress in mitochondria. Antioxid Redox Signal. 2010, 12: 1-13. 10.1089/ars.2008.2282.
Minamishima S, Bougaki M, Sips PY, Yu JD, Minamishima YA, Elrod JW, Lefer DJ, Bloch KD, Ichinose F: Hydrogen sulfide improves survival after cardiac arrest and cardiopulmonary resuscitation via a nitric oxide synthase 3-dependent mechanism in mice. Circulation. 2009, 120: 888-896. 10.1161/CIRCULATIONAHA.108.833491.
Ryter S, Morse D, Choi A: Heme oxygenase-1/carbon monoxide:from basic science to therapeutic applications. Antioxid Red Siganl. 2010, 12: 1-13. 10.1089/ars.2008.2282.
Katori M, Busuttil RW, Kupiec-Weglinski JW: Heme oxygenase-1 system in organ transplantation. Transplantation. 2002, 74: 905-912. 10.1097/00007890-200210150-00001.
Akamatsu Y, Haga M, Tyagi S, Yamashita K, Graca-Souza AV, Ollinger R, Czismadia E, May GA, Ifedigbo E, Otterbein LE, et al: Heme oxygenase-1-derived carbon monoxide protects hearts from transplant associated ischemia reperfusion injury. FASEB J. 2004, 18: 771-772.
Yoshida Y, Hatayama N, Seki K: Study on the preservation with CO (PCO = 200-2,000 hPa), resuscitation, and heterotopic transplantation of an isolated rat heart. Cell Transplant. 2009, 18: 535-540.
Sato K, Balla J, Otterbein L, Smith RN, Brouard S, Lin Y, Csizmadia E, Sevigny J, Robson SC, Vercellotti G, et al: Carbon monoxide generated by heme oxygenase-1 suppresses the rejection of mouse-to-rat cardiac transplants. J Immunol. 2001, 166: 4185-4194.
Zhou HC, Ding WG, Cui XG, Pan P, Zhang B, Li WZ: Carbon monoxide inhalation ameliorates conditions of lung grafts from rat brain death donors. Chin Med J (Engl). 2008, 121: 1411-1419.
Williams JW, Mital D, Chong A, Kottayil A, Millis M, Longstreth J, Huang W, Brady L, Jensik S: Experiences with leflunomide in solid organ transplantation. Transplantation. 2002, 73: 358-366. 10.1097/00007890-200202150-00008.
Hartsfield CL: Cross talk between carbon monoxide and nitric oxide. Antioxid Redox Signal. 2002, 4: 301-307. 10.1089/152308602753666352.
Boehning D, Snyder S: Novel neural modulators. Ann Rev Neurosci. 2003, 26: 105-131. 10.1146/annurev.neuro.26.041002.131047.
Maejima Y, Adachi S, Morikawa K, Ito H, Isobe M: Nitric oxide inhibits myocardial apoptosis by preventing caspase-3 activity via S-nitrosylation. J Mol Cell Cardiol. 2005, 38: 163-174. 10.1016/j.yjmcc.2004.10.012.
Rakhit R, Mojet M, Marber M, Duchen M: Mitochondria as targets for nitric oxide-induced protection during simulated ischemia and reoxygenation in isolated neonatal cardiomyocytes. Ciruculation. 2001, 103: 2617-2623.
Korhonen R, Lahti A, Kankaanranta H, Moilanen E: Nitric oxide production and signaling in inflammation. Curr Drug Targets Inflamm Allergy. 2005, 4: 471-479. 10.2174/1568010054526359.
Wang R, Wang Z, Wu L: Carbon monoxide-induced vasorelaxation and the underlying mechanisms. Br J Pharmacol. 1997, 121: 927-934. 10.1038/sj.bjp.0701222.
Otterbein L: The evolution of carbon monoxide into medicine. Respir Care. 2009, 54: 925-932. 10.4187/002013209793800394.
Zhou H, Liu J, Pan P, Jin D, Ding W, Li W: Carbon monoxide inhalation decreased lung injury via anti-inflammatory and anti-apoptotic effects in brain death rats. Exp Biol Med (Maywood). 2010, 235: 1236-1243. 10.1258/ebm.2010.010147.
Shibuya N, Mikami Y, Kimura Y, Nagahara N, Kimura H: Vascular endothelium expresses 3-mercaptopyruvate sulfurtransferase and produces hydrogen sulfide. J Biochem. 2009, 146: 623-626. 10.1093/jb/mvp111.
Wagner CA: Hydrogen sulfide: a new gaseous signal molecule and blood pressure regulator. J Nephrol. 2009, 22: 173-176.
Kasparek MS, Linden DR, Kreis ME, Sarr MG: Gasotransmitters in the gastrointestinal tract. Surgery. 2008, 143: 455-459. 10.1016/j.surg.2007.10.017.
Ryter SW, Morse D, Choi AM: Carbon monoxide: to boldly go where NO has gone before. Sci STKE. 2004, 2004: RE6-10.1126/stke.2302004re6.
Nathan C: Nitric oxide as a secretory product of mammalian cells. FASEB J. 1992, 6: 3051-3064.
Durante W: Targeting heme oxygenase-1 in vascular disease. Curr Drug Targets. 2010, 11: 1504-1516.
Li XH, Du JB, Bu DF, Tang XY, Tang CS: Sodium hydrosulfide alleviated pulmonary vascular structural remodeling induced by high pulmonary blood flow in rats. Acta Pharmacol Sin. 2006, 27: 971-980. 10.1111/j.1745-7254.2006.00353.x.
Wanstall J, Jeffery T, Gambino A, Lovren F, Triggle C: Vascular smooth muscle relaxation mediated by nitric oxide donors: a comparison with acetylcholine, nitric oxide and nitroxyl ion. Br J Pharmaco. 2001, 134: 463-472. 10.1038/sj.bjp.0704269.
Zhang QY, Du JB, Zhang CY, Tang CS: The regulation of carbon monoxide/heme oxygenase system by hydrogen sulfide in rats with hypoxic pulmonary hypertension. Zhonghua Jie He He Hu Xi Za Zhi. 2004, 27: 659-663.
Burger D, Xiang H, Lu X, Feng Q: Role of heme oxygenase-1 in the cardioprotective effects of erythorpoietin during myocardial ischemia. Am J Physiol Heart Circ Physiol. 2009, 1: H84-93.
Acknowledgements
Supported by grants from the American Diabetes Association (7-09-BS-26) and the National Institutes of Health (NIH) National Heart Lung and Blood Institute (1R01HL098481-01) to JWC. This work was also supported by funding from the Carlyle Fraser Heart Center (CFHC) of Emory University Hospital Midtown.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
BFM and JWC wrote the manuscript. All authors have read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Moody, B.F., Calvert, J.W. Emergent role of gasotransmitters in ischemia-reperfusion injury. Med Gas Res 1, 3 (2011). https://doi.org/10.1186/2045-9912-1-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/2045-9912-1-3