
Abstract
Background
Infantile hemangiomas are benign vascular tumors primarily found on the skin in 10% of the pediatric population. The etiology of this disease is largely unknown and while large scale genomic studies have examined the transcriptomes of infantile hemangioma tumors as a whole, no study to date has compared the global gene expression profiles of pure infantile hemangioma endothelial cells (HEMECs) to that of normal human dermal microvascular endothelial cells (HDMVECs).
Methods
To shed light on the molecular differences between these normal and aberrant dermal endothelial cell types, we performed whole genome microarray analysis on purified cultures of HEMECs and HDMVECs. We then utilized qPCR and immunohistochemistry to confirm our microarray results.
Results
Our array analysis identified 125 genes whose expression was upregulated and 104 genes whose expression was downregulated by greater than two fold in HEMECs compared to HDMVECs. Bioinformatics analysis revealed three major classifications of gene functions that were altered in HEMECs including cell adhesion, cell cycle, and arachidonic acid production. Several of these genes have been reported to be critical regulators and/or mutated in cancer, vascular tumors, and vascular malformations. We confirmed the expression of a subset of these differentially expressed genes (ANGPT2, ANTXR1, SMARCE1, RGS5, CTAG2, LTBP2, CLDN11, and KISS1) using qPCR and utilized immunohistochemistry on a panel of paraffin embedded infantile hemangioma tumor tissues to demonstrate that the cancer/testis antigen CTAG2 is highly abundant in vessel-dense proliferating infantile hemangiomas and with significantly reduced levels during tumor involution as vascular density decreases.
Conclusion
Our data reveal that the transcriptome of HEMECs is reflective of a pro-proliferative cell type with altered adhesive characteristics. Moveover, HEMECs show altered expression of many genes that are important in the progression and prognosis of metastatic cancers.
Similar content being viewed by others


Introduction
Infantile hemangiomas are benign tumors of vascular origin that affect approximately 10% of the pediatric population. These tumors are characterized by a rapid proliferation phase over the first 1–2 years of the child’s life, followed by a slow and steady decline over the next 5–7 years leading to the complete involution of the tumor mass. Approximately 90% of all infantile hemangiomas remain small and are best left alone to naturally involute. However in about 10% of the cases the tumors exhibit aggressive characteristics based on their size, location, number, etc. and must be actively treated to avoid patient disfigurement and/or mortality.
The etiology of infantile hemangiomas is largely unknown, particularly with regard to the cellular origin of the tumor. Circumstantial evidence suggests that these lesions are of aberrant placental origin as evidenced by upregulated Glut1 expression [1], and some labs have ventured to hypothesize that they may be formed from metastatic invasion of placenta-derived chorangioma cells [2]. Indeed, transcriptional profiling of human placenta, infantile hemangioma, and eight normal and diseased vascularized tissues suggests that high transcriptome similarity is shared between placenta and hemangioma tissues, more so than any of the other tissues tested [3]. Global gene expression analysis of infantile hemangioma tumors has been previously performed by two labs. Ritter et al. [4] utilized microarray analysis on whole tumors and identified immune regulators and indoleamine 2,3 dioxygenase as key regulators of infantile hemangioma involution. Calicchio et al. [5] utilized laser capture microdissection and genome-wide transcriptional profiling of vessels from proliferating and involuting hemangiomas. The authors strongly associated proliferating hemangioma vessels with increased expression of genes involved in endothelial-pericyte interactions and neuronal/vascular patterning, and involuting hemangiomas with chronic inflammatory mediators and angiogenic inhibitors. Given the high density of tightly associated pericytes in infantile hemangiomas and the inevitable collateral capture of intraluminal white cells, fibroblasts, mast cells, and perivascular collagen with laser microdissection, these data represent changes from numerous cell types within the infantile hemangioma tumor, but are not reflective specifically of the aberrant endothelial cells which contribute to disease. While these genomics studies have provided great mechanistic insight into the etiology and progression of the disease, they have not addressed the unique differences between abnormal infantile hemangioma endothelial cells and the normal dermal endothelial cells that are resident in the surrounding skin area of the patient. Understanding these differences could identify targetable pathways that could be exploited to preferentially block hemangioma growth and spread, but spare normal endothelial cells.
To date, no direct whole genome comparison of pure cultures of human dermal microvascular endothelial cells (HDMVECs) and infantile hemangioma endothelial cells (HEMECs) has been reported. To address this, we performed whole genome microarray profiling of the gene expression alterations between low passage pure cultures of HEMECs and HDMVECs. We identified a number of transcriptional alterations that are likely to contribute to the aggressive phenotype of infantile hemangiomas and that could potentially be utilized in immunotherapy against particularly aggressive hemangiomas tumors.
Materials and methods
Cell culture and chemicals
The HEMEC cell line was previously isolated from a proliferating-phase infantile hemangioma specimen collected from a female infant and generously donated to us by Joyce Bischoff (Harvard Medical School) [6]. The primary culture of neonatal HDMVECs was purchased from ATCC. Both cell lines were cultured as previously reported [7]. For all experiments, cell lines were used at <5 passages.
Proliferation assay
Cells were plated at equivalent sub-confluent densities and maintained in a Nikon Biostation CT time lapse imaging station. Cell proliferation was measured by counting cells per vision field from 5 independent areas over a 96 hour time course. Data presented is the average of the counts plus or minus the standard deviation. Student’s t-test was used to evaluate statistical significance. Data with p<0.05 was considered significant.
Migration assay
Confluent cultures were scratch wounded and the progress of “wound healing” was monitored using a Nikon Biostation CT time lapse imaging station over a 9 hour period. Data presented is the average migration speed plus or minus the standard deviation. Student’s t-test was used to evaluate statistical significance (p<0.05). Data with p<0.05 was considered significant.
Immunofluorescence
Cells were plated onto collagen type I coated glass coverslips, fixed in 4% paraformaldehyde, and incubated with antibodies against phospho-focal adhesion kinase (p-FAK; 1:1000; Cell Signaling #3283), rhodamine conjugated phalloidin (1:350; Cytoskeleton Inc.), or DAPI and imaged via a Nikon Eclipse Ti laser scanning confocal microscope.
Microarray analysis
Total RNA was amplified and biotin-labeled using Illumina TotalPrep RNA Amplification Kit (Ambion). 750 ng of biotinylated aRNA was then briefly heat-denatured and loaded onto expression arrays to hybridize overnight. Following hybridization, arrays were labeled with Cy3-streptavidin and imaged on the Illumina ISCAN. Intensity values were transferred to Agilent GeneSpring GX microarray analysis software and data was filtered based on quality of each call. Statistical relevance was determined using ANOVA with a Benjamini Hochberg FDR multiple testing correction (p-value < 0.05). Data were then limited by fold change analysis to statistically relevant data points demonstrating a 2-fold or more change in expression. Pathway analysis was performed using Metacore software. The microarray data from this experiment is publically available on the Gene Expression Omnibus (GEO Accession #GSE43742).
Quantitative real time PCR analysis
RNA was isolated from cells using the Ambion Purelink Minikit according to the manufacturer’s directions. qRT-PCR was performed on an ABI7900HT RT-PCR system using TaqMan Assays with predesigned primer sets for the genes of interest (Invitrogen). All RT-PCR experiments were performed in triplicate.
Immunohistochemistry
Paraffinized infantile hemangioma tissues were labeled with CTAG2 antibody (1:200, Santa Cruz Biotechnology #sc99243) and quantified using Alkaline Phosphatase detection (CellMarque). Positive and negative controls from breast carcinoma tissues were stained with CTAG2 antibody or sham, respectively. Use of de-identified human tissues was approved by the Texas Tech University Health Sciences Center Institutional Review Board for the Protection of Human Subjects (IRB E13029). Waiver of informed consent was approved by IRB.
Results and discussion
A comparison of the proliferation and migration rates of HEMECs and HDMVECs under standard growth conditions revealed no significant difference between normal and hemangioma endothelial cell types, however HEMECs grown under reduced serum conditions (0.5% fetal bovine serum) exhibited an approximately 30% increase in proliferation and an approximately 18% increase in migration relative to HDMVECs grown under the same conditions (Figure 1A & B). This suggests the higher serum concentrations were likely masking any phenotypic advantage attributed to the HEMECs. Moreover, it indicates the proliferative and migratory capacity of HEMECs are unique from that observed in HDMVECs and agrees with earlier reports suggesting advantages in these areas for HEMECs [6]. Comparisons of fluorescent images of the actin cytoskeleton and active focal adhesion complexes obtained with confocal microscopy revealed that HDMVECs display primarily peripheral membrane localized p-FAK, indicating sites of cellular attachment to the extracellular matrix (ECM) (Figure 1C). In contrast, p-FAK localization in HEMECs was observed along the entirety of the actin stress fibers, suggesting cellular adhesion to its substrate is markedly altered in HEMECs. Indeed, it has previously been reported that HEMECs display unique expression of genes involved in cellular adhesion [8].

Analysis of HDMVEC and HEMEC phenotypes. (A) Analysis of proliferation rates between HDMVECs and HEMECs over a 48 hr time course. (B) Analysis of the migration rates of HDMVECs and HEMECs nine hours after initial scratch from a micropipette. (C) Immunofluorescent imaging of actin (red), p-FAK (green), and nucleus (blue). (red asterisks for panels A &B represent statistically significant values [p<0.05] as determined by Student’s t-test).
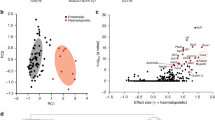
Whole genome microarray analysis reveals large scale alterations in gene expression between HEMECs and HDMVECs
Given the phenotypic differences observed between HEMECs and HDMVECs, we compared the global gene expression patterns between pure cultures of these cells using Illumina high density BeadArrays to elucidate which molecular factors are deregulated in HEMECs. Our array analysis identified 125 genes whose expression was upregulated and 104 genes whose expression was downregulated (2 fold or greater, p<0.05) in HEMECs compared to HDMVECs (Table 1). Metacore analysis of the 2 fold or greater gene expression changes revealed three major classifications of gene functions that are altered in HEMECs including cell adhesion (TIMP1, COL1A1, COL1A2, MMP1, MMP13, SERPINE2, COL4A6, LAMC2, MMP2, CD44, CAV1, CCL2, JAM3, CLDN11, LYVE1), cell cycle (CCND2, CDKN2A, CCNA1, NCAPD2), and arachidonic acid production (ACSL5, FAP, LIPG, PLA2G4C). Given the number of adhesion genes whose expression is altered in HEMECs compared to HDMVECs, it is no surprise that we observed altered subcellular localization of p-FAK in HEMECs (Figure 1C), reflecting a unique adhesive phenotype in these cells. Our data reflect altered cell cycle regulation in HEMECs, with a downregulation of CCND2 (cyclin D2) and CDKN2A (p16Ink4A) and a potent 6.6 fold increase in CCNA1 (cyclin A1), and these changes may contribute to the enhanced proliferation rates in HEMECs and the uncontrolled cell growth observed in infantile hemangiomas tumors. Alterations in the expression of genes involved in arachidonic acid production were unique in that this polyunsaturated fatty acid can serve as a lipid second messenger in the regulation of phospholipase-C and protein kinase-C signaling, is a key inflammatory intermediate, and can act as a vasodilator [9].
We confirmed a small subset of these gene expression changes utilizing qPCR, revealing equivocal trends in gene expression between the microarray and qPCR data for ANGPT2, ANTXR1, SMARCE1, RGS5, CTAG2, LTBP2, CLDN11, and KISS1 (Table 2). Each of these genes has been firmly established to play critical roles in regulating angiogenesis and/or tumor progression [10–17]. Missense mutations in ANTXR1 have been reported in several infantile hemangiomas and contribute to the constitutive VEGFR2 signaling associated with these tumors [18]. Mutations and signaling aberrations in Tie2, the cognate receptor for ANGPT2, play central roles in the development of various vascular disorders [19, 20]. ANGPT2 has previously been shown to be down-regulated in response to serum in HEMECs [19]. Interestingly, ANGPT2 expression is higher in HEMECs compared to normal placental endothelial cells and is increased in proliferative infantile hemangioma tumors relative to involuting ones [5]. Virtually undetectable in normal vasculature, RGS5 is greatly upregulated in the vasculature of solid tumors and may have the potential to serve as a tumor biomarker [12]. The downregulation of the metastasis suppressor KISS1 that we observed in HEMECs may partially explain the locally aggressive properties of infantile hemangiomas, as this gene encodes an angiogenic suppressor [16, 21]. Moreover, the expression of KISS1 is markedly reduced in aggressive metastatic melanomas and breast cancers, and this loss of expression contributes to the metastatic phenotype of these cells [17, 22]. It is intriguing that such genes (particularly the cancer-specific genes) are aberrantly expressed in HEMECs, and undoubtedly their deregulation could potentiate aberrant vascular tumor states. As it has been proposed that infantile hemangiomas may be derived from motile placental-derived chorangioma cells [2], future genomics analysis should compare the transcriptomes of each tumor type to identify if aberrant expression of tumor-related genes is shared between the tissues.
Overexpression of the CTAG2 cancer/testis antigen in a panel of infantile hemangioma tumors
In our microarray analysis, the cancer/testis antigen CTAG2 displayed the highest upregulation of mRNA expression in HEMECs compared to the HDMECs. This gene, whose function is completely unknown, has been shown to be significantly increased in several metastatic cancers, and is actively being researched as a target of immune therapy for aggressive cancers [23–29]. If CTAG2 is preferentially upregulated in infantile hemangiomas, it is possible that treatment of disfiguring or life threatening infantile hemangioma tumors could employ immune therapy against this antigen. Furthermore, CTAG2 is reported to be a target for antigen-specific T-cells in patients with various metastatic tumors [29, 30]. A recent study has shown that nearly half of the patients with spontaneous CTAG2-specific CD4(+) T cell responses had circulating CTAG2-specific antibodies that recognized epitopes located in the C-terminal portion of CTAG2 [30]. As involution of infantile hemangiomas is believed to be due in part to an immune mediated attack on the tumor itself [4], it is possible that T-cell targeting of the overexpressed CTAG2 protein could contribute to this process. We confirmed our microarray data at the protein level by performing immunohistochemistry on a panel of 16 paraffin embedded infantile hemangioma tumors representing both the proliferating and involuting stages of the disease and 4 normal neonatal dermal tissues. A limited amount of CTAG2 expression was observed in the normal dermal tissues (a few nerve cells and bundles present staining, whereas the fibroblasts and collagen fibers are negative), and despite this gene being coined a “cancer/testis specific antigen”, analysis of publically available microarray datasets suggests this gene is expressed at a low level across a large number of tissues (http://www.biogps.org) and it has been reported in the literature to be expressed in the placenta and ovary [31]. In proliferating tumors (composed of densely proliferating endothelial cells), we observed intense CTAG2 staining in the endothelial cells for all sections analyzed (Figure 2). In contrast, involuting tumors (marked by substantial adipocyte deposits—a characteristic of the later stages in the development of this tumor [32]) exhibited significantly reduced levels of CTAG2 staining. As Calicchio et al. did not detect significant differences in CTAG2 expression between microdissected endothelial cells from proliferating and involuting infantile hemangiomas and the staining intensity of individual blood vessels appears relatively constant between proliferating and involuting hemangiomas, we suspect that the reduced CTAG2 staining in involuting tumors is most likely due to reductions in tumor vascular density but not changes in gene transcription.

Detection of CTAG2 protein levels in infantile hemangioma tissues. Proliferating and involuting infantile hemangioma tissues as well as normal neonatal foreskin tissues were cut from paraffin blocks, incubated with antibodies against CTAG2, and detected using alkaline phosphatase staining (red). Immunohistochemistry (IHC) controls included incubations without CTAG2 antibody (negative control) and with CTAG2 antibody (positive control) in thin sections from metastatic breast cancer. All images were obtained at 100X total magnification.
Conclusion
Our data indicate that global transcriptional expression patterns are markedly unique between pure cultures of HDMVECs and HEMECs with major alterations in cell cycle, adhesion, and arachidonic acid metabolism genes. Though considered benign, HEMECs showed surprising aberrant regulation in the expression of several genes involved in tumor progression. Our finding that CTAG2 is highly expressed in infantile hemangiomas may lead to the development of immune-mediated therapies against infantile hemangiomas.
References
North PE, Waner M, Mizeracki A, Mihm MC: GLUT1: a newly discovered immunohistochemical marker for juvenile hemangiomas. Hum Pathol. 2000, 31: 11-22. 10.1016/S0046-8177(00)80192-6.
Mihm MC, Nelson JS: Hypothesis: the metastatic niche theory can elucidate infantile hemangioma development. J Cutan Pathol. 2010, 37 (Suppl 1): 83-87.
Barnes CM, Huang S, Kaipainen A, Sanoudou D, Chen EJ, Eichler GS, Guo Y, Yu Y, Ingber DE, Mulliken JB, Beggs AH, Folkman J, Fishman SJ: Evidence by molecular profiling for a placental origin of infantile hemangioma. Proc Natl Acad Sci USA. 2005, 102: 19097-19102. 10.1073/pnas.0509579102.
Ritter MR, Moreno SK, Dorrell MI, Rubens J, Ney J, Friedlander DF, Bergman J, Cunningham BB, Eichenfield L, Reinisch J, Cohen S, Veccione T, Holmes R, Friedlander SF, Friedlander M: Identifying potential regulators of infantile hemangioma progression through large-scale expression analysis: a possible role for the immune system and indoleamine 2,3 dioxygenase (IDO) during involution. Lymphat Res Biol. 2003, 1: 291-299. 10.1089/153968503322758094.
Calicchio ML, Collins T, Kozakewich HP: Identification of signaling systems in proliferating and involuting phase infantile hemangiomas by genome-wide transcriptional profiling. Am J Pathol. 2009, 174: 1638-1649. 10.2353/ajpath.2009.080517.
Boye E, Yu Y, Paranya G, Mulliken JB, Olsen BR, Bischoff J: Clonality and altered behavior of endothelial cells from hemangiomas. J Clin Invest. 2001, 107: 745-752. 10.1172/JCI11432.
Stiles J, Amaya C, Pham R, Rowntree RK, Lacaze M, Mulne A, Bischoff J, Kokta V, Boucheron LE, Mitchell DC, Bryan BA: Propranolol treatment of infantile hemangioma endothelial cells: A molecular analysis. Exp Ther Med. 2012, 4: 594-604.
Khan ZA, Melero-Martin JM, Wu X, Paruchuri S, Boscolo E, Mulliken JB, Bischoff J: Endothelial progenitor cells from infantile hemangioma and umbilical cord blood display unique cellular responses to endostatin. Blood. 2006, 108: 915-921. 10.1182/blood-2006-03-006478.
Pfister SL, Gauthier KM, Campbell WB: Vascular pharmacology of epoxyeicosatrienoic acids. Adv Pharmacol. 2010, 60: 27-59.
Fagiani E, Christofori G: Angiopoietins in angiogenesis. Cancer Lett. 2013, 328: 18-26. 10.1016/j.canlet.2012.08.018.
Chaudhary A, St Croix B: Selective blockade of tumor angiogenesis. Cell Cycle. 2012, 11: 2253-2259. 10.4161/cc.20374.
Silini A, Ghilardi C, Figini S, Sangalli F, Fruscio R, Dahse R, Pedley RB, Giavazzi R, Bani M: Regulator of G-protein signaling 5 (RGS5) protein: a novel marker of cancer vasculature elicited and sustained by the tumor's proangiogenic microenvironment. Cell Mol Life Sci. 2012, 69: 1167-1178. 10.1007/s00018-011-0862-8.
Garcia-Pedrero JM, Kiskinis E, Parker MG, Belandia B: The SWI/SNF chromatin remodeling subunit BAF57 is a critical regulator of estrogen receptor function in breast cancer cells. J Biol Chem. 2006, 281: 22656-22664. 10.1074/jbc.M602561200.
Lethe B, Lucas S, Michaux L, De Smet C, Godelaine D, Serrano A, De Plaen E, Boon T: LAGE-1, a new gene with tumor specificity. Int J Cancer. 1998, 76: 903-908. 10.1002/(SICI)1097-0215(19980610)76:6<903::AID-IJC22>3.0.CO;2-1.
Wessells H, Sullivan CJ, Tsubota Y, Engel KL, Kim B, Olson NE, Thorner D, Chitaley K: Transcriptional profiling of human cavernosal endothelial cells reveals distinctive cell adhesion phenotype and role for claudin 11 in vascular barrier function. Physiol Genomics. 2009, 39: 100-108. 10.1152/physiolgenomics.90354.2008.
Cho SG, Yi Z, Pang X, Yi T, Wang Y, Luo J, Wu Z, Li D, Liu M: Kisspeptin-10, a KISS1-derived decapeptide, inhibits tumor angiogenesis by suppressing Sp1-mediated VEGF expression and FAK/Rho GTPase activation. Cancer Res. 2009, 69: 7062-7070. 10.1158/0008-5472.CAN-09-0476.
Mitchell DC, Stafford LJ, Li D, Bar-Eli M, Liu M: Transcriptional regulation of KiSS-1 gene expression in metastatic melanoma by specificity protein-1 and its coactivator DRIP-130. Oncogene. 2007, 26: 1739-1747. 10.1038/sj.onc.1209963.
Jinnin M, Medici D, Park L, Limaye N, Liu Y, Boscolo E, Bischoff J, Vikkula M, Boye E, Olsen BR: Suppressed NFAT-dependent VEGFR1 expression and constitutive VEGFR2 signaling in infantile hemangioma. Nat Med. 2008, 14: 1236-1246. 10.1038/nm.1877.
Yu Y, Varughese J, Brown LF, Mulliken JB, Bischoff J: Increased Tie2 expression, enhanced response to angiopoietin-1, and dysregulated angiopoietin-2 expression in hemangioma-derived endothelial cells. Am J Pathol. 2001, 159: 2271-2280. 10.1016/S0002-9440(10)63077-5.
London NR, Whitehead KJ, Li DY: Endogenous endothelial cell signaling systems maintain vascular stability. Angiogenesis. 2009, 12: 149-158. 10.1007/s10456-009-9130-z.
Ramaesh T, Logie JJ, Roseweir AK, Millar RP, Walker BR, Hadoke PW, Reynolds RM: Kisspeptin-10 inhibits angiogenesis in human placental vessels ex vivo and endothelial cells in vitro. Endocrinology. 2010, 151: 5927-5934. 10.1210/en.2010-0565.
Mitchell DC, Abdelrahim M, Weng J, Stafford LJ, Safe S, Bar-Eli M, Liu M: Regulation of KiSS-1 metastasis suppressor gene expression in breast cancer cells by direct interaction of transcription factors activator protein-2alpha and specificity protein-1. J Biol Chem. 2006, 281: 51-58.
Odunsi K, Jungbluth AA, Stockert E, Qian F, Gnjatic S, Tammela J, Intengan M, Beck A, Keitz B, Santiago D, Williamson B, Scanlan MJ, Ritter G, Chen YT, Driscoll D, Sood A, Lele S, Old LJ: NY-ESO-1 and LAGE-1 cancer-testis antigens are potential targets for immunotherapy in epithelial ovarian cancer. Cancer Res. 2003, 63: 6076-6083.
Zeng G, Aldridge ME, Wang Y, Pantuck AJ, Wang AY, Liu YX, Han Y, Yuan YH, Robbins PF, Dubinett SM, deKernion JB, Belldegrun AS: Dominant B cell epitope from NY-ESO-1 recognized by sera from a wide spectrum of cancer patients: implications as a potential biomarker. Int J Cancer. 2005, 114: 268-273. 10.1002/ijc.20716.
Kan T, Yamasaki S, Kondo K, Teratani N, Kawabe A, Kaganoi J, Meltzer SJ, Imamura M, Shimada Y: A new specific gene expression in squamous cell carcinoma of the esophagus detected using representational difference analysis and cDNA microarray. Oncology. 2006, 70: 25-33. 10.1159/000091183.
Shao Y, Sun ZY, Sun SW, Zhao Y, Sin WY, Yuan YH, Simpson AJ, Old LJ, Sang XT, Mao YL, Xie Y, Huang JF, Zhao HT: Identification and expression analysis of novel LAGE-1 alleles with single nucleotide polymorphisms in cancer patients. J Cancer Res Clin Oncol. 2008, 134: 495-502. 10.1007/s00432-007-0312-z.
Andrade VC, Vettore AL, Felix RS, Almeida MS, Carvalho F, Oliveira JS, Chauffaille ML, Andriolo A, Caballero OL, Zago MA, Colleoni GW: Prognostic impact of cancer/testis antigen expression in advanced stage multiple myeloma patients. Cancer Immun. 2008, 8: 2-
Wang XY, Chen HS, Luo S, Zhang HH, Fei R, Cai J: Comparisons for detecting NY-ESO-1 mRNA expression levels in hepatocellular carcinoma tissues. Oncol Rep. 2009, 21: 713-719.
Pollack SM, Li Y, Blaisdell MJ, Farrar EA, Chou J, Hoch BL, Loggers ET, Rodler E, Eary JF, Conrad EU, Jones RL, Yee C: NYESO-1/LAGE-1s and PRAME are targets for antigen specific T cells in chondrosarcoma following treatment with 5-Aza-2-deoxycitabine. PLoS One. 2012, 7: e32165-10.1371/journal.pone.0032165.
Kudela P, Sun Z, Fourcade J, Janjic B, Kirkwood JM, Maillere B, Zarour HM: Epitope hierarchy of spontaneous CD4+ T cell responses to LAGE-1. J Immunol. 2011, 186: 312-322. 10.4049/jimmunol.1001989.
Kalejs M, Erenpreisa J: Cancer/testis antigens and gametogenesis: a review and "brain-storming" session. Cancer Cell Int. 2005, 5: 4-10.1186/1475-2867-5-4.
Kleiman A, Keats EC, Chan NG, Khan ZA: Evolution of hemangioma endothelium. Exp Mol Pathol. 2012, 93: 264-272. 10.1016/j.yexmp.2012.04.020.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
JMS Performed proliferation and migration assays, confocal microscopy, and qPCR RKR Cultured the HDMVEC and HIHECs and prepared samples for microarray analysis. CA Generated and analyzed tables of statistically significant genes, performed bioinformatics analysis to identify key gene networks. DD Performed histology and immunohistochemistry. VK Provided human tissues and expertise in histological and immunohistochemistry analysis. DCM Performed microarray analysis and bioinformatics analysisc. BAB Wrote manuscript, analyzed data, coordinated project.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Stiles, J.M., Rowntree, R.K., Amaya, C. et al. Gene expression analysis reveals marked differences in the transcriptome of infantile hemangioma endothelial cells compared to normal dermal microvascular endothelial cells. Vasc Cell 5, 6 (2013). https://doi.org/10.1186/2045-824X-5-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/2045-824X-5-6