
Abstract
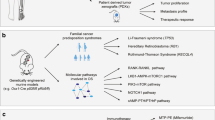
Osteosarcoma is the most common form of bone cancer. Pivotal insight into the genes involved in human osteosarcoma has been provided by the study of rare familial cancer predisposition syndromes. Three kindreds stand out as predisposing to the development of osteosarcoma: Li-Fraumeni syndrome, familial retinoblastoma and RecQ helicase disorders, which include Rothmund-Thomson Syndrome in particular. These disorders have highlighted the important roles of P53 and RB respectively, in the development of osteosarcoma. The association of OS with RECQL4 mutations is apparent but the relevance of this to OS is uncertain as mutations in RECQL4 are not found in sporadic OS. Application of the knowledge or mutations of P53 and RB in familial and sporadic OS has enabled the development of tractable, highly penetrant murine models of OS. These models share many of the cardinal features associated with human osteosarcoma including, importantly, a high incidence of spontaneous metastasis. The recent development of these models has been a significant advance for efforts to improve our understanding of the genetics of human OS and, more critically, to provide a high-throughput genetically modifiable platform for preclinical evaluation of new therapeutics.
Similar content being viewed by others


Review
Osteosarcoma
Osteosarcoma (OS) is the most common primary tumour of bone. It is most frequent in children and adolescents with an incidence of 7.3 per 1 million of the population [1]. Although OS is mainly classified as a childhood disease, a second peak of incidence is reported in the elderly population [1]. The majority of OS tumours are situated in the long bones with a small proportion located in the pelvis and axial skeleton [2, 3]. OS has a relatively high metastatic rate, with the lung being the most common site of spread.
The current treatment for OS revolves around the use of chemotherapy, radiotherapy and the surgical removal of the tumour. The chemotherapeutic regimen for OS patients combines cisplatin, doxorubicin and high doses of methotrexate [4]. Surgical resection is coupled with limb salvage procedures to remove malignant tissue and minimize the impact on quality of life.
The lack of new therapeutic options for the management of OS has translated to a stagnation of patient outcomes [5, 6]. Survival and prognosis rates have remained largely unchanged in two decades despite increased detection and monitoring afforded by advances in clinical imaging modalities [7–9]. Furthermore, there are difficulties associated with the study of OS in humans, such as recruiting sufficient patients to allow clinical insights in trialing new treatment options. A key component to improving patient outcome will be the development and application of faithful experimental models of human OS. Such models can serve as a preclinical platform for the identification of new therapeutic targets and the in vivo testing and triaging of those proposed for human trials. Experimentally derived interventions could then be developed in in vivo models where therapies can be rigorously evaluated side by side prior to human evaluation. Equally importantly, experimental OS models serve as a means to further understand the genetics and biology of OS with an emphasis on metastatic disease.
Animal models of osteosarcoma
Robust animal models have the capacity to preclinically evaluate therapeutic interventions derived from the extensive basic research efforts underway in OS. To date, the major species used to deliberately generate experimental OS are the mouse and the rat [10, 11]. The lineage and temporal specificity afforded by murine genetic engineering has lead to a rapid increase in the quality and fidelity of murine OS models when compared to the human condition. Spontaneous disease arising in large breed pet dogs is also of note as a model of human OS and is useful to understanding OS in humans and veterinary practice. It is also gaining prominence in the research environment as a validated model of spontaneous OS [12–14].
Rodent models of OS have been established for many decades and were originally generated through the exposure to chemical and radioactive carcinogens. [15–17]. These models demonstrated the principle of high-penetrance OS models that histologically resemble human OS. However, they possessed several caveats regarding their application to preclinical studies. The majority of OS in humans is sporadic, while the carcinogen-induced murine OS are more representative of therapy-induced disease rather than the primary lesions arising in the majority of human OS [18, 19]. Radiation induced OS models generally have a longer latency than alternate strategies and can result in a range of non-mesenchymal tumours due to its non-specific nature. Furthermore it has not been clearly defined what genetic lesions occur during the initiation and maintenance of these tumours. Nonetheless, these radiation-induced OS models have yielded robust experimental data and gave rise to valuable reagents such as cell lines to complement human OS studies. Further characterization of these tumours would enable the rational application of these alongside the recently generated tractable genetically engineered models.
Human hereditary disorders: insight into the genetics of human OS
Rare human hereditary disorders offer powerful insights into genes that play critical roles in human cancer biology in vivo. This is because they offer unequivocal evidence of defined genetic lesions and their importance in human disease pathogenesis. There is a cluster of familial syndromes that predispose to the development of OS and are of relevance to understanding the underlying genetics of OS. Li-Fraumeni syndrome, familial Retinoblastoma and RecQ helicase disorders such as Rothmund-Thomson Syndrome (RTS) are caused by germ-line mutations of P53, RB and RECQL4 respectively. These three kindreds have a greatly enhanced incidence of OS compared to the general population as documented in a range of clinical studies in affected families. In particular, Li-Fraumeni Syndrome patients are highly prone to develop OS, while OS is the second most common tumour type in Retinoblastoma patients [20–22]. OS tumours are a frequent feature of the tumour spectrum affecting RTS patients, however unlike mutations in p53 and the Rb pathway, RECQL4 mutations are not observed in sporadic OS [23].
A range of approaches has been used to incorporate information from clinical human OS to model the disease in the mouse. In particular, transgenic and germ-line loss of function alleles have demonstrated important roles for p53 mutations in generating experimental OS. More recently, lineage-restricted somatic deletion models that generate high penetrant metastatic disease have been described [24, 25]. These models will provide a definitive assessment on the roles of genes in the initiation and maintenance of OS. Furthermore they can be exploited to reveal new therapeutic avenues that can be targeted for the development of new therapies, with a particular emphasis on metastatic disease.
Human hereditary disorders and osteosarcoma
Li-fraumeni syndrome (LFS)
Li-Fraumeni syndrome is an autosomal dominant disorder with germ-line heterozygous mutation in P53. It is characterized by a predisposition to a range of cancers [26, 27]. LFS patients have a highly elevated risk of developing soft tissue sarcoma and osteosarcoma [28], and mutations in the “p53-pathway” are thought to be essential for the formation of human cancer.
Mutations in components of the p53 pathway are found in both familial and sporadic OS. Interestingly, the P53 allele itself is found to be mutated in human OS, most commonly as missense mutations [29, 30]. P53 mutations are not associated with therapeutic response or metastatic status [31, 32]. Other reported lesions in the p53 pathway in human OS include amplification of MDM2 and loss of p19ARF[33–37].
Hereditary retinoblastoma
Patients with familial retinoblastoma possess germline mutations in the Retinoblastoma (RB) gene [38]. Rb is a critical co-ordinator of G1-S phase cell cycle progression through its interaction with E2F and has been implicated in a wide range of cellular processes [39].
OS represents the second most frequent tumour in this kindred after retinoblastoma itself, with nearly half of all patients developing OS [40]. Most cases of sporadic OS present with modifications in at least one allele in the Rb locus [41, 42]. The contribution of therapy to OS development in retinoblastoma patients may be more significant than that occurring in LFS. In particular, OS arising from hereditary retinoblastoma is often located at the site of prior radiotherapy. Studies of radiation induced OS has observed mutation of P53 and retention of the intact RB allele in hereditary retinoblastoma patients [43]. As with the p53 pathway, mutations in the members of the Rb pathway occur frequently in OS with known mutations including amplifications of Cyclin E and CDK4 [44–48].
OS mouse models based on p53 and Rb mutations
The majority of murine OS models to date have been developed based on knowledge of the mutation of p53 and Rb pathways in both familial and sporadic human OS. Mice with germline mutations of p53 developed OS, but also succumbed to a wide range of tumours [49, 50]. Mice with tumour-associated p53 variants presented with a higher incidence of OS than germ-line p53 null animals, amongst the tumour spectrum these animals develop [51]. Mice with homozygous deletions of RB are embryonic lethal and their heterozygous counterparts are not predisposed to OS [50, 52]. The role of genetic compensation by other family members is apparent with the Rb related p107 and p130 in certain circumstance [53]. However, neither p107−/− nor p130−/− mice (or compound mutants that are viable) have a reported susceptibility to OS and these genes are not frequently mutated in human cancers based on data available through the COSMIC database [54].
The move to conditional lineage-restricted alleles of both p53 and pRb has allowed the development of new and more faithful models of OS. Utilising Prx1-Cre, which deletes LoxP flanked alleles in the early budding mesenchymal tissue of the limbs, 22% of mice with p53 heterozygosity develop OS. Homozygous deletion of p53 had a three-fold increase in OS occurrence. However, the deletion of Rb alone in mesenchymal progenitors failed to produce OS tumours [55]. Interestingly the conditional deletion of both p53 and Rb using Prx1-Cre resulted in approximately 70% of animals developing a poorly differentiated soft tissue sarcoma (PD-STS). This result suggests that the cell of origin is strongly influencing the arising tumour phenotype, with primitive multipotential cells favoring the development of PD-STS whilst committed osteoblast precursors give rise to OS at high incidence.
A separate group utilized the same transgenic system and yielded similar results. Over 60% of Prx1-Cre-p53fl/fl mice developed OS, while the homozygous deletion of Rb in isolation again yielded no tumours. The compound deletion of one Rb allele with homozygous p53 deletion increased the OS incidence rate to 92%. However, homozygous deletion of both genes yielded only 18% of OS tumours with a strong preference for hibernomas [56].
Rb has been proposed to have a role in influencing late osteoblast differentiation by interacting with Runx2 [57]. However, the removal of Rb alone is not sufficient to induce OS in a number of independent studies. Rb mutation does show a profound synergy with p53 mutation in the induction of experimental OS [24, 25]. Similarly, shRNAs that reduced Rb expression in p53-deficient OS cell lines (prior to allografts) gave rise to more aggressive and multilineage tumours [56]. The experimental approaches strongly suggest that mutation on the p53 pathway can serve as an initiating event in OS with mutation in the Rb pathway strongly synergizing in the immortalisation of osteoblastic cells.
Rothmund Thomson syndrome (RTS) and RecQ disorders
RTS is a rare autosomal disorder that consists of epithelial features (skin atrophy, hyper/hypo-pigmentation), congenital skeletal malformations (leading to short stature), premature ageing and increased malignant disease [58]. Most RTS patients have germ-line mutations in the RECQL4 DNA helicase [59–63]. RTS patients often present with multiple malignancies. In two separate studies, significant portions of RTS patients developed OS with median ages below 11 yrs [23, 64]. Conversely, overexpression of Recql4 was reported in human OS tumours with chromosomal abberations and instabilities in the 8q24 locus, which also contains c-Myc [65, 66]. RTS patients with truncating Recql4 mutations associate with a higher risk of developing OS as compared to non-truncated mutations [67, 68].
RECQL4 is a member of a family of DNA helicases including Bloom (BLM) and Werner (WRN) helicases, All three members are associated with familial cancer predisposition syndromes with high frequencies of mesenchymal derived tumours, with RTS in particular developing OS at approximately 30% frequency. As an ATP-dependent DNA helicase, Recql4 is recruited at the G1 and S phases of the cell cycle and plays a critical role in regulating DNA replication. Recql4 deficiency in mice is associated with karyotypic abnormalities and increased rates of aneuploidy [69, 70]. Strikingly in contrast to p53 and Rb mutations, Recql4 mutations are not associated with sporadic human OS and appear restricted to familial RTS OS. The failure to find RECQL4 mutations in sporadic OS raises several questions regarding the nature of the disease and whether it represents a distinct entity or subtype of OS. Further efforts characterizing the RTS- related OS are needed to clarify this and efforts to model RTS mutations in mouse may be informative. The contribution of prior chemotherapy/radiotherapy for other cancers arising in RTS patients may be a confounding factor in RTS-associated OS.
Recql4 Mutation in the mouse
Of the familial OS syndromes, the least is known about the role of Recql4. The expression of Recql4 shares an inverse relationship with Rb, although telomere-lengthening activities are enhanced in cells lacking both genes [71, 72]. Interestingly, Recql4 expression plays a role in osteoblast proliferation but its reduction is reported to be needed for full differentiation [73].
The attempts at modeling of Recql4 deficiency in mice has led to confounding results. Three non-conditional alleles have been reported. The first allele replaced exons 5 through 8 with a LacZ cassette. The homozygous deficient animals were reported as very early embryonic lethal between embryonic days 3–6 [74]. The second reported allele involved deletion of exon 13. The homozygous mutants were viable but exhibited severe growth retardation and multiple abnormalities and 95% of the mice died within 2 weeks of birth [75]. Hetrozygous Recql4 mutants were viable and had a decreased bone mass [73]. The third allele involved replacement of part of exon 9 through to exon 13 with a PGK-Hprt mini gene cassette [76]. These mice were viable and homozygous Recql4 deficient animals presented with a range of defects reminiscent of the human RTS alleles. Approximately 16% of mice with homozygous Recql4 mutations died within 24hrs of birth. 5.8% of animals displayed skeletal defects of the animals that survived past 24hrs. Cancers were detected in 5% of Recql4−/− animals in an aged cohort of 100 animals compared to 43 age matched controls, and of these 2 animals developed OS and 3 animals developed lymphoma. This low rate of tumour formation contrasts with the clinical presentation of RTS. The development and characterization of new targeted alleles will be needed to resolve the role of Recql4 in the initiation and maintenance of OS.
Werner & bloom syndromes
Werner syndrome is characterized by premature ageing and cancer predisposition that occurs during adolescence, whereas Bloom syndrome is characterized by short statures and photosensitive skin [77]. Both disorders are inherited in an autosomal recessive manner, and are attributed to germ-line mutations of the WRN and BLM genes respectively.
BLM plays a major role in maintaining genomic stability in cells [78]. Likewise, WRN acts against DNA breakages during chromatin structural modifications [79]. It is interesting to note that the expression of BLM and WRN is induced by the loss of Rb. Also, cells that lack the normal expression of all 3 genes presented with enhanced telomere lengthening [71, 72]. When treated with chemotherapeutics, cells that were deficient for BLM or WRN had decreased cell proliferation with impaired cell viability [80].
Werner Syndrome patients present with a range of cancers including OS [81, 82]. Similarly, patients with Bloom Syndrome are predisposed to various cancers, coupled with an early onset of these tumours [83, 84]. As for RTS, the relevance of these mutations to sporadic OS is also unclear and further work is needed to clarify the relationship between these OS and their sporadic counterpart.
BLM & WRN mouse models
Genetically engineered mice that habour null mutations of BLM were generated by 3 separate groups. Mice with homozygous deletion of BLM were embryonic lethal by day 13.5 and presented with an increased level of apoptosis and anaemia [85]. However, viable BLM-null mice were generated with the removal of neomycin plasmid sequence, of which 30% of these mice presented with a wide spectrum of spontaneous tumours [86]. Heterozygous mutant mice were also viable, with a predisposition to develop tumours [87].
Mice with homozygous deficiency for WRN were viable and developed tumours by 2 years of age. Interestingly, the combined deletions of p53 and WRN in mice resulted in various soft tissue sarcomas, where half of these mice developed tumours by 3 months of age [88]. However, its strongest link to OS was evident when WRN and Telomerase RNA Component (Terc) deficiency were combined in mice, with 50% of these mice developing OS [89]. Of note, these were not lineage-restricted alleles suggesting that these pathways co-operate specifically in osteoblasts and strongly synergise in the development of OS.
Paget’s Disease and p62
Paget’s disease of the bone is characterized by abnormalities in bone growth and destruction, resulting in limb deformities [90]. It is autosomal dominant in nature, and affects mainly adults over the age of 55 [91, 92]. It is also often asymptomatic until patients present with fracture or bone pain [93].
Sequestosome1 (SQSTM1) is the only gene currently identified and associated with Paget’s disease of the bone [94]. Also known as p62, this gene contributes to autophagy and removal of abnormal cells [95]. Interestingly, p62 expression needs to be repressed to suppress tumourigenesis [96].
The fraction of patients with Paget’s disease presenting with OS does not exceed 1% [97–101]. This cohort coincide with the second peak of OS incidence rates in the elderly [1, 102]. The survival rate of Paget’s disease-associated OS is 5% at 5 years [103].
Insights from p62 mouse models
Two separate groups generated transgenic mice that possessed the p62 mutation present in patients with Paget’s disease. There were conflicting results with regards to the histological bone features. However, mice from both groups presented with increased osteoclasts in response to RANKL stimulation, reminiscent of Paget’s disease patients [104, 105]. No OS was reported in these mice.
Other genes associated with osteosarcoma
A range of other genes have been implicated in OS pathogenesis based on studies of human OS samples and cell lines (Table 1). These mutations appear to be cooperative to the defects in the p53 and Rb pathways. Their involvement in OS pathogenesis is also supported by evidence derived from a range of genetically engineered mouse approaches.
c-Fos
The overexpression of c-Fos was first noted in human OS tumour samples, particularly in metastasized tumours [124, 125]. Its expression was also detected in mouse sporadic and radiation-induced OS [123]. In addition, genetically engineered mice that overexpressed c-Fos developed OS, thus suggestive of its role in OS pathogenesis [126, 127]. However, the overexpression of c-Fos in humans is linked to fibrous dysplasia, of which less than 2% of patients develop OS [143, 144]. Also, a recent study detected no change in c-Fos gene expression between human osteoblasts and OS tumours, which is in conflict with findings from Gamberi and Wu [66]. Therefore, the role of c-Fos in OS requires further studies to close the gap between transgenic mouse biology and human clinical studies.
c-MYC
Amplification of the c-MYC gene is more prominent in Paget’s disease-related OS as compared to primary OS, although genetic rearrangement does not appear to be the cause [145, 146]. Clinically, c-MYC expression levels in OS tumour samples was linked to resistance to methotrexate, with high c-MYC expression correlating to worse outcomes in OS patients [147].
A small cohort of transgenic mice developed OS when c-MYC expression was turned on with a tetracycline regulated transgene in haematopoietic cells [148]. The OS arising in these studies was most likely a result of ectopic expression of the transgene in osteoblastic cells. When c-MYC expression was inactivated by doxycycline administration, tumours transplanted into syngeneic mice regressed as OS cells differentiated into mature osteocytes [149]. In a subsequent report from the same group, the tumour regression from c-MYC inactivation in OS cells was attributed to the induction of senescence [150]. The development of OS was also reported in retrovirally transdcued c-MYC-overexpressing mesenchymal progenitor cells derived from Ink4a/Arf mutant mice [115].
Osteoblast lineage restricted expression of Simian Virus 40 (SV40) T antigen
Antigens of the SV40 virus interact with and inactivate tumour suppressor genes including both Rb and p53 [151, 152]. Interestingly, the SV40 gene was detected in a portion of human OS tumours, of which the sequence revealed viral integration in half of these tumours [153]. Early studies of transgenic mice that expressed SV40 antigens presented with OS and other tumours [154, 155]. A recent study of mice that expressed the SV40 T antigen in mature osteoblasts using the osteocalcin promoter presented with bone tumours and were morbid by 21 weeks of age. This timeframe for tumour development is strikingly similar to that observed with Osx-Cre p53fl/flpRbfl/fl animals. The tumours in Ocn-SV40Tag animals were histologically confirmed as OS and possessed various levels of calcification. Also, the OS tumours metastasized at high frequency and were found predominantly in the lung and spleen [156].
Further analysis of tumours derived in this model revealed a recurrent genomic deletion of the Prkar1a gene [156]. Correspondingly, deletion of 1 allele of Prkar1a dramatically accelerated OS formation in mice with Ocn-SV40 T antigen with tumours arising within 5 weeks of birth. The analysis of human tumours found a subset of human OS also habour a Prkar1a deletion, demonstrating the power of mouse models to uncover new information into the complex genetics of human OS.
Cell cycle genes: p15INK4b, p16INK4a
Several negative regulators of the G1-S cell cycle phase transition have been implicated in human OS. These fall into the “Rb pathway” and provide further support to the near obligate nature of this pathway disruption in the genesis of OS. p15INK4b was demonstrated to be repressed by c-MYC expression [157]. Mice deficient for p15INK4b (along with p14ARF and p16INK4a) developed a wide spectrum of cancers, including soft tissue sarcomas [111]. Genetic alterations were found in human patient-derived OS cell lines in the p15INK4b locus [112]. Deletions of the p16 genomic locus were apparent in samples from OS patients [158]. Loss of p16INK4A expression was found in pediatric OS samples, with its expression level correlating to survival rates [114].
Translating human cancer into animal models: issues & challenges
Human cell lines vs animal models?
Experimental studies of OS have involved the use of cell lines and animal disease models [159, 160]. However, cytogenetic complexity in human OS has confounded the efforts [161]. In particular, some human OS cell lines such as U2OS and SAOS-2 have been in use and passaged for many decades [162, 163]. The extended passage and tissue culture can result in the acquisition of adaptive mutations from cell-culture conditions, as seen in long-term culturing of embryonic stem cells and lung cancer cell lines [164–166]. As such, the drift in gene expression signatures may make it less representative of the original tumour tissue and also lead to heterogeneity of the cell line populations held by different investigators [167, 168]. The recent establishment and description of new OS cell lines opens up new avenues of study and hopefully improves the fidelity of tissue culture studies when referenced back to the human disease.
Murine and canine primary OS-derived cells have an advantage in this aspect. As a result of the relatively large amounts of primary, non-treated tumour tissue being available it is possible to establish early-passage cell lines for studies. Also, as mice on pure genetic backgrounds can be used, this will eliminate a significant source of intra-sample variation. The gene signatures from these lines would be expected to more closely mimic their primary tumour counterpart [165]. Also, the issues of over-passaging and culture-adaptation would be avoided as a result [169, 170]. Most importantly, paired primary and metastatic disease samples from untreated mice can be isolated for robust comparisons of paired disease. This research aspect would not be readily possible from available human samples and canine OS cell lines.
The recent study in the identification of the Prkar1a gene performed by Khokha and colleagues highlights the power of genetically engineered murine models to gain new insights into human OS genetics [156]. In particular, the use of high-resolution comparative genomic hybridization (cGH) in primary tumours among other complementary analytical techniques was utilized in this project. This allows biologically relevant genetic changes during OS pathogenesis to be isolated, defined and validated from aneuploidy- associated “noise”. Such approaches coupled with the developed murine models may allow significant advances in our understanding of the complexity of OS.
The comparison of primary and metastatic disease from as many of these models as possible would be a novel approach to develop a better understanding of metastatic disease. This will be very useful for understanding the genetics and cell biology of metastatic OS, and the epigenetic processes that drive these mechanisms. The experimental approach focused on by analysis of paired primary and metastatic tumours and cell lines derived from the same animal should provide a strong basis for identifying key drivers of the progression and maintenance of metastatic disease. Such an approach could be a starting point to develop better therapeutic strategies for treating metastatic disease, the primary cause of mortality in OS patients.
Different mouse models for different OS conditions
Various technological advancements have been incorporated into generating transgenic cancer mouse models. This includes germline & conditional knockouts, alleles bearing point mutations and tissue/region-specific gene expression [171, 172]. These technologies have allowed for multiple paradigms in exploring targeted gene expression and its role in OS pathogenesis. For instance, the Cre-Lox system is widely used to turn off the expression of targeted genes [173]. The turning off of desired genes using Cre-Lox is most often an irreversible step and is useful for modelling OS related to the partial and complete loss of gene function. For instance, the occurrence of OS in mice with homozygous p53 and Rb deletions mimics the clinical scenario of patients with autosomal-dominant hereditary disorders as well as lesions found in the sporadic OS population [24, 25].
The mouse models employed by two separate groups produced varying OS incidence rates, which was correlated with pRb and p53 status [24, 25]. This observation is concordant with various sporadic-OS patient reports where allelic alterations for both genes were reported retrospectively [42, 174–176]. The murine models have suggested strongly that deficiency for p53 is a strong initiating event for the development of OS and that disruption of the Rb pathway is a strongly synergistic mutation. The recent work from the Lees group provides an elegant model for the interaction and relative contribution of the p53 and pRb pathway mutations to the biological aspects of OS [56]. An unresolved question which will require analysis of human OS is to determine if the genetic alterations in OS could be different between sporadic and those associated with hereditary disorders.
An outstanding question is do mutations in all members of the p53 and Rb pathways contribute equally to tumour formation? For example, null mutation of the cyclin-dependent kinase p27Kip1, which results in deregulation of the “Rb pathway” did not result in OS in these mice [122, 177]. When coupled with a p53 mutation would p27Kip1 or p21Cip1 deficiency recapitulate all or only partial aspects of the loss of Rb? This is intriguing in light of the spectrum of mutations that have been reported in human OS. It provides an opportunity to compare mutations in distinct components of these pathways directly in the murine models that have been developed.
The emerging use of RNA interference (RNAi) in transgenic cancer models presents an exciting avenue to explore OS genetics and therapeutics. This is because the expression of targeted genes can be manipulated reversibly in a temporally controlled fashion to elucidate its biological purpose [178–180]. Also, this model provides the attractive prospect of exploring therapeutic target inhibition and resistance. As siRNA/shRNA represents a loss of function allele that are efficient but rarely complete this technology could be harnessed for the rapid and large scale in vivo screening of putative therapeutic targets. As small molecule inhibitors, like siRNA/shRNA, provide efficient but rarely complete target inactivation the testing of candidate therapeutic targets is highly suited to this approach.
The OS cell of origin
The OS cell of origin has been widely discussed in the research literature. Its identity was proposed to be mesenchymal stem cells due to its potential to give rise to osteoblasts [181–183]. It also aligns with the notion that OS is differentiation-defective, due to the lack of terminally differentiated osteoblastic cells [120, 184, 185]. Identified by expression of Runx2, these mesenchymal progenitors are purported to be the source of OS initiating cells [186–189]. It is important to note that the cancer cell of origin is not necessarily related to the origin of the cancer stem cell [190]. Likewise, the OS cell of origin need not be mesenchymal stem cells, despite the various postulations suggesting this. In particular, the deletion of p53 in mesenchymal progenitor cells only yielded 61% of OS, with the rest being poorly differentiated soft-tissue sarcoma [55]. Also, soft-tissue sarcomas seem most likely to arise from mesenchymal stem/progenitor cells [191, 192]. As the multipotent mesenchymal/skeletal stem cells can give rise to bone, cartilage and adipose cell lineages, perhaps it plays a more realistic purpose as a pan-sarcoma cell of origin.
Data derived from a range of genetic approaches most strongly favours the OS cell of origin to be found within the committed osteoblast lineage. In particular, accumulating experimental evidence is most consistent with OS arising from the osteoblastic progenitor population [24, 25]. For instance, the deletion of p53 in pre-osteoblasts and osteoblast progenitors resulted in significantly higher OS incidence rates than early multi-lineage potential cells (Figure 1 and Table 2). As osteoblast progenitors are more committed than their mesenchymal counterparts, this would correlate to decreased occurrence of other sarcoma types. As such, these studies strongly propose that OS arises from the osteoblast lineage-committed progenitor population and that the resulting tumour phenotype is a result of the accumulated genetic mutations that are present.

Schematic representation of osteoblastic lineage commitment and differentiation from mesenchymal progenitors and the models developed in genetically engineered murine models . A) Normal osteoblast development from mesenchymal stem cells. Genes associated with the commitment and differentiation of osteoblasts are listed along with an approximation of the developmental state of the osteoblastic cells. B) Using the differentiation schematic, the different Cre lines that have been described are in bold with an approximation of the putative cells expressing the Cre. The alleles that are disrupted (loss of function for p53 and pRb; over-expression for SV40TAg) are shown on the arrows. Other tumour types associated with the different models are also highlighted. For further information on these alleles see Table 2.
Metastatic disease – high fidelity and high penetrant models
The use of cancer mouse models with high penetrance allows a substantial population of mice with metastatic disease to be established. In particular, the mice generated by 3 separate groups developed OS with significant metastasis to soft tissues [24, 25, 156]. These models will be valuable in pre-clinical studies, as primary and metastasized tumours could be procured for the comparative studies. Advances in small animal imaging techniques such as μPET and μCT coupled with serology for alkaline phosphatase make possible the establishment of cohorts of animals with primary and a small metastatic disease burden. This strategy makes possible an assessment of therapeutic interventions in the context of primary and metastatic disease which are the most pressing clinical need. Longitudinal studies using such approaches would be an effective means to test and triage candidate therapeutic approaches in a controlled and reproducible manner. When coupled with xenografts of human material it may facilitate translation into rational clinical trials. Also, untreated paired tumour tissue will be useful as it is not readily collected in humans.
Conclusion
Li-Fraumeni, Retinoblastoma and Rothmund-Thomson Syndrome are three human familial cancer syndromes that present with the strongest association to OS. Amongst sporadic OS, a wider range of genes and members of the p53 and Rb pathways are also implicated in OS pathogenesis. These mutations fulfill a range of the prerequisite requirements associated with the hallmarks of cancer, however the genes do not carry equal importance in tumour biology nor fully account for the pathogenesis of OS [194]. The integration of genetically engineered murine models based on familial human genetics of OS and additional experimental models such as the spontaneous OS arising in large breed dogs combine to form the basis of a preclinical platform that can serve to translate the extensive basic research efforts associated with OS to a clinically meaningful advantage. The use of primary human xenografts, in contrast to approaches using established human OS cell lines, adds an important component to the preclinical assessment phase of any new therapeutic options [195]. The underlying genetics in OS covers a wide spectrum, ranging from complete loss of gene function to hypomorphic mutations and gain of function. Various genetically modified mouse models of OS are now available and have demonstrated clearly that these are able to recapitulate the clinical spectrum of human OS.
Abbreviations
- BLM:
-
Bloom
- LFS:
-
Li-Fraumeni Syndrome
- L-MTP-PE:
-
Liposomal Muramyl-Tripeptide Phosphatidyl Ethanolamine
- Ocn:
-
Osteocalcin
- OS:
-
Osteosarcoma
- PD-STS:
-
Poorly differentiated soft tissue sarcoma
- Rb:
-
Retinoblastoma
- shRNA:
-
Short hairpin RNA
- siRNA:
-
Small interfering RNA
- SQSTM1:
-
Sequestosome1
- SV40:
-
Simian Virus 40
- RNAi:
-
RNA interference
- Tag:
-
T antigen
- Terc:
-
Telomerase RNA Component
- WRN:
-
Werner.
References
Mirabello L, Troisi RJ, Savage SA: International osteosarcoma incidence patterns in children and adolescents, middle ages and elderly persons. Int J Cancer. 2009, 125: 229-234.
Klein MJ, Siegal GP: Osteosarcoma: anatomic and histologic variants. Am J Clin Pathol. 2006, 125: 555-581.
Lamoureux F, Trichet V, Chipoy C, Blanchard F, Gouin F, Redini F: Recent advances in the management of osteosarcoma and forthcoming therapeutic strategies. Expert Rev Anticancer Ther. 2007, 7: 169-181.
Chou AJ, Gorlick R: Chemotherapy resistance in osteosarcoma: current challenges and future directions. Expert Rev Anticancer Ther. 2006, 6: 1075-1085.
Ferguson PC, McLaughlin CE, Griffin AM, Bell RS, Deheshi BM, Wunder JS: Clinical and functional outcomes of patients with a pathologic fracture in high-grade osteosarcoma. J Surg Oncol. 2010, 102: 120-124.
Jawad MU, Cheung MC, Clarke J, Koniaris LG, Scully SP: Osteosarcoma: improvement in survival limited to high-grade patients only. J Cancer Res Clin Oncol. 2011, 137: 597-607.
Arndt V, Lacour B, Steliarova-Foucher E, Spix C, Znaor A, Pastore G, Stiller C, Brenner H: Up-to-date monitoring of childhood cancer long-term survival in Europe: tumours of the sympathetic nervous system, retinoblastoma, renal and bone tumours, and soft tissue sarcomas. Ann Oncol. 2007, 18: 1722-1733.
Gatta G, Capocaccia R, Stiller C, Kaatsch P, Berrino F, Terenziani M: Childhood cancer survival trends in Europe: a EUROCARE Working Group study. J Clin Oncol. 2005, 23: 3742-3751.
Mirabello L, Troisi RJ, Savage SA: Osteosarcoma incidence and survival rates from 1973 to 2004: data from the Surveillance, Epidemiology, and End Results Program. Cancer. 2009, 115: 1531-1543.
Heymann D, Ory B, Blanchard F, Heymann MF, Coipeau P, Charrier C, Couillaud S, Thiery JP, Gouin F, Redini F: Enhanced tumor regression and tissue repair when zoledronic acid is combined with ifosfamide in rat osteosarcoma. Bone. 2005, 37: 74-86.
Ko SC, Cheon J, Kao C, Gotoh A, Shirakawa T, Sikes RA, Karsenty G, Chung LW: Osteocalcin promoter-based toxic gene therapy for the treatment of osteosarcoma in experimental models. Cancer Res. 1996, 56: 4614-4619.
Johnson AS, Couto CG, Weghorst CM: Mutation of the p53 tumor suppressor gene in spontaneously occurring osteosarcomas of the dog. Carcinogenesis. 1998, 19: 213-217.
Lascelles BD, Dernell WS, Correa MT, Lafferty M, Devitt CM, Kuntz CA, Straw RC, Withrow SJ: Improved survival associated with postoperative wound infection in dogs treated with limb-salvage surgery for osteosarcoma. Ann Surg Oncol. 2005, 12: 1073-1083.
Paoloni M, Davis S, Lana S, Withrow S, Sangiorgi L, Picci P, Hewitt S, Triche T, Meltzer P, Khanna C: Canine tumor cross-species genomics uncovers targets linked to osteosarcoma progression. BMC Genomics. 2009, 10: 625.
Ingleton PM, Coulton LA, Preston CJ, Martin TJ: Alkaline phosphatase in serum and tumour of rats bearing a hormone-responsive transplantable osteogenic sarcoma. Eur J Cancer. 1979, 15: 685-691.
Martin TJ, Ingleton PM, Underwood JC, Michelangeli VP, Hunt NH, Melick RA: Parathyroid hormone-responsive adenylate cyclase in induced transplantable osteogenic rat sarcoma. Nature. 1976, 260: 436-438.
Underwood JC, Melick RA, Loomes RS, Dangerfield VM, Crawford A, Coulton L, Ingleton PM, Martin TJ: Structural and functional correlations in parathyroid hormone responsive transplantable osteogenic sarcomas. Eur J Cancer. 1979, 15: 1151-1158.
Jones KB: Osteosarcomagenesis: modeling cancer initiation in the mouse. Sarcoma. 2011, 2011: 694136.
McHugh JB, Thomas DG, Herman JM, Ray ME, Baker LH, Adsay NV, Rabah R, Lucas DR: Primary versus radiation-associated craniofacial osteosarcoma: Biologic and clinicopathologic comparisons. Cancer. 2006, 107: 554-562.
Birch JM, Blair V, Kelsey AM, Evans DG, Harris M, Tricker KJ, Varley JM: Cancer phenotype correlates with constitutional TP53 genotype in families with the Li-Fraumeni syndrome. Oncogene. 1998, 17: 1061-1068.
Garber JE, Goldstein AM, Kantor AF, Dreyfus MG, Fraumeni JF, Li FP: Follow-up study of twenty-four families with Li-Fraumeni syndrome. Cancer Res. 1991, 51: 6094-6097.
Gurney JG, Severson RK, Davis S, Robison LL: Incidence of cancer in children in the United States. Sex-, race-, and 1-year age-specific rates by histologic type. Cancer. 1995, 75: 2186-2195.
Wang LL, Levy ML, Lewis RA, Chintagumpala MM, Lev D, Rogers M, Plon SE: Clinical manifestations in a cohort of 41 Rothmund-Thomson syndrome patients. Am J Med Genet. 2001, 102: 11-17.
Berman SD, Calo E, Landman AS, Danielian PS, Miller ES, West JC, Fonhoue BD, Caron A, Bronson R, Bouxsein ML: Metastatic osteosarcoma induced by inactivation of Rb and p53 in the osteoblast lineage. Proc Natl Acad Sci U S A. 2008, 105: 11851-11856.
Walkley CR, Qudsi R, Sankaran VG, Perry JA, Gostissa M, Roth SI, Rodda SJ, Snay E, Dunning P, Fahey FH: Conditional mouse osteosarcoma, dependent on p53 loss and potentiated by loss of Rb, mimics the human disease. Genes Dev. 2008, 22: 1662-1676.
Li FP, Fraumeni JF, Mulvihill JJ, Blattner WA, Dreyfus MG, Tucker MA, Miller RW: A cancer family syndrome in twenty-four kindreds. Cancer Res. 1988, 48: 5358-5362.
Malkin D, Li FP, Strong LC, Fraumeni JF, Nelson CE, Kim DH, Kassel J, Gryka MA, Bischoff FZ, Tainsky MA: Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science. 1990, 250: 1233-1238.
Schneider K, Garber J: Li-Fraumeni Syndrome. Gene Reviews [Internet]. Edited by: Pagon R, Bird T, Dolan C. 1999, University of Washington, Seattle, Seattle,
Gokgoz N, Wunder JS, Mousses S, Eskandarian S, Bell RS, Andrulis IL: Comparison of p53 mutations in patients with localized osteosarcoma and metastatic osteosarcoma. Cancer. 2001, 92: 2181-2189.
Overholtzer M, Rao PH, Favis R, Lu XY, Elowitz MB, Barany F, Ladanyi M, Gorlick R, Levine AJ: The presence of p53 mutations in human osteosarcomas correlates with high levels of genomic instability. Proc Natl Acad Sci U S A. 2003, 100: 11547-11552.
Pakos EE, Kyzas PA, Ioannidis JP: Prognostic significance of TP53 tumor suppressor gene expression and mutations in human osteosarcoma: a meta-analysis. Clin Cancer Res. 2004, 10: 6208-6214.
Wunder JS, Gokgoz N, Parkes R, Bull SB, Eskandarian S, Davis AM, Beauchamp CP, Conrad EU, Grimer RJ, Healey JH: TP53 mutations and outcome in osteosarcoma: a prospective, multicenter study. J Clin Oncol. 2005, 23: 1483-1490.
Ladanyi M, Cha C, Lewis R, Jhanwar SC, Huvos AG, Healey JH: MDM2 gene amplification in metastatic osteosarcoma. Cancer Res. 1993, 53: 16-18.
Miller CW, Aslo A, Won A, Tan M, Lampkin B, Koeffler HP: Alterations of the p53, Rb and MDM2 genes in osteosarcoma. J Cancer Res Clin Oncol. 1996, 122: 559-565.
Miller CW, Yeon C, Aslo A, Mendoza S, Aytac U, Koeffler HP: The p19INK4D cyclin dependent kinase inhibitor gene is altered in osteosarcoma. Oncogene. 1997, 15: 231-235.
Rauch DA, Hurchla MA, Harding JC, Deng H, Shea LK, Eagleton MC, Niewiesk S, Lairmore MD, Piwnica-Worms D, Rosol TJ: The ARF tumor suppressor regulates bone remodeling and osteosarcoma development in mice. PLoS One. 2011, 5: e15755.
Smida J, Baumhoer D, Rosemann M, Walch A, Bielack S, Poremba C, Remberger K, Korsching E, Scheurlen W, Dierkes C: Genomic alterations and allelic imbalances are strong prognostic predictors in osteosarcoma. Clin Cancer Res. 2010, 16: 4256-4267.
Lohmann DR, Gerick M, Brandt B, Oelschlager U, Lorenz B, Passarge E, Horsthemke B: Constitutional RB1-gene mutations in patients with isolated unilateral retinoblastoma. Am J Hum Genet. 1997, 61: 282-294.
Hiebert SW: Regions of the retinoblastoma gene product required for its interaction with the E2F transcription factor are necessary for E2 promoter repression and pRb-mediated growth suppression. Mol Cell Biol. 1993, 13: 3384-3391.
Wong FL, Boice JD, Abramson DH, Tarone RE, Kleinerman RA, Stovall M, Goldman MB, Seddon JM, Tarbell N, Fraumeni JF, Li FP: Cancer incidence after retinoblastoma. Radiation dose and sarcoma risk. JAMA. 1997, 278: 1262-1267.
Hansen MF, Koufos A, Gallie BL, Phillips RA, Fodstad O, Brogger A, Gedde-Dahl T, Cavenee WK: Osteosarcoma and retinoblastoma: a shared chromosomal mechanism revealing recessive predisposition. Proc Natl Acad Sci U S A. 1985, 82: 6216-6220.
Wadayama B, Toguchida J, Shimizu T, Ishizaki K, Sasaki MS, Kotoura Y, Yamamuro T: Mutation spectrum of the retinoblastoma gene in osteosarcomas. Cancer Res. 1994, 54: 3042-3048.
Gonin-Laurent N, Hadj-Hamou NS, Vogt N, Houdayer C, Gauthiers-Villars M, Dehainault C, Sastre-Garau X, Chevillard S, Malfoy B: RB1 and TP53 pathways in radiation-induced sarcomas. Oncogene. 2007, 26: 6106-6112.
Benassi MS, Molendini L, Gamberi G, Ragazzini P, Sollazzo MR, Merli M, Asp J, Magagnoli G, Balladelli A, Bertoni F, Picci P: Alteration of pRb/p16/cdk4 regulation in human osteosarcoma. Int J Cancer. 1999, 84: 489-493.
Janeway KA, Walkley CR: Modeling human osteosarcoma in the mouse: From bedside to bench. Bone. 2010, 47: 859-865.
Lockwood WW, Stack D, Morris T, Grehan D, O'Keane C, Stewart GL, Cumiskey J, Lam WL, Squire JA, Thomas DM, O'Sullivan MJ: Cyclin E1 is amplified and overexpressed in osteosarcoma. J Mol Diagn. 2011, 13: 289-296.
Molendini L, Benassi MS, Magagnoli G, Merli M, Sollazzo MR, Ragazzini P, Gamberi G, Ferrari C, Balladelli A, Bacchini P, Picci P: Prognostic significance of cyclin expression in human osteosarcoma. Int J Oncol. 1998, 12: 1007-1011.
Wunder JS, Eppert K, Burrow SR, Gokgoz N, Bell RS, Andrulis IL: Co-amplification and overexpression of CDK4, SAS and MDM2 occurs frequently in human parosteal osteosarcomas. Oncogene. 1999, 18: 783-788.
Jacks T, Remington L, Williams BO, Schmitt EM, Halachmi S, Bronson RT, Weinberg RA: Tumor spectrum analysis in p53-mutant mice. Curr Biol. 1994, 4: 1-7.
Williams BO, Remington L, Albert DM, Mukai S, Bronson RT, Jacks T: Cooperative tumorigenic effects of germline mutations in Rb and p53. Nat Genet. 1994, 7: 480-484.
Lavigueur A, Maltby V, Mock D, Rossant J, Pawson T, Bernstein A: High incidence of lung, bone, and lymphoid tumors in transgenic mice overexpressing mutant alleles of the p53 oncogene. Mol Cell Biol. 1989, 9: 3982-3991.
Lee EY, Chang CY, Hu N, Wang YC, Lai CC, Herrup K, Lee WH, Bradley A: Mice deficient for Rb are nonviable and show defects in neurogenesis and haematopoiesis. Nature. 1992, 359: 288-294.
Viatour P, Somervaille TC, Venkatasubrahmanyam S, Kogan S, McLaughlin ME, Weissman IL, Butte AJ, Passegue E, Sage J: Hematopoietic stem cell quiescence is maintained by compound contributions of the retinoblastoma gene family. Cell Stem Cell. 2008, 3: 416-428.
Forbes SA, Bindal N, Bamford S, Cole C, Kok CY, Beare D, Jia M, Shepherd R, Leung K, Menzies A: COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2011, 39: D945-D950.
Lin PP, Pandey MK, Jin F, Raymond AK, Akiyama H, Lozano G: Targeted mutation of p53 and Rb in mesenchymal cells of the limb bud produces sarcomas in mice. Carcinogenesis. 2009, 30: 1789-1795.
Calo E, Quintero-Estades JA, Danielian PS, Nedelcu S, Berman SD, Lees JA: Rb regulates fate choice and lineage commitment in vivo. Nature. 2010, 466: 1110-1114.
Thomas DM, Carty SA, Piscopo DM, Lee JS, Wang WF, Forrester WC, Hinds PW: The retinoblastoma protein acts as a transcriptional coactivator required for osteogenic differentiation. Mol Cell. 2001, 8: 303-316.
Simon T, Kohlhase J, Wilhelm C, Kochanek M, De Carolis B, Berthold F: Multiple malignant diseases in a patient with Rothmund-Thomson syndrome with RECQL4 mutations: Case report and literature review. Am J Med Genet A. 2010, 152A: 1575-1579.
Anbari KK, Ierardi-Curto LA, Silber JS, Asada N, Spinner N, Zackai EH, Belasco J, Morrissette JD, Dormans JP: Two primary osteosarcomas in a patient with Rothmund-Thomson syndrome. Clin Orthop Relat Res. 2000, 378: 213-223.
Der Kaloustian VM, McGill JJ, Vekemans M, Kopelman HR: Clonal lines of aneuploid cells in Rothmund-Thomson syndrome. Am J Med Genet. 1990, 37: 336-339.
Kitao S, Lindor NM, Shiratori M, Furuichi Y, Shimamoto A: Rothmund-thomson syndrome responsible gene, RECQL4: genomic structure and products. Genomics. 1999, 61: 268-276.
Siitonen HA, Kopra O, Kaariainen H, Haravuori H, Winter RM, Saamanen AM, Peltonen L, Kestila M: Molecular defect of RAPADILINO syndrome expands the phenotype spectrum of RECQL diseases. Hum Mol Genet. 2003, 12: 2837-2844.
Vennos EM, Collins M, James WD: Rothmund-Thomson syndrome: review of the world literature. J Am Acad Dermatol. 1992, 27: 750-762.
Stinco G, Governatori G, Mattighello P, Patrone P: Multiple cutaneous neoplasms in a patient with Rothmund-Thomson syndrome: case report and published work review. J Dermatol. 2008, 35: 154-161.
Maire G, Yoshimoto M, Chilton-MacNeill S, Thorner PS, Zielenska M, Squire JA: Recurrent RECQL4 imbalance and increased gene expression levels are associated with structural chromosomal instability in sporadic osteosarcoma. Neoplasia. 2009, 11: 260-268. 263p following 268,
Sadikovic B, Thorner P, Chilton-Macneill S, Martin JW, Cervigne NK, Squire J, Zielenska M: Expression analysis of genes associated with human osteosarcoma tumors shows correlation of RUNX2 overexpression with poor response to chemotherapy. BMC Cancer. 2010, 10: 202.
Wang LL, Gannavarapu A, Kozinetz CA, Levy ML, Lewis RA, Chintagumpala MM, Ruiz-Maldanado R, Contreras-Ruiz J, Cunniff C, Erickson RP: Association between osteosarcoma and deleterious mutations in the RECQL4 gene in Rothmund-Thomson syndrome. J Natl Cancer Inst. 2003, 95: 669-674.
Wang W, Seki M, Narita Y, Nakagawa T, Yoshimura A, Otsuki M, Kawabe Y, Tada S, Yagi H, Ishii Y, Enomoto T: Functional relation among RecQ family helicases RecQL1, RecQL5, and BLM in cell growth and sister chromatid exchange formation. Mol Cell Biol. 2003, 23: 3527-3535.
Kitao S, Ohsugi I, Ichikawa K, Goto M, Furuichi Y, Shimamoto A: Cloning of two new human helicase genes of the RecQ family: biological significance of multiple species in higher eukaryotes. Genomics. 1998, 54: 443-452.
Thangavel S, Mendoza-Maldonado R, Tissino E, Sidorova JM, Yin J, Wang W, Monnat RJ, Falaschi A, Vindigni A: Human RECQ1 and RECQ4 helicases play distinct roles in DNA replication initiation. Mol Cell Biol. 2010, 30: 1382-1396.
Garcia-Cao M, Gonzalo S, Dean D, Blasco MA: A role for the Rb family of proteins in controlling telomere length. Nat Genet. 2002, 32: 415-419.
Liu Y, El-Naggar S, Clem B, Chesney J, Dean DC: The Rb/E2F pathway and Ras activation regulate RecQ helicase gene expression. Biochem J. 2008, 412: 299-306.
Yang J, Murthy S, Winata T, Werner S, Abe M, Prahalad AK, Hock JM: Recql4 haploinsufficiency in mice leads to defects in osteoblast progenitors: Implications for low bone mass phenotype. Biochem Biophys Res Commun. 2006, 344: 346-352.
Ichikawa K, Noda T, Furuichi Y: [Preparation of the gene targeted knockout mice for human premature aging diseases, Werner syndrome, and Rothmund-Thomson syndrome caused by the mutation of DNA helicases]. Nippon Yakurigaku Zasshi. 2002, 119: 219-226.
Hoki Y, Araki R, Fujimori A, Ohhata T, Koseki H, Fukumura R, Nakamura M, Takahashi H, Noda Y, Kito S, Abe M: Growth retardation and skin abnormalities of the Recql4-deficient mouse. Hum Mol Genet. 2003, 12: 2293-2299.
Mann MB, Hodges CA, Barnes E, Vogel H, Hassold TJ, Luo G: Defective sister-chromatid cohesion, aneuploidy and cancer predisposition in a mouse model of type II Rothmund-Thomson syndrome. Hum Mol Genet. 2005, 14: 813-825.
German J: Bloom's syndrome. Dermatol Clin. 1995, 13: 7-18.
Ellis NA, Groden J, Ye TZ, Straughen J, Lennon DJ, Ciocci S, Proytcheva M, German J: The Bloom's syndrome gene product is homologous to RecQ helicases. Cell. 1995, 83: 655-666.
Turaga RV, Massip L, Chavez A, Johnson FB, Lebel M: Werner syndrome protein prevents DNA breaks upon chromatin structure alteration. Aging Cell. 2007, 6: 471-481.
Mao FJ, Sidorova JM, Lauper JM, Emond MJ, Monnat RJ: The human WRN and BLM RecQ helicases differentially regulate cell proliferation and survival after chemotherapeutic DNA damage. Cancer Res. 2010, 70: 6548-6555.
Goto M, Miller RW, Ishikawa Y, Sugano H: Excess of rare cancers in Werner syndrome (adult progeria). Cancer Epidemiol Biomarkers Prev. 1996, 5: 239-246.
Ishikawa Y, Miller RW, Machinami R, Sugano H, Goto M: Atypical osteosarcomas in Werner Syndrome (adult progeria). Jpn J Cancer Res. 2000, 91: 1345-1349.
Bachrati CZ, Hickson ID: RecQ helicases: suppressors of tumorigenesis and premature aging. Biochem J. 2003, 374: 577-606.
German J: Bloom syndrome: a mendelian prototype of somatic mutational disease. Medicine (Baltimore). 1993, 72: 393-406.
Chester N, Kuo F, Kozak C, O'Hara CD, Leder P: Stage-specific apoptosis, developmental delay, and embryonic lethality in mice homozygous for a targeted disruption in the murine Bloom's syndrome gene. Genes Dev. 1998, 12: 3382-3393.
Luo G, Santoro IM, McDaniel LD, Nishijima I, Mills M, Youssoufian H, Vogel H, Schultz RA, Bradley A: Cancer predisposition caused by elevated mitotic recombination in Bloom mice. Nat Genet. 2000, 26: 424-429.
Goss KH, Risinger MA, Kordich JJ, Sanz MM, Straughen JE, Slovek LE, Capobianco AJ, German J, Boivin GP, Groden J: Enhanced tumor formation in mice heterozygous for Blm mutation. Science. 2002, 297: 2051-2053.
Lebel M, Cardiff RD, Leder P: Tumorigenic effect of nonfunctional p53 or p21 in mice mutant in the Werner syndrome helicase. Cancer Res. 2001, 61: 1816-1819.
Chang S, Multani AS, Cabrera NG, Naylor ML, Laud P, Lombard D, Pathak S, Guarente L, DePinho RA: Essential role of limiting telomeres in the pathogenesis of Werner syndrome. Nat Genet. 2004, 36: 877-882.
Roodman GD, Windle JJ: Paget disease of bone. J Clin Invest. 2005, 115: 200-208.
Eekhoff ME, van der Klift M, Kroon HM, Cooper C, Hofman A, Pols HA, Papapoulos SE: Paget's disease of bone in The Netherlands: a population-based radiological and biochemical survey–the Rotterdam Study. J Bone Miner Res. 2004, 19: 566-570.
Hocking L, Slee F, Haslam SI, Cundy T, Nicholson G, van Hul W, Ralston SH: Familial Paget's disease of bone: patterns of inheritance and frequency of linkage to chromosome 18q. Bone. 2000, 26: 577-580.
Selby PL, Davie MW, Ralston SH, Stone MD: Guidelines on the management of Paget's disease of bone. Bone. 2002, 31: 366-373.
Laurin N, Brown JP, Lemainque A, Duchesne A, Huot D, Lacourciere Y, Drapeau G, Verreault J, Raymond V, Morissette J: Paget disease of bone: mapping of two loci at 5q35-qter and 5q31. Am J Hum Genet. 2001, 69: 528-543.
Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T, Mizushima N, Iwata J, Ezaki J, Murata S: Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell. 2007, 131: 1149-1163.
Mathew R, Karp CM, Beaudoin B, Vuong N, Chen G, Chen HY, Bray K, Reddy A, Bhanot G, Gelinas C: Autophagy suppresses tumorigenesis through elimination of p62. Cell. 2009, 137: 1062-1075.
Hansen MF, Seton M, Merchant A: Osteosarcoma in Paget's disease of bone. J Bone Miner Res. 2006, 21 (Suppl 2): P58-P63.
Mangham DC, Davie MW, Grimer RJ: Sarcoma arising in Paget's disease of bone: declining incidence and increasing age at presentation. Bone. 2009, 44: 431-436.
Rousiere M, Michou L, Cornelis F, Orcel P: Paget's disease of bone. Best Pract Res Clin Rheumatol. 2003, 17: 1019-1041.
Wermers RA, Tiegs RD, Atkinson EJ, Achenbach SJ, Melton LJ: Morbidity and mortality associated with Paget's disease of bone: a population-based study. J Bone Miner Res. 2008, 23: 819-825.
Yochum TR: Paget's sarcoma of bone. Radiologe. 1984, 24: 428-433.
Longhi A, Errani C, Gonzales-Arabio D, Ferrari C, Mercuri M: Osteosarcoma in patients older than 65 years. J Clin Oncol. 2008, 26: 5368-5373.
Huvos AG, Butler A, Bretsky SS: Osteogenic sarcoma associated with Paget's disease of bone. A clinicopathologic study of 65 patients. Cancer. 1983, 52: 1489-1495.
Hiruma Y, Kurihara N, Subler MA, Zhou H, Boykin CS, Zhang H, Ishizuka S, Dempster DW, Roodman GD, Windle JJ: A SQSTM1/p62 mutation linked to Paget's disease increases the osteoclastogenic potential of the bone microenvironment. Hum Mol Genet. 2008, 17: 3708-3719.
Kurihara N, Hiruma Y, Zhou H, Subler MA, Dempster DW, Singer FR, Reddy SV, Gruber HE, Windle JJ, Roodman GD: Mutation of the sequestosome 1 (p62) gene increases osteoclastogenesis but does not induce Paget disease. J Clin Invest. 2007, 117: 133-142.
Bates S, Phillips AC, Clark PA, Stott F, Peters G, Ludwig RL, Vousden KH: p14ARF links the tumour suppressors RB and p53. Nature. 1998, 395: 124-125.
Yuan XW, Zhu XF, Huang XF, Sheng PY, He AS, Yang ZB, Deng R, Feng GK, Liao WM: P14ARF sensitizes human osteosarcoma cells to cisplatin-induced apoptosis in a p53-independent manner. Cancer Biol Ther. 2007, 6: 1074-1080.
Tsuchiya T, Sekine K, Hinohara S, Namiki T, Nobori T, Kaneko Y: Analysis of the p16INK4, p14ARF, p15, TP53, and MDM2 genes and their prognostic implications in osteosarcoma and Ewing sarcoma. Cancer Genet Cytogenet. 2000, 120: 91-98.
Benassi MS, Molendini L, Gamberi G, Magagnoli G, Ragazzini P, Gobbi GA, Sangiorgi L, Pazzaglia L, Asp J, Brantsing C, Picci P: Involvement of INK4A gene products in the pathogenesis and development of human osteosarcoma. Cancer. 2001, 92: 3062-3067.
Oh JH, Kim HS, Kim HH, Kim WH, Lee SH: Aberrant methylation of p14ARF gene correlates with poor survival in osteosarcoma. Clin Orthop Relat Res. 2006, 442: 216-222.
Krimpenfort P, Ijpenberg A, Song JY, van der Valk M, Nawijn M, Zevenhoven J, Berns A: p15Ink4b is a critical tumour suppressor in the absence of p16Ink4a. Nature. 2007, 448: 943-946.
Miller CW, Aslo A, Campbell MJ, Kawamata N, Lampkin BC, Koeffler HP: Alterations of the p15, p16, and p18 genes in osteosarcoma. Cancer Genet Cytogenet. 1996, 86: 136-142.
Nielsen GP, Burns KL, Rosenberg AE, Louis DN: CDKN2A gene deletions and loss of p16 expression occur in osteosarcomas that lack RB alterations. Am J Pathol. 1998, 153: 159-163.
Maitra A, Roberts H, Weinberg AG, Geradts J: Loss of p16(INK4a) expression correlates with decreased survival in pediatric osteosarcomas. Int J Cancer. 2001, 95: 34-38.
Shimizu T, Ishikawa T, Sugihara E, Kuninaka S, Miyamoto T, Mabuchi Y, Matsuzaki Y, Tsunoda T, Miya F, Morioka H: c-MYC overexpression with loss of Ink4a/Arf transforms bone marrow stromal cells into osteosarcoma accompanied by loss of adipogenesis. Oncogene. 2010, 29: 5687-5699.
Sharpless NE, Bardeesy N, Lee KH, Carrasco D, Castrillon DH, Aguirre AJ, Wu EA, Horner JW, DePinho RA: Loss of p16Ink4a with retention of p19Arf predisposes mice to tumorigenesis. Nature. 2001, 413: 86-91.
Agiostratidou G, Derventzi A, Gonos ES: Over-expression of CDKIs p15INK4b, p16INK4a and p21CIP1/WAF1 genes mediate growth arrest in human osteosarcoma cell lines. In Vivo. 2001, 15: 443-446.
Kawaguchi K, Oda Y, Sakamoto A, Saito T, Tamiya S, Iwamoto Y, Tsuneyoshi M: Molecular analysis of p53, MDM2, and H-ras genes in osteosarcoma and malignant fibrous histiocytoma of bone in patients older than 40 years. Mod Pathol. 2002, 15: 878-888.
Liao WM, Zhang CL, Li FB, Zeng BF, Zeng YX: p21WAF1/CIP1 gene DNA sequencing and its expression in human osteosarcoma. Chin Med J (Engl). 2004, 117: 936-940.
Thomas DM, Johnson SA, Sims NA, Trivett MK, Slavin JL, Rubin BP, Waring P, McArthur GA, Walkley CR, Holloway AJ: Terminal osteoblast differentiation, mediated by runx2 and p27KIP1, is disrupted in osteosarcoma. J Cell Biol. 2004, 167: 925-934.
Deng C, Zhang P, Harper JW, Elledge SJ, Leder P: Mice lacking p21CIP1/WAF1 undergo normal development, but are defective in G1 checkpoint control. Cell. 1995, 82: 675-684.
Martin-Caballero J, Flores JM, Garcia-Palencia P, Serrano M: Tumor susceptibility of p21(Waf1/Cip1)-deficient mice. Cancer Res. 2001, 61: 6234-6238.
Schon A, Michiels L, Janowski M, Merregaert J, Erfle V: Expression of protooncogenes in murine osteosarcomas. Int J Cancer. 1986, 38: 67-74.
Gamberi G, Benassi MS, Bohling T, Ragazzini P, Molendini L, Sollazzo MR, Pompetti F, Merli M, Magagnoli G, Balladelli A, Picci P: C-myc and c-fos in human osteosarcoma: prognostic value of mRNA and protein expression. Oncology. 1998, 55: 556-563.
Wu JX, Carpenter PM, Gresens C, Keh R, Niman H, Morris JW, Mercola D: The proto-oncogene c-fos is over-expressed in the majority of human osteosarcomas. Oncogene. 1990, 5: 989-1000.
Sunters A, McCluskey J, Grigoriadis AE: Control of cell cycle gene expression in bone development and during c-Fos-induced osteosarcoma formation. Dev Genet. 1998, 22: 386-397.
Wang ZQ, Liang J, Schellander K, Wagner EF, Grigoriadis AE: c-fos-induced osteosarcoma formation in transgenic mice: cooperativity with c-jun and the role of endogenous c-fos. Cancer Res. 1995, 55: 6244-6251.
Bialek P, Kern B, Yang X, Schrock M, Sosic D, Hong N, Wu H, Yu K, Ornitz DM, Olson EN: A twist code determines the onset of osteoblast differentiation. Dev Cell. 2004, 6: 423-435.
Maestro R, Dei Tos AP, Hamamori Y, Krasnokutsky S, Sartorelli V, Kedes L, Doglioni C, Beach DH, Hannon GJ: Twist is a potential oncogene that inhibits apoptosis. Genes Dev. 1999, 13: 2207-2217.
Entz-Werle N, Lavaux T, Metzger N, Stoetzel C, Lasthaus C, Marec P, Kalifa C, Brugieres L, Pacquement H, Schmitt C: Involvement of MET/TWIST/APC combination or the potential role of ossification factors in pediatric high-grade osteosarcoma oncogenesis. Neoplasia. 2007, 9: 678-688.
Entz-Werle N, Stoetzel C, Berard-Marec P, Kalifa C, Brugiere L, Pacquement H, Schmitt C, Tabone MD, Gentet JC, Quillet R: Frequent genomic abnormalities at TWIST in human pediatric osteosarcomas. Int J Cancer. 2005, 117: 349-355.
Entz-Werle N, Choquet P, Neuville A, Kuchler-Bopp S, Clauss F, Danse JM, Simo-Noumbissie P, Guerin E, Gaub MP, Freund JN: Targeted apc;twist double-mutant mice: a new model of spontaneous osteosarcoma that mimics the human disease. Transl Oncol. 2010, 3: 344-353.
Bergmann C, Senderek J, Anhuf D, Thiel CT, Ekici AB, Poblete-Gutierrez P, van Steensel M, Seelow D, Nurnberg G, Schild HH: Mutations in the gene encoding the Wnt-signaling component R-spondin 4 (RSPO4) cause autosomal recessive anonychia. Am J Hum Genet. 2006, 79: 1105-1109.
Blaydon DC, Ishii Y, O'Toole EA, Unsworth HC, Teh MT, Ruschendorf F, Sinclair C, Hopsu-Havu VK, Tidman N, Moss C: The gene encoding R-spondin 4 (RSPO4), a secreted protein implicated in Wnt signaling, is mutated in inherited anonychia. Nat Genet. 2006, 38: 1245-1247.
Chen K, Fallen S, Abaan HO, Hayran M, Gonzalez C, Wodajo F, MacDonald T, Toretsky JA, Uren A: Wnt10b induces chemotaxis of osteosarcoma and correlates with reduced survival. Pediatr Blood Cancer. 2008, 51: 349-355.
Cleton-Jansen AM, Anninga JK, Briaire-de Bruijn IH, Romeo S, Oosting J, Egeler RM, Gelderblom H, Taminiau AH, Hogendoorn PC: Profiling of high-grade central osteosarcoma and its putative progenitor cells identifies tumourigenic pathways. Br J Cancer. 2009, 101: 1909-1918.
Matushansky I, Hernando E, Socci ND, Mills JE, Matos TA, Edgar MA, Singer S, Maki RG, Cordon-Cardo C: Derivation of sarcomas from mesenchymal stem cells via inactivation of the Wnt pathway. J Clin Invest. 2007, 117: 3248-3257.
Kuroki T, Trapasso F, Shiraishi T, Alder H, Mimori K, Mori M, Croce CM: Genetic alterations of the tumor suppressor gene WWOX in esophageal squamous cell carcinoma. Cancer Res. 2002, 62: 2258-2260.
Ried K, Finnis M, Hobson L, Mangelsdorf M, Dayan S, Nancarrow JK, Woollatt E, Kremmidiotis G, Gardner A, Venter D: Common chromosomal fragile site FRA16D sequence: identification of the FOR gene spanning FRA16D and homozygous deletions and translocation breakpoints in cancer cells. Hum Mol Genet. 2000, 9: 1651-1663.
Paige AJ, Taylor KJ, Taylor C, Hillier SG, Farrington S, Scott D, Porteous DJ, Smyth JF, Gabra H, Watson JE: WWOX: a candidate tumor suppressor gene involved in multiple tumor types. Proc Natl Acad Sci U S A. 2001, 98: 11417-11422.
Kurek KC, Del Mare S, Salah Z, Abdeen S, Sadiq H, Lee SH, Gaudio E, Zanesi N, Jones KB, DeYoung B: Frequent attenuation of the WWOX tumor suppressor in osteosarcoma is associated with increased tumorigenicity and aberrant RUNX2 expression. Cancer Res. 2010, 70: 5577-5586.
Aqeilan RI, Trapasso F, Hussain S, Costinean S, Marshall D, Pekarsky Y, Hagan JP, Zanesi N, Kaou M, Stein GS: Targeted deletion of Wwox reveals a tumor suppressor function. Proc Natl Acad Sci U S A. 2007, 104: 3949-3954.
Candeliere GA, Glorieux FH, Prud'homme J, St-Arnaud R: Increased expression of the c-fos proto-oncogene in bone from patients with fibrous dysplasia. N Engl J Med. 1995, 332: 1546-1551.
Ruggieri P, Sim FH, Bond JR, Unni KK: Malignancies in fibrous dysplasia. Cancer. 1994, 73: 1411-1424.
Ladanyi M, Park CK, Lewis R, Jhanwar SC, Healey JH, Huvos AG: Sporadic amplification of the MYC gene in human osteosarcomas. Diagn Mol Pathol. 1993, 2: 163-167.
Ueda T, Healey JH, Huvos AG, Ladanyi M: Amplification of the MYC Gene in Osteosarcoma Secondary to Paget's Disease of Bone. Sarcoma. 1997, 1: 131-134.
Scionti I, Michelacci F, Pasello M, Hattinger CM, Alberghini M, Manara MC, Bacci G, Ferrari S, Scotlandi K, Picci P, Serra M: Clinical impact of the methotrexate resistance-associated genes C-MYC and dihydrofolate reductase (DHFR) in high-grade osteosarcoma. Ann Oncol. 2008, 19: 1500-1508.
Felsher DW, Bishop JM: Reversible tumorigenesis by MYC in hematopoietic lineages. Mol Cell. 1999, 4: 199-207.
Jain M, Arvanitis C, Chu K, Dewey W, Leonhardt E, Trinh M, Sundberg CD, Bishop JM, Felsher DW: Sustained loss of a neoplastic phenotype by brief inactivation of MYC. Science. 2002, 297: 102-104.
Wu CH, van Riggelen J, Yetil A, Fan AC, Bachireddy P, Felsher DW: Cellular senescence is an important mechanism of tumor regression upon c-Myc inactivation. Proc Natl Acad Sci U S A. 2007, 104: 13028-13033.
Ahuja D, Saenz-Robles MT, Pipas JM: SV40 large T antigen targets multiple cellular pathways to elicit cellular transformation. Oncogene. 2005, 24: 7729-7745.
Chen W, Possemato R, Campbell KT, Plattner CA, Pallas DC, Hahn WC: Identification of specific PP2A complexes involved in human cell transformation. Cancer Cell. 2004, 5: 127-136.
Mendoza SM, Konishi T, Miller CW: Integration of SV40 in human osteosarcoma DNA. Oncogene. 1998, 17: 2457-2462.
Knowles BB, McCarrick J, Fox N, Solter D, Damjanov I: Osteosarcomas in transgenic mice expressing an alpha-amylase-SV40 T-antigen hybrid gene. Am J Pathol. 1990, 137: 259-262.
Wilkie TM, Schmidt RA, Baetscher M, Messing A: Smooth muscle and bone neoplasms in transgenic mice expressing SV40 T antigen. Oncogene. 1994, 9: 2889-2895.
Molyneux SD, Di Grappa MA, Beristain AG, McKee TD, Wai DH, Paderova J, Kashyap M, Hu P, Maiuri T, Narala SR: Prkar1a is an osteosarcoma tumor suppressor that defines a molecular subclass in mice. J Clin Invest. 2010, 120: 3310-3325.
Staller P, Peukert K, Kiermaier A, Seoane J, Lukas J, Karsunky H, Moroy T, Bartek J, Massague J, Hanel F, Eilers M: Repression of p15INK4b expression by Myc through association with Miz-1. Nat Cell Biol. 2001, 3: 392-399.
Mohseny AB, Tieken C, van der Velden PA, Szuhai K, de Andrea C, Hogendoorn PC, Cleton-Jansen AM: Small deletions but not methylation underlie CDKN2A/p16 loss of expression in conventional osteosarcoma. Genes Chromosomes Cancer. 2010, 49: 1095-1103.
Ek ET, Dass CR, Choong PF: Commonly used mouse models of osteosarcoma. Crit Rev Oncol Hematol. 2006, 60: 1-8.
Fan TM: Animal models of osteosarcoma. Expert Rev Anticancer Ther. 2010, 10: 1327-1338.
Fletcher JA, Gebhardt MC, Kozakewich HP: Cytogenetic aberrations in osteosarcomas. Nonrandom deletions, rings, and double-minute chromosomes. Cancer Genet Cytogenet. 1994, 77: 81-88.
Fogh J, Fogh JM, Orfeo T: One hundred and twenty-seven cultured human tumor cell lines producing tumors in nude mice. J Natl Cancer Inst. 1977, 59: 221-226.
Lind M, Eriksen EF, Bunger C: Bone morphogenetic protein-2 but not bone morphogenetic protein-4 and −6 stimulates chemotactic migration of human osteoblasts, human marrow osteoblasts, and U2-OS cells. Bone. 1996, 18: 53-57.
Baker DE, Harrison NJ, Maltby E, Smith K, Moore HD, Shaw PJ, Heath PR, Holden H, Andrews PW: Adaptation to culture of human embryonic stem cells and oncogenesis in vivo. Nat Biotechnol. 2007, 25: 207-215.
Daniel VC, Marchionni L, Hierman JS, Rhodes JT, Devereux WL, Rudin CM, Yung R, Parmigiani G, Dorsch M, Peacock CD, Watkins DN: A primary xenograft model of small-cell lung cancer reveals irreversible changes in gene expression imposed by culture in vitro. Cancer Res. 2009, 69: 3364-3373.
Imreh MP, Gertow K, Cedervall J, Unger C, Holmberg K, Szoke K, Csoregh L, Fried G, Dilber S, Blennow E, Ahrlund-Richter L: In vitro culture conditions favoring selection of chromosomal abnormalities in human ES cells. J Cell Biochem. 2006, 99: 508-516.
Fernando A, Glaysher S, Conroy M, Pekalski M, Smith J, Knight LA, Di Nicolantonio F, Cree IA: Effect of culture conditions on the chemosensitivity of ovarian cancer cell lines. Anticancer Drugs. 2006, 17: 913-919.
Lisle JW, Choi JY, Horton JA, Allen MJ, Damron TA: Metastatic osteosarcoma gene expression differs in vitro and in vivo. Clin Orthop Relat Res. 2008, 466: 2071-2080.
Esquenet M, Swinnen JV, Heyns W, Verhoeven G: LNCaP prostatic adenocarcinoma cells derived from low and high passage numbers display divergent responses not only to androgens but also to retinoids. J Steroid Biochem Mol Biol. 1997, 62: 391-399.
Wenger SL, Senft JR, Sargent LM, Bamezai R, Bairwa N, Grant SG: Comparison of established cell lines at different passages by karyotype and comparative genomic hybridization. Biosci Rep. 2004, 24: 631-639.
Frese KK, Tuveson DA: Maximizing mouse cancer models. Nat Rev Cancer. 2007, 7: 645-658.
Sahin E, Depinho RA: Linking functional decline of telomeres, mitochondria and stem cells during ageing. Nature. 2010, 464: 520-528.
Bouvier J, Cheng JG: Recombineering-based procedure for creating Cre/loxP conditional knockouts in the mouse. Curr Protoc Mol Biol. 2009, Chapter 23: Unit 23 13.
Sztan M, Papai Z, Szendroi M, Looij M, Olah E: Allelic Losses from Chromosome 17 in Human Osteosarcomas. Pathol Oncol Res. 1997, 3: 115-120.
Wunder JS, Czitrom AA, Kandel R, Andrulis IL: Analysis of alterations in the retinoblastoma gene and tumor grade in bone and soft-tissue sarcomas. J Natl Cancer Inst. 1991, 83: 194-200.
Yamaguchi T, Toguchida J, Yamamuro T, Kotoura Y, Takada N, Kawaguchi N, Kaneko Y, Nakamura Y, Sasaki MS, Ishizaki K: Allelotype analysis in osteosarcomas: frequent allele loss on 3q, 13q, 17p, and 18q. Cancer Res. 1992, 52: 2419-2423.
Fero ML, Rivkin M, Tasch M, Porter P, Carow CE, Firpo E, Polyak K, Tsai LH, Broudy V, Perlmutter RM: A syndrome of multiorgan hyperplasia with features of gigantism, tumorigenesis, and female sterility in p27(Kip1)-deficient mice. Cell. 1996, 85: 733-744.
Gupta S, Schoer RA, Egan JE, Hannon GJ, Mittal V: Inducible, reversible, and stable RNA interference in mammalian cells. Proc Natl Acad Sci U S A. 2004, 101: 1927-1932.
Premsrirut PK, Dow LE, Kim SY, Camiolo M, Malone CD, Miething C, Scuoppo C, Zuber J, Dickins RA, Kogan SC: A rapid and scalable system for studying gene function in mice using conditional RNA interference. Cell. 2011, 145: 145-158.
Takiguchi M, James C, Josefsson EC, Carmichael CL, Premsrirut PK, Lowe SW, Hamilton JR, Huang DC, Kile BT, Dickins RA: Transgenic, inducible RNAi in megakaryocytes and platelets in mice. J Thromb Haemost. 2010, 8: 2751-2756.
Conget PA, Minguell JJ: Phenotypical and functional properties of human bone marrow mesenchymal progenitor cells. J Cell Physiol. 1999, 181: 67-73.
Deng ZL, Sharff KA, Tang N, Song WX, Luo J, Luo X, Chen J, Bennett E, Reid R, Manning D: Regulation of osteogenic differentiation during skeletal development. Front Biosci. 2008, 13: 2001-2021.
Pittenger MF, Mackay AM, Beck SC, Jaiswal RK, Douglas R, Mosca JD, Moorman MA, Simonetti DW, Craig S, Marshak DR: Multilineage potential of adult human mesenchymal stem cells. Science. 1999, 284: 143-147.
Haydon RC, Luu HH, He TC: Osteosarcoma and osteoblastic differentiation: a new perspective on oncogenesis. Clin Orthop Relat Res. 2007, 454: 237-246.
Tataria M, Quarto N, Longaker MT, Sylvester KG: Absence of the p53 tumor suppressor gene promotes osteogenesis in mesenchymal stem cells. J Pediatr Surg. 2006, 41: 624-632. discussion 624–632,
Komori T: Regulation of osteoblast differentiation by transcription factors. J Cell Biochem. 2006, 99: 1233-1239.
Li N, Yang R, Zhang W, Dorfman H, Rao P, Gorlick R: Genetically transforming human mesenchymal stem cells to sarcomas: changes in cellular phenotype and multilineage differentiation potential. Cancer. 2009, 115: 4795-4806.
Mohseny AB, Szuhai K, Romeo S, Buddingh EP, Briaire-de Bruijn I, de Jong D, van Pel M, Cleton-Jansen AM, Hogendoorn PC: Osteosarcoma originates from mesenchymal stem cells in consequence of aneuploidization and genomic loss of Cdkn2. J Pathol. 2009, 219: 294-305.
Tang N, Song WX, Luo J, Haydon RC, He TC: Osteosarcoma development and stem cell differentiation. Clin Orthop Relat Res. 2008, 466: 2114-2130.
Visvader JE: Cells of origin in cancer. Nature. 2011, 469: 314-322.
Choi J, Curtis SJ, Roy DM, Flesken-Nikitin A, Nikitin AY: Local mesenchymal stem/progenitor cells are a preferential target for initiation of adult soft tissue sarcomas associated with p53 and Rb deficiency. Am J Pathol. 2010, 177: 2645-2658.
Rubio R, Garcia-Castro J, Gutierrez-Aranda I, Paramio J, Santos M, Catalina P, Leone PE, Menendez P, Rodriguez R: Deficiency in p53 but not retinoblastoma induces the transformation of mesenchymal stem cells in vitro and initiates leiomyosarcoma in vivo. Cancer Res. 2010, 70: 4185-4194.
Lengner CJ, Steinman HA, Gagnon J, Smith TW, Henderson JE, Kream BE, Stein GS, Lian JB, Jones SN: Osteoblast differentiation and skeletal development are regulated by Mdm2-p53 signaling. J Cell Biol. 2006, 172: 909-921.
Hanahan D, Weinberg RA: The hallmarks of cancer. Cell. 2000, 100: 57-70.
Houghton PJ, Morton CL, Tucker C, Payne D, Favours E, Cole C, Gorlick R, Kolb EA, Zhang W, Lock R: The pediatric preclinical testing program: description of models and early testing results. Pediatr Blood Cancer. 2007, 49: 928-940.
Acknowledgements
This work is supported by an NHMRC Career Development Award (C.R.W) and NHMRC Project grants (C.R.W); a CRC for Cancer Therapeutics (CTx) graduate student award (A.J.M.N). Research was supported in part by the Victorian Government’s Operational Infrastructure Support Program (to SVI); C.R.W. is the Leukaemia Foundation Philip Desbrow Senior Research Fellow.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors would like to declare that there are no competing financial, professional or personal interests that might have influenced the performance or presentation of the work described in this manuscript.
Authors' contributions
AJN, AJM, and EKB acquired data and drafted the manuscript. CRW conceived, participated in the design, coordination and helped to draft the manuscript. All authors read and approved the final manuscript.
Alvin JM Ng, Anthony J Mutsaers contributed equally to this work.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Ng, A.J., Mutsaers, A.J., Baker, E.K. et al. Genetically engineered mouse models and human osteosarcoma. Clin Sarcoma Res 2, 19 (2012). https://doi.org/10.1186/2045-3329-2-19
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/2045-3329-2-19