
Abstract
Children diagnosed with the hereditary form of retinoblastoma (Rb), a rare eye cancer caused by a germline mutation in the RB1 tumor suppressor gene, have excellent survival, but face an increased risk of bone and soft tissue sarcomas. This predisposition to sarcomas has been attributed to genetic susceptibility due to inactivation of the RB1 gene as well as past radiotherapy for Rb. The majority of bone and soft tissue sarcomas among hereditary Rb survivors occur in the head, within the radiation field, but they also occur outside the radiation field. Sarcomas account for almost half of the second primary cancers in hereditary Rb survivors, but they are very rare following non-hereditary Rb. Sarcomas among hereditary Rb survivors arise at ages similar to the pattern of occurrence in the general population. There has been a trend over the past two decades to replace radiotherapy with chemotherapy and other focal therapies (laser or cryosurgery), and most recently, chemosurgery in order to reduce the incidence of sarcomas and other second cancers in Rb survivors. Given the excellent survival of most Rb patients treated in the past, it is important for survivors, their families and health care providers to be aware of the heightened risk for sarcomas in hereditary patients.
Similar content being viewed by others


Introduction
Children diagnosed with the hereditary form of retinoblastoma (Rb), a rare eye cancer caused by a germline mutation in the RB1 tumor suppressor gene, have excellent survival, but face an increased risk for the development of sarcomas, both soft tissue (STS) and bone. This predisposition to sarcomas in retinoblastoma survivors has been attributed to genetic susceptibility as well as past radiation treatment for Rb.
Retinoblastoma epidemiology
Retinoblastoma is a rare pediatric cancer of the eye with an autosomal dominant inheritance pattern. It is caused by mutations in the RB1 tumor suppressor gene, located on chromosome 13q14 with very high penetrance and expressivity[1]. Approximately 80%-90% of RB1 gene carriers develop ocular tumors. This gene encodes the cell cycle regulatory retinoblastoma gene protein (pRb), controls cellular differentiation during both embryogenesis and in adult tissues, regulates apoptotic cell death, maintains cell cycle arrest and preserves chromosome stability[2].
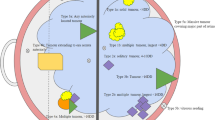
Retinoblastoma occurs in two forms: hereditary (30-40%) and non-hereditary (60-70%). Hereditary retinoblastoma is caused by a germline mutation in one allele of the RB1 gene and an acquired somatic mutation in the other allele, whereas the non-hereditary form is caused by somatic mutations in both alleles. The hereditary form is characterized by disease in both eyes (bilateral Rb) and is typically diagnosed before 12 months of age, whereas, the non-hereditary form affects one eye (unilateral Rb) and is diagnosed between 2–5 years of age. About 10-15% of patients with unilateral Rb, however, carry a germline mutation and are considered hereditary. This difference in diagnosis age led Knudson to develop the two-hit theory[3], in which only one additional mutation is needed for hereditary Rb and two hits or somatic mutations are needed for non-hereditary Rb[4]. The age-adjusted annual incidence rate of retinoblastoma is 3.1 per 107 with a 5-year relative survival of 97.5% in the U.S.[5]. Treatment for Rb has historically consisted primarily of radiotherapy (both external beam and radioactive plaques), enucleation, chemotherapy, focal therapies such as laser or cryotherapy, or a combination of these modalities.
Subsequent malignancies after retinoblastoma
Long-term survivors of hereditary retinoblastoma are at an increased 20-fold risk of developing and dying from a subsequent non-ocular cancer, primarily bone and soft tissue sarcomas, melanoma and brain tumors[6, 7]. Survivors of non-hereditary Rb are at much lower risk of a subsequent primary cancer, similar to the risk in the general population[8–10]. The risk for sarcomas in hereditary patients has been attributed to genetic susceptibility and past treatment with radiation[8, 11, 12]. In addition to radiotherapy, chemotherapy, specifically alkylating agents, has been associated with the risk of bone cancer after Rb[6, 13, 14], but less so for soft tissue sarcomas[15].
Bone sarcomas after retinoblastoma
Patterns of risk
Bone sarcomas are one of the most common second primary cancers occurring after hereditary retinoblastoma accounting for 25%-30% of all second primary cancers[6–8, 16, 17]. Bone sarcomas are typically diagnosed in Rb survivors between 10 and 20 years of age, similar to the incidence pattern in the general population[5]. In these studies, the majority of bone sarcomas occurred within the radiation field in the head region, but up to 40% was diagnosed outside the treatment field, primarily in the lower legs[8, 11, 17].
Table1 presents risks for bone sarcomas from epidemiologic cohort studies including at least 100 hereditary Rb survivors. The standardized incidence and mortality rates for bone sarcomas are increased several hundred-fold compared to population rates, due to the rarity of these tumors in the general population. A much lower risk for bone sarcomas was observed in the one cohort study that included non-irradiated survivors and began follow-up 25 years after Rb diagnosis[10]. It has been estimated that the cumulative incidence of bone sarcoma following retinoblastoma is 7% at 20 years[13, 18]. Osteosarcoma is the most common type of bone sarcoma reported after Rb, but both chondrosarcoma and Ewing sarcoma have been reported as well[19, 20], although risk estimates are not available for these other two types.
Treatment for Rb and risk of bone sarcomas
Both high-dose radiation and increasing cumulative dose of chemotherapy, mainly alkylating agents (cyclophosphamide and triethylenemelamine or TEM), have been linked to the occurrence of bone sarcomas following hereditary Rb[13, 14]. Higher risks have been noted for the combination of radiotherapy and chemotherapy compared to either treatment alone[6–8, 13, 14]. An earlier study of British Rb patients provided some evidence that cyclophosphamide may increase the effect of radiotherapy on the risk of bone sarcoma[8].
In a case–control study of bone and soft tissue sarcomas after hereditary Rb, risk increased with increasing dose up to 10.7-fold at doses greater than 60 Gy[11]. The mean dose to the head among cases was 32.8 Gy, whereas the lower limbs had received virtually no radiation (<0.1 Gy). In an update of that study, the location of 75 bone sarcomas was skull and face (61%), lower limbs (29%), trunk (7.6%), and unknown location (3.8%)[7].
Based on a series of 155 osteosarcomas following hereditary Rb identified from the literature and one institute, investigators reported that the mean age of onset was related to the osteosarcoma location[21]. Sarcomas occurring in the radiation field were diagnosed one year earlier compared to those diagnosed outside the field (mean age = 12.2 years [range 3–35] vs. mean age = 13.4 years [range 4–22]. This age difference suggested to the investigators that different biologic mechanisms may be associated with the development of bone sarcomas depending upon the location in the body.
Studies of other pediatric malignancies have also reported an increased risk for second osteosarcomas following radiation and chemotherapy treatment for a first cancer (for a detailed review of radiation-related sarcomas, see Berrington de Gonzalez et al. in this issue).
Soft Tissue Sarcomas
Patterns of risk
Soft tissue sarcomas (STS) are also one of the most common subsequent cancers following hereditary Rb accounting for 12% up to 32% of all second cancers[6, 7, 16]. In one large cohort study, an increased risk for STS was first observed within 10 years of Rb diagnosis and continued through adult life up to 50 years after Rb, with specific subtypes occurring at similar ages as in the general population[22, 23]. Fifty years after radiation treatment for hereditary Rb, the cumulative risk of developing a STS was 13.1%, and the cumulative incidence for a STS inside the radiation field was higher than outside the field (8.9% vs. 5.1%)[22]. Table2 presents the incidence and mortality due to STS after Rb in cohort studies of at least 100 hereditary Rb survivors.
Subtype heterogeneity
STS diagnosed in Rb patients comprise a heterogeneous group of tumors of fat, cartilage and muscle; however, only one study has evaluated the risk of STS by histology after hereditary Rb[22]. Leiomyosarcoma (LMS) constituted the most common type of STS after Rb, with the majority diagnosed 30 and more years after Rb. This is consistent with LMS being one of the most common STS in the general population[23]. Although many LMS occurred in the head and neck region, the majority of LMS in females were diagnosed in the uterus[24]. Loss of heterozygosity in RB1 has been reported in uterine LMS[25], which may confer an increased susceptibility to this tumor in this population. LMS of other pelvic sites have also been reported after Rb[26], and there have been several case reports of LMS diagnosed in the bladder[27, 28].
Very high risks have also been observed for fibrosarcomas, rhabdomyosarcomas and pleomorphic sarcomas within the first 10 years after Rb[22, 29]. These histologic types comprised the majority of STS that were diagnosed in or near the field of radiation, in contrast to LMS, which were more likely to occur outside the radiation field (Table3). Only 10% of rhabdomyosarcomas arise in the soft tissue of the head, neck or face in the general population, whereas all of the rhabdomyosarcomas arose in the head following radiation for Rb[22].
An increased risk for liposarcomas that began 10 years after diagnosis of hereditary Rb was observed in the study by Kleinerman et al.[22]. Lipomas, a benign tumor of fat tissue, have also been reported to be increased in that cohort, and the investigators noted a possible association between lipomas and subsequent risk of a soft tissue sarcoma[30]. Following this observation, a RB1 mutation was identified in lipomas from hereditary Rb patients[31, 32].
It has been suggested that females may be at higher risk of STS after hereditary Rb[9], but studies of Rb survivors have not consistently reported a higher risk among females. Males have a higher rate of Rb in the general population and all liposarcomas and lipomas occurred in males in the cohort in which they were evaluated[22, 30].
Treatment for Rb and risk of STS
Although both radiotherapy and chemotherapy for hereditary Rb have been associated with an increased risk for STS, the evidence is more consistent for radiotherapy. (For a detailed review of radiation-related sarcoma, see Berrington de Gonzalez et al. in this issue). Wong et al. demonstrated a radiation dose–response for STS whereby risk increased with dose up to a significant 11-fold increased risk at ≥60 Gy[11]. The risk for STS was not associated with increasing alkylating agent score in the same cohort[22], whereas in another study of STS after all types of pediatric malignancies, including Rb, the risk for STS increased significantly with cumulative dose of alkylating agents, adjusted for radiation exposure[15]. Increased risks of STS have also been noted following surgery only for hereditary Rb[6, 10].
Molecular evidence for an association of sarcomas with RB1
In additional to the epidemiologic evidence of an excess risk for both bone and STS in hereditary Rb patients, structural alterations of the RB1 gene are well documented in primary bone sarcomas[33] and soft tissue sarcomas[34–36]. Most of the bone and soft tissue sarcomas diagnosed in hereditary Rb patients have complex karyotypes, including fibrosarcoma, LMS, pleomorphic sarcoma, liposarcoma and osteosarcoma that are all related to inherited defects in the RB pathway[37]. A comprehensive review by Burkhart and Sage of cellular mechanisms of tumor suppression by the retinoblastoma gene discusses the loss of RB1 function and cancer progression[2].
Conclusion
Hereditary Rb patients are at significant risk of developing a sarcoma due to past radiation treatment and genetic susceptibility. Sarcomas account for approximately 40% to 60% of second cancers in hereditary Rb survivors. There is convincing epidemiologic evidence linking past radiotherapy with sarcomas in hereditary patients. Risk of bone and STS begins within 10 years of treatment for hereditary Rb and continues throughout adulthood, most notably for STS.
Recognition of the increased risk for sarcomas associated with past radiotherapy has influenced the current treatment of retinoblastoma with a trend towards greater use of chemotherapy, focal therapies, and most recently, chemosurgery[38–40]. In addition, guidelines for imaging children for pre-treatment diagnostic evaluation of Rb without the use of ionizing radiation have been recommended to reduce the risk of second cancers in Rb patients[41]. However, the risk for bone sarcomas and STS remains, reflecting the genetic predisposition to these sarcomas due to loss of heterozygosity in the RB1 gene. Patients who were treated in 1960s and 1970s with radiotherapy are still at risk in their adult years for the development of STS. Given the excellent survival of most retinoblastoma patients, it is important for survivors, their families and health care providers to be aware of these risks, especially for hereditary patients[42]. There is on-going research to try to identify whether specific RB1 mutations or location of mutations predispose to sarcomas, which could lead to identification of those survivors at greatest risk[43]. The development of comprehensive guidelines for long-term follow-up that are specifically tailored for detection of sarcomas and other second primary cancers in retinoblastoma survivors are also needed, especially for those patients who received radiotherapy in the past.
Author contributions
RK and SS participated in the review of existing data, RK, SS and MT contributed to the interpretation of the data, and all participated in the draft of the manuscript. All authors read and approved the final manuscript.
Abbreviations
- Rb:
-
retinoblastoma
- STS:
-
soft tissue sarcoma
- LMS:
-
leiomyosarcoma.
References
Harbour JW: Molecular basis of low-penetrance retinoblastoma. Arch Ophthalmol. 2001, 119: 1699-1704.
Burkhart DL, Sage J: Cellular mechanisms of tumour suppression by the retinoblastoma gene. Nat Rev Cancer. 2008, 8: 671-682. 10.1038/nrc2399
Knudson AG: Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci U S A. 1971, 68: 820-823. 10.1073/pnas.68.4.820
Little MP, Kleinerman RA, Stiller CA, Li G: Kroll ME. 2011, Murphy MF: Analysis of retinoblastoma age incidence data using a fully stochastic cancer model. Int J Cancer,
SEER: Cancer Statistics Review. 2008, [http://seer.cancer.gov/csr/1975_2008], []
Marees T, Moll AC, Imhof SM, de Boer MR, Ringens PJ, van Leeuwen FE: Risk of second malignancies in survivors of retinoblastoma: more than 40 years of follow-up. J Natl Cancer Inst. 2008, 100: 1771-1779. 10.1093/jnci/djn394
Kleinerman RA, Tucker MA, Tarone RE, Abramson DH, Seddon JM, Stovall M, Li FP, Fraumeni JF: Risk of new cancers after radiotherapy in long-term survivors of retinoblastoma: an extended follow-up. J Clin Oncol. 2005, 23: 2272-2279. 10.1200/JCO.2005.05.054
Draper GJ, Sanders BM, Kingston JE: Second primary neoplasms in patients with retinoblastoma. Br J Cancer. 1986, 53: 661-671. 10.1038/bjc.1986.110
Eng C, Li FP, Abramson DH, Ellsworth RM, Wong FL, Goldman MB, Seddon J, Tarbell N, Boice JD: Mortality from second tumors among long-term survivors of retinoblastoma. J Natl Cancer Inst. 1993, 85: 1121-1128. 10.1093/jnci/85.14.1121
Fletcher O, Easton D, Anderson K, Gilham C, Jay M, Peto J: Lifetime risks of common cancers among retinoblastoma survivors. J Natl Cancer Inst. 2004, 96: 357-363. 10.1093/jnci/djh058
Wong FL, Boice JD, Abramson DH, Tarone RE, Kleinerman RA, Stovall M, Goldman MB, Seddon JM, Tarbell N, Fraumeni JF, Li FP: Cancer incidence after retinoblastoma. Radiation dose and sarcoma risk. JAMA. 1997, 278: 1262-1267.
Yu CL, Tucker MA, Abramson DH, Furukawa K, Seddon JM, Stovall M, Fraumeni JF, Kleinerman RA: Cause-specific mortality in long-term survivors of retinoblastoma. J Natl Cancer Inst. 2009, 101: 581-591. 10.1093/jnci/djp046
Hawkins MM, Wilson LM, Burton HS, Potok MH, Winter DL, Marsden HB, Stovall MA: Radiotherapy, alkylating agents, and risk of bone cancer after childhood cancer. J Natl Cancer Inst. 1996, 88: 270-278. 10.1093/jnci/88.5.270
Tucker MA, D'Angio GJ, Boice JD, Strong LC, Li FP, Stovall M, Stone BJ, Green DM, Lombardi F, Newton W: Bone sarcomas linked to radiotherapy and chemotherapy in children. N Engl J Med. 1987, 317: 588-593. 10.1056/NEJM198709033171002
Jenkinson HC, Winter DL, Marsden HB, Stovall MA, Stevens MC, Stiller CA, Hawkins MM: A study of soft tissue sarcomas after childhood cancer in Britain. Br J Cancer. 2007, 97: 695-699. 10.1038/sj.bjc.6603908
Reulen RC, Frobisher C, Winter DL, Kelly J, Lancashire ER, Stiller CA, Pritchard-Jones K, Jenkinson HC, Hawkins MM: Long-term risks of subsequent primary neoplasms among survivors of childhood cancer. JAMA. 2011, 305: 2311-2319. 10.1001/jama.2011.747
Woo KI, Harbour JW: Review of 676 second primary tumors in patients with retinoblastoma: association between age at onset and tumor type. Arch Ophthalmol. 2010, 128: 865-870. 10.1001/archophthalmol.2010.126
Kleinerman R, Yu CL, Little MP, Abramson DH, Seddon JH, Tucker MA: Variation of second cancer risk by family history of retinoblastoma among long-term survivors. Journal of Clin Oncol. 2012, 30: 950-957. 10.1200/JCO.2011.37.0239. in press,
Cope JU, Tsokos M, Miller RW: Ewing sarcoma and sinonasal neuroectodermal tumors as second malignant tumors after retinoblastoma and other neoplasms. Med Pediatr Oncol. 2001, 36: 290-294. 10.1002/1096-911X(20010201)36:2<290::AID-MPO1067>3.0.CO;2-5
Moll AC, Imhof SM, Schouten-Van Meeteren AY, Kuik DJ, Hofman P, Boers M: Second primary tumors in hereditary retinoblastoma: a register-based study, 1945–1997: is there an age effect on radiation-related risk?. Ophthalmology. 2001, 108: 1109-1114. 10.1016/S0161-6420(01)00562-0
Chauveinc L, Mosseri V, Quintana E, Desjardins L, Schlienger P, Doz F, Dutrillaux B: Osteosarcoma following retinoblastoma: age at onset and latency period. Ophthalmic Genet. 2001, 22: 77-88. 10.1076/opge.22.2.77.2228
Kleinerman RA, Tucker MA, Abramson DH, Seddon JM, Tarone RE, Fraumeni JF: Risk of soft tissue sarcomas by individual subtype in survivors of hereditary retinoblastoma. J Natl Cancer Inst. 2007, 99: 24-31. 10.1093/jnci/djk002
Toro JR, Travis LB, Wu HJ, Zhu K, Fletcher CD, Devesa SS: Incidence patterns of soft tissue sarcomas, regardless of primary site, in the surveillance, epidemiology and end results program, 1978–2001: An analysis of 26, 758 cases. Int J Cancer. 2006, 119: 2922-2930. 10.1002/ijc.22239
Francis JH, Kleinerman RA: Seddon JM. 2011, Abramson DH: Increased risk of secondary uterine leiomyosarcoma in hereditary retinoblastoma. Gynecol Oncol,
Dei Tos AP, Maestro R, Doglioni C, Piccinin S, Libera DD, Boiocchi M, Fletcher CD: Tumor suppressor genes and related molecules in leiomyosarcoma. Am J Pathol. 1996, 148: 1037-1045.
Venkatraman L, Goepel JR, Steele K, Dobbs SP, Lyness RW, McCluggage WG: Soft tissue, pelvic, and urinary bladder leiomyosarcoma as second neoplasm following hereditary retinoblastoma. J Clin Pathol. 2003, 56: 233-236. 10.1136/jcp.56.3.233
Bleoo SL, Godbout R, Rayner D, Tamimi Y, Moore RB: Leiomyosarcoma of the bladder in a retinoblastoma patient. Urol Int. 2003, 71: 118-121. 10.1159/000071109
Brucker B, Ernst L, Meadows A, Zderic S: A second leiomyosarcoma in the urinary bladder of a child with a history of retinoblastoma 12 years following partial cystectomy. Pediatr Blood Cancer. 2006, 46: 811-814. 10.1002/pbc.20506
Cebulla CM, Kleinerman RA, Alegret A, Kulak A, Dubovy SR, Hess DJ, Murray TG: Rapid Appearance of Rhabdomyosarcoma after Radiation and Chemotherapy for Retinoblastoma: A Clinicopathologic Correlation. Retin Cases Brief Rep. 2009, 3: 343-346. 10.1097/ICB.0b013e31817377a5
Li FP, Abramson DH, Tarone RE, Kleinerman RA, Fraumeni JF, Boice JD: Hereditary retinoblastoma, lipoma, and second primary cancers. J Natl Cancer Inst. 1997, 89: 83-84. 10.1093/jnci/89.1.83
Rieder H, Lohmann D, Poensgen B, Fritz B, Aslan M, Drohm D, Strombach Angersbach FJ, Rehder H: Loss of heterozygosity of the retinoblastoma (RB1) gene in lipomas from a retinoblastoma patient. J Natl Cancer Inst. 1998, 90: 324-326. 10.1093/jnci/90.4.324
Genuardi M, Klutz M, Devriendt K, Caruso D, Stirpe M, Lohmann DR: Multiple lipomas linked to an RB1 gene mutation in a large pedigree with low penetrance retinoblastoma. Eur J Hum Genet. 2001, 9: 690-694. 10.1038/sj.ejhg.5200694
Friend SH, Bernards R, Rogelj S, Weinberg RA, Rapaport JM, Albert DM, Dryja TP: A human DNA segment with properties of the gene that predisposes to retinoblastoma and osteosarcoma. Nature. 1986, 323: 643-646. 10.1038/323643a0
Stratton MR, Williams S, Fisher C, Ball A, Westbury G, Gusterson BA, Fletcher CD, Knight JC, Fung YK, Reeves BR: Structural alterations of the RB1 gene in human soft tissue tumours. Br J Cancer. 1989, 60: 202-205. 10.1038/bjc.1989.251
Friend SH, Horowitz JM, Gerber MR, Wang XF, Bogenmann E, Li FP, Weinberg RA: Deletions of a DNA sequence in retinoblastomas and mesenchymal tumors: organization of the sequence and its encoded protein. Proc Natl Acad Sci U S A. 1987, 84: 9059-9063. 10.1073/pnas.84.24.9059
Bookstein R, Lee WH: Molecular genetics of the retinoblastoma suppressor gene. Crit Rev Oncog. 1991, 2: 211-227.
Helman LJ, Meltzer P: Mechanisms of sarcoma development. Nat Rev Cancer. 2003, 3: 685-694. 10.1038/nrc1168
Gobin YP, Dunkel IJ, Marr BP, Brodie SE, Abramson DH: Intra-arterial chemotherapy for the management of retinoblastoma: four-year experience. Arch Ophthalmol. 2011, 129: 732-737. 10.1001/archophthalmol.2011.5
Turaka K, Shields CL, Meadows AT, Leahey A: Second malignant neoplasms following chemoreduction with carboplatin, etoposide, and vincristine in 245 patients with intraocular retinoblastoma. Pediatr Blood Cancer. 2012, 59: 121-125. 10.1002/pbc.23278
Abramson DH: Chemosurgery for retinoblastoma: what we know after 5 years. Arch Ophthalmol. 2011, 129: 1492-1494. 10.1001/archophthalmol.2011.354
de Graaf P, Goricke S, Rodjan F, Galluzzi P, Maeder P, Castelijns JA, Brisse HJ: Guidelines for imaging retinoblastoma: imaging principles and MRI standardization. Pediatr Radiol. 2012, 42: 2-14. 10.1007/s00247-011-2201-5
Meadows AT: Retinoblastoma survivors: sarcomas and surveillance. J Natl Cancer Inst. 2007, 99: 3-5. 10.1093/jnci/djk014
Dommering CJ, Marees T, van der Hout AH, Imhof SM, Meijers-Heijboer H, Ringens PJ, van Leeuwen FE, Moll AC: RB1 mutations and second primary malignancies after hereditary retinoblastoma. Fam Cancer. 2011, 11: 225-233.
MacCarthy A, Bayne AM, Draper GJ, Eatock EM, Kroll ME, Stiller CA, Vincent TJ, Hawkins MM, Jenkinson HC, Kingston JE: Non-ocular tumours following retinoblastoma in Great Britain 1951 to 2004. Br J Ophthalmol. 2009, 93: 1159-1162. 10.1136/bjo.2008.146035
Marees T, van Leeuwen FE, de Boer MR, Imhof SM, Ringens PJ, Moll AC: Cancer mortality in long-term survivors of retinoblastoma. Eur J Cancer. 2009, 45: 3245-3253. 10.1016/j.ejca.2009.05.011
Acquaviva A, Ciccolallo L, Rondelli R, Balistreri A, Ancarola R, Cozza R, Hadjistilianou D, Francesco SD, Toti P, Pastore G: Mortality from second tumour among long-term survivors of retinoblastoma: a retrospective analysis of the Italian retinoblastoma registry. Oncogene. 2006, 25: 5350-5357. 10.1038/sj.onc.1209786
Acknowledgement
This research was supported by the Intramural Research Program of the National Institutes of Health, National Cancer Institute.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Kleinerman, R.A., Schonfeld, S.J. & Tucker, M.A. Sarcomas in hereditary retinoblastoma. Clin Sarcoma Res 2, 15 (2012). https://doi.org/10.1186/2045-3329-2-15
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/2045-3329-2-15