
Abstract
Stress-related psychiatric disorders, such as unipolar depression and post-traumatic stress disorder (PTSD), occur more frequently in women than in men. Emerging research suggests that sex differences in receptors for the stress hormones, corticotropin releasing factor (CRF) and glucocorticoids, contribute to this disparity. For example, sex differences in CRF receptor binding in the amygdala of rats may predispose females to greater anxiety following stressful events. Additionally, sex differences in CRF receptor signaling and trafficking in the locus coeruleus arousal center combine to make females more sensitive to low levels of CRF, and less adaptable to high levels. These receptor differences in females could lead to hyperarousal, a dysregulated state associated with symptoms of depression and PTSD. Similar to the sex differences observed in CRF receptors, sex differences in glucocorticoid receptor (GR) function also appear to make females more susceptible to dysregulation after a stressful event. Following hypothalamic pituitary adrenal axis activation, GRs are critical to the negative feedback process that inhibits additional glucocorticoid release. Compared to males, female rats have fewer GRs and impaired GR translocation following chronic adolescent stress, effects linked to slower glucocorticoid negative feedback. Thus, under conditions of chronic stress, attenuated negative feedback in females would result in hypercortisolemia, an endocrine state thought to cause depression. Together, these studies suggest that sex differences in stress-related receptors shift females more easily into a dysregulated state of stress reactivity, linked to the development of mood and anxiety disorders. The implications of these receptor sex differences for the development of novel pharmacotherapies are also discussed.
Similar content being viewed by others


Introduction
Many mood and anxiety disorders are considered stress-related, because of the crucial role stress plays in their etiology and symptomology. For example, stress is associated with the onset and severity of unipolar depression, panic disorder, and generalized anxiety disorder [1–7]. In the case of post-traumatic stress disorder (PTSD), the disease is, by definition, precipitated by a stressful event [4, 8]. Additionally, early life stress can sensitize later responses to stress and is associated with the development of mood and anxiety disorders in adulthood [9–11].
A common feature shared by most stress-related psychiatric disorders is that they occur more frequently in women than in men. Women are, in fact, twice as likely as men to suffer from unipolar depression and several anxiety disorders, including PTSD [1, 12–15]. This disparity is often attributed to gender differences in psychological factors, including affect, cognitive style, and social role identification [16–23]. Some commonly presumed causes for the gender gap, such as differences in the types of trauma experienced, preexisting psychiatric disorders, and sex differences in reporting, are unlikely to play a significant role because the male–female disparity remains, even after these variables have been controlled for [4, 14, 24, 25]. Nonetheless, it is likely that some psychological factors contribute to the sex bias in stress-related disease. New research investigating molecular sex differences in stress response systems, on the other hand, indicates that biological factors also contribute to sex differences in the prevalence of stress-related psychiatric disorders. This review will discuss emerging data that has identified sex differences in receptors for the stress hormones, corticotropin releasing factor (CRF) and glucocorticoids. The reviewed studies suggest that receptor sex differences can cause stress responses in females to shift more easily into a dysregulated state and likely play an important role in the disparity seen between males and females in mood and anxiety rates.
Review
Endocrine and central effects of CRF
CRF is best known for its role in activating the hypothalamic pituitary adrenal (HPA) axis. This endocrine response is initiated during a stressful event when CRF released from neurons in the paraventricular nucleus of the hypothalamus stimulates pituitary corticotropes to secrete adrenocorticotropic hormone (ACTH) [26]. ACTH released into the bloodstream acts on the cortex of the adrenal glands causing them to release glucocorticoids (cortisol in humans and corticosterone in rodents). Glucocorticoids then feedback at the levels of the hypothalamus and pituitary to terminate the HPA axis activation [27]. Additionally, these hormones act on the hippocampus, which in turn provides negative feedback to the hypothalamus in order to aid in the termination of stress-induced activation of the HPA axis [28, 29].
High levels of CRF immunoreactivity also are observed outside of the hypothalamus in other forebrain regions, such as the olfactory bulb, cerebral cortex, nucleus accumbens, septum, bed nucleus of the stria terminalis (BNST), preoptic nuclei, amygdala, and hippocampus [30–32]. Mid- and hindbrain regions that contain CRF include the periaqueductal gray, raphe nuclei, lateral tegmental nucleus, locus coeruleus (LC), parabrachial nucleus, cerebellum, and the nucleus of the solitary tract [30–32]. This widespread extra-hypothalamic expression means that CRF is in a position to centrally modulate a variety of cognitive and behavioral responses to stress. For example, CRF is known to activate the amygdala, BNST, and prefrontal cortex to mediate a number of behaviors, including those related to anxiety, defensive responses, and fear conditioning [31, 33–39]. Additionally, monoaminergic neuronal transmission is regulated by CRF [40–43], further underscoring the pivotal role of CRF in coordinating a broad range of responses to stressful events.
CRF receptor distribution and function
CRF exerts its actions via two G-protein coupled receptors: CRF1 receptor type (CRF1) and CRF2 receptor type (CRF2) [44–46]. Both CRF receptors are found in regions involved in regulating stress, mood, and anxiety. Specifically, CRF1 is predominantly found in the cerebral cortex, cerebellum, pons, and anterior lobe of the pituitary, while CRF2 is more prominent in the septum, choroid plexus, and posterior lobe of the pituitary [47–49]. In many brain regions, like the hypothalamus, hippocampus, amygdala, and BNST, there is considerable variation between the expression of CRF1 and CRF2 (e.g., CRF1 expression is greater in the periventricular zone of the hypothalamus, while CRF2 expression is greater in the ventromedial nucleus of the hypothalamus) [47–49]. This heterogeneous distribution of CRF receptor subtypes suggests that each subtype has a unique role in mediating CRF function. Genetic and pharmacological manipulations support this idea. Activation of CRF1 initiates the endocrine stress response and mediates anxiety-related behavior, while CRF1 antagonism or receptor knockout reduces anxiety [50–57]. The role of CRF2 in mediating stress-related behavior is more complex. Data from CRF2 knockout mice suggest that activation CRF2 attenuates activity of the HPA axis, thereby counteracting the stimulating effect of CRF1 [58–60]. The role of CRF2 in mediating anxiety is less consistent with some, but not all, studies implicating these receptors in the reduction of anxiety-related behavior [50, 59, 61–64]. These inconsistencies may arise from the fact that some effects of CRF2 are region specific [65–67]. Nevertheless, the sometimes opposing actions of CRF1 and CRF2 have led to proposal that generally CRF1 increases while CRF2 decreases stress responsivity [50].
Given the widespread distribution of CRF1 and CRF2, it is clear that CRF is critically poised to modulate a number of brain regions. Thus, it is not surprising that dysregulation of CRF and its receptors can result in a number of adverse consequences. CRF hypersecretion is linked to the pathophysiology of depression and certain anxiety disorders [68–70]. High levels of CRF are found in the cerebral spinal fluid and brain regions of patients with depression and PTSD, and these levels are reduced with antidepressant treatment [70–83]. Multiple genomic studies have identified single nucleotide polymorphisms on the CRF1 gene that are linked to depression, panic disorder, and PTSD [84–89]. Further evidence for CRF1 in mediating depression comes from a clinical trial in which CRF1 antagonist treatment of depressed patients reduced depressed mood and anxiety symptoms [90]. Taken together, these studies suggest a causal role for CRF dysregulation in the development of stress-related pathology.
Sex differences in CRF receptors: Evidence from knockout mice and binding studies
The psychiatric disorders linked to CRF dysregulation occur more frequently in women. Thus, it is likely that sex differences in the CRF system underlie this disparity. Indeed, sex differences in CRF expression have been observed in the paraventricular nucleus of the hypothalamus, amygdala, and BNST, with females typically having greater CRF expression [91–94]. In addition to sex differences in the expression of CRF, sex difference in CRF receptors could contribute to sex differences in stress vulnerability. Evidence for sex differences in CRF receptors comes, in part, from CRF2 knockout mice that have a phenotype attributable to unimpeded CRF1 activity [59, 95]. Female CRF2-deficint mice display more depressive-like behaviors on the forced swim test than their male counterparts, an effect blocked by a CRF1 receptor antagonist [95]. Not only does this finding reveal that depressive-like behavior is mediated, at least in part, by CRF1, but this result suggests that sex differences at the level of the CRF1 contribute to increased depressive-like behavior in females. Despite this evidence for sex differences in CRF1, few studies have directly evaluated sex differences in CRF receptor expression and function.
Recently, Weathington and Cooke (2012) identified sex differences in CRF receptor binding in the amygdala. CRF1 binding was significantly greater in the basolateral and posteroventral nuclei of the amygdala in female than male rats. Interestingly, this sex difference was not identified until after puberty. Sex differences in CRF2 binding also emerged following puberty [96]. Compared to juvenile males, adult males had increased CRF2 binding in the four subnuclei of the amygdala examined (i.e., basolateral, posterodorsal, posteroventral, and central), while CRF2 levels remained low following puberty in females. Similarly, in the BNST, which is considered part of the extended amygdala, adult male voles had greater CRF2 binding than their female counterparts [97]. Sex differences in CRF receptor binding in the amygdala could contribute to sex differences in anxiety. Blocking or reducing CRF1 receptors in the basolateral nucleus of the amygdala decreases anxiety-related behavior [98, 99]. If the role of CRF2 in the amygdala is to counteract the effects of CRF1 activation, like it is in other regions, CRF2 activation could decrease CRF1-mediated anxiety. Compared to males, female rats have a greater ratio of CRF1:CRF2 binding in the basolateral nucleus of the amygdala. This sex difference could translate into increased anxiety following stressful events in females, which, if true in humans, would predispose women anxiety disorders such as PTSD.
The fact that sex differences in CRF receptor binding emerge after puberty, implicates the gonadal hormone surges that occur during puberty in these effects [96]. Estrogen, androgen, and progesterone receptors are found in CRF producing regions [49, 100–104]. Moreover, androgen treatment increases CRF2 mRNA in several brain regions of male rats [105]. In the human myometrium, estrogen and progesterone increase CRF1 expression, although this has not yet been demonstrated in the brain [106]. The mechanism underlying gonadal hormone regulation of CRF receptor expression is not fully understood, but putative estrogen, progesterone, and androgen response elements have been identified on the promoter regions of CRF receptor genes [105, 107, 108]. Thus, direct regulation of gene transcription by gonadal hormones is possible. Together these data suggest that sex difference in CRF1 and CRF2 binding are likely due to gonadal hormone regulation of CRF receptor number, rather than other mechanisms (e.g., sex differences in differences in receptor affinity for the CRF or neuronal number).
Sex differences in CRF1 receptor signaling
In addition to sex differences in CRF receptor binding, sex differences in CRF1 signaling have been identified that can render neurons of females more sensitive to CRF [109–111]. The CRF1 is a G protein-coupled receptor that changes conformation upon CRF binding to promote the coupling of GTP binding proteins, which initiate intracellular signaling cascades [51, 112]. Although CRF1 can promiscuously couple several G proteins, it preferentially binds Gs [112, 113]. CRF-induced Gs activation stimulates adenylyl cyclase to form cyclic adenosine monophosphate (cAMP), increasing protein kinase A (PKA) signaling [114, 115]. Sex differences in CRF1 coupling to Gs have been identified [109]. Specifically, receptor immunoprecipitation of cortical tissue revealed that more Gs was pulled down with the CRF1 of females compared to males under unstressed conditions, indicating that the CRF1 in female rat cortex is more highly coupled to Gs than in males. Using the same immunoprecipitation technique, it was found that acute stressor exposure (15 min of forced swimming) enhanced CRF1-Gs coupling in males to female levels. Stressed females had similarly high levels of CRF1-Gs binding as unstressed females. Additionally, these effects were not modulated by circulating ovarian hormones as coupling was comparable in both ovariectomized and intact female rats. The increased binding of CRF1 to Gs in females would translate into greater cAMP-PKA signaling in this group. Mice genetically modified to have either greater Gs activation or increased PKA-signaling have an anxiogenic phenotype [116, 117]. Thus, the sex difference in CRF1-Gs coupling and subsequent signaling could increase anxiety in response to stressful events in females.
Interestingly, sex differences in CRF1-Gs coupling may also occur in other brain regions. Although the receptor immunoprecipitation technique is limited to the cortex because of the amount of protein required, the sex difference in CRF1-Gs coupling found in cortex mirrors the sex difference in the physiological response of LC neurons to CRF. During a stressful event, CRF released in the LC activates noradrenergic neurons to coordinate arousal, attention, and vigilance [42, 118–120]. LC neurons of female rats are more sensitive to CRF than males, such that a dose of CRF that is too low to activate neuronal firing in males increases neuronal firing in females [121]. Acute swim stress enhances LC neuronal sensitivity in males to female levels, but does not alter sensitivity of female neurons [121]. Notably, these sex differences parallel the direction of the sex differences in CRF1-Gs coupling in cortex, such that stressed males and females (regardless of stress history) had neurons that were more sensitive to CRF and CRF1 receptors that were more highly coupled to Gs than unstressed males. The CRF-induced increase in LC neuronal firing is mediated by the cAMP-PKA signaling cascade [122]. Thus, the sex difference in coupling and subsequent signaling likely causes the sex difference in LC physiology. Consistent with this idea, pretreatment of LC neurons with a cAMP antagonist revealed that the majority of the LC physiological response to CRF in unstressed females and stressed males was cAMP mediated, whereas in unstressed males, only 50% of the LC response to CRF was cAMP-mediated [109]. Together these data suggest that sex differences in CRF1-Gs coupling and cAMP-PKA signaling render LC neurons of females more sensitive to CRF. This effect can have consequences for LC-mediated stress responses in females. Typically, activation of the LC-arousal system is thought to be adaptive, as it promotes cognition and behavior aimed at coping with stress [119, 123]. However, activation of this system to stress levels by stimuli that should be subthreshold for stress reactivity would be disruptive. Given that female LC neurons are more sensitive to CRF, LC activation is more likely to be inappropriately elicited in females.
Sex differences in CRF1 receptor trafficking
Sex differences in CRF1 trafficking also are observed in LC neurons. Like most G-protein coupled receptors, in response to saturating concentrations of ligand, the CRF1 receptor is desensitized and internalized (i.e., trafficked from the membrane to the cytosol) [51, 124–126]. This is considered a cellular adaptation to high levels of CRF because the internalized receptors can no longer be activated. Both CRF agonist treatment and acute swim stress (15 min) induce CRF1 internalization in LC neurons of male rats [109, 126, 127]. Interestingly, swim stress fails to internalize these receptors in female rats [109]. A failure of CRF1 internalization in females could result from sex differences in CRF1 binding to β-arrestin, the protein that initiates the internalization process [51, 128, 129]. Stress increases CRF1-β-arrestin binding in cortical tissue in males, but fails to do so in females [109]. If the sex difference in CRF1 association with β-arrestin is also present in LC neurons, it would account for sex differences in CRF1 trafficking.
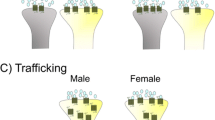
The lack of CRF1 internalization observed following stress in females could result in female LC neurons that are more sensitive to conditions of CRF hypersecretion, which as noted, occurs in depression and PTSD. This idea was tested by using CRF overexpressing mice that model conditions of chronic stress and display anxiety-related behavior [130–132]. High levels of CRF expression in the LC region were observed in CRF overexpressing mice, regardless of sex [133]. The firing rates of LC neurons were evaluated in these mice and compared to wild type controls. LC firing rates were similar in male and female wild type mice. However, LC neurons of female CRF overexpressing mice fired roughly 3× faster than male and female wild type controls [133]. Interestingly, the firing rates of male overexpressing mice were maintained at wild type levels. Because CRF expression was high in the LC of male overexpressing mice, this result suggested the presence of some compensatory cellular mechanism in this group. As noted, internalization is a cellular adaptation that can reduce cellular responding to high levels of CRF, so CRF1 internalization was evaluated. Much like the trafficking pattern following acute stress in rats, CRF1 receptors were internalized in male but not female overexpressing mice [133]. The inability of female CRF1 to internalize under conditions of CRF hypersecretion can explain the high LC neuronal firing rate in this group (Figure 1). High levels of LC activation are linked to hyperarousal, a symptom of stress-related disorders that leads to increased agitation, restlessness, re-experiencing, and sleep disturbance [69, 134–136]. The aforementioned results suggest that sex differences CRF1 modulation of the LC-arousal systems could more easily shift females into this dysregulated state of hyperarousal.

This schematic illustrates sex differences in CRF 1 trafficking in LC dendrites [133]. Under conditions of excessive release of CRF (green), CRF1 receptors (blue) internalize in male neurons (left). This effect reduces neuronal responses to CRF in males. In females (right), CRF1 receptors fail to internalize under conditions of CRF hypersecretion, resulting in greater neuronal responses to CRF in this group.
CRF binding protein (CRF-BP)
In addition to the mechanisms already discussed, the activity of CRF is also regulated by CRF-BP [137–139]. CRF-BP is expressed in the pituitary and in several brain regions including the olfactory bulb, amygdala, BNST, the ventral premammillary and dorsomedial nuclei of the hypothalamus, and sensory relay nuclei in midbrain and brain stem [140]. CRF-BP inhibits CRF-induced ACTH release in pituitary corticotropes in vitro, suggesting CRF-BP sequesters free CRF [141]. Centrally, both acute stress and CRF administration increase the expression of CRF-BP in the basolateral nucleus of the amygdala, an effect that may facilitate recovery from stressor exposure [142–144]. However, in the ventral tegmental area, CRF-BP potentiates the signaling of CRF receptors, thereby amplifying the effects of CRF [145]. Interestingly, sex differences in expression of CRF-BP have been observed in the pituitary, with female mice having higher levels of CRF-BP than their male counterparts [146]. Thus, the complex role of CRF-BP in modulating CRF, and its potential to contribute to sex differences in the development of stress-related disorders, certainly warrants further study.
Glucocorticoids regulate responses to stress
Activation of the HPA axis leads to the release of glucocorticoids. These stress hormones promote physiological changes to adapt to acute stress, such as metabolizing glucose, catabolizing proteins and lipids, and decreasing inflammation [29]. As noted earlier, glucocorticoids also feedback centrally and at the level of the pituitary to terminate the endocrine response to stress. However, if the HPA stress response becomes persistently activated due to impaired negative feedback or other mechanisms, then the adverse state of hypercortisolemia can occur. Hypercortisolemia contributes to the pathophysiology of a number of psychiatric and medical disorders including depression [147–154].
Glucocorticoids act through two receptors: mineralocorticoid receptors (MRs) and glucocorticoid receptors (GRs). MRs are highly expressed in the hippocampus and septum, while GRs are found throughout the brain in regions including the hippocampus, septum, amygdala, hypothalamus, and the nucleus of the solitary tract [155–157]. The affinity of these receptors is different, such that glucocorticoids bind to MRs with higher affinity (Kd, 1 nM), but to GRs with lower affinity (Kd, 2.5-5 nM) [155, 156, 158]. The differential distribution and affinity of these receptors has led to the proposal that MRs maintain the tone of the stress system under normal conditions [155]. In contrast, GRs, because of their lower affinity for the ligand, respond to high levels of glucocorticoids to mediate the physiological recovery from stressful events and restore the system to homeostasis [155, 159]. In support of this, there are two phases of glucocorticoid negative feedback, fast and delayed, and both appear to be regulated by GRs. Specifically, fast feedback is thought to be mediated by membrane bound GRs that, when activated, induce endocannabinoid suppression of the hypothalamus [160, 161]. Delayed feedback involves the translocation of nuclear GRs from the cytosol to the nucleus where they can repress gene transcription of CRF, CRF1, and precursors of ACTH [162–167]. These studies highlight the fact that GRs are the critical receptor subtype that coordinates the recovery of the HPA axis response to stress.
Glucocorticoid negative feedback can be assessed by the dexamethasone suppression test [168–171]. This test was developed to diagnose Cushing’s syndrome, a disorder characterized by excessive endogenous glucocorticoid secretion, but also has been used to assess HPA axis dysregulation in several psychiatric disorders, including depression [168, 169, 171]. Dexamethasone is a synthetic glucocorticoid that binds with high affinity for GRs [172]. In normal subjects, dexamethasone suppresses the release of cortisol, but it fails to suppress cortisol in 20-50% of depressed patients, indicating impaired negative feedback [169–171]. When the dexamethasone suppression test is combined with the CRF stimulation test, the sensitivity to differentiate between normal and pathological states increases to 80%, suggesting the combined test is a better assessment of the HPA axis dysregulation observed in depression [173–175]. Decreased negative feedback in depression is thought to result in the hypercortisolemia that is often observed in these patients [147–153, 176]. Interestingly, there is evidence that HPA axis dysregulation precedes depression, and that cortisol levels and negative feedback normalize with antidepressant treatment or clinical recovery [148, 149, 151]. Additionally, patients with Cushing’s syndrome often experience depression, and removal of their adrenal glands relieves depressive symptoms [177, 178]. Taken together, these finding indicate that changes in glucocorticoid negative feedback may cause depressive symptoms [148, 149, 151].
Sex differences in glucocorticoid negative feedback
The most widely reported sex difference in stress response systems is that female rodents have higher levels of HPA axis hormones than males. Specifically, basal levels of glucocorticoids are elevated in female rats relative to their male counterparts [93, 179, 180]. Stressor exposure induces greater corticosterone release for a longer duration in female than male rodents [91, 93, 179–185]. In contrast to the consistent findings in rodents, reports of sex differences in cortisol levels in humans are equivocal [186–196]. Despite these discrepancies, mechanisms that can lead to higher levels of glucocorticoids in females have been identified. One such mechanism is greater CRF expression in the hypothalamus of female compared to male rodents, which can translate into greater HPA axis activation [91–94, 197]. Another mechanism involves sex differences in glucocorticoid negative feedback. As noted, in response to acute stress, female rodents have a protracted elevation of corticosterone, which is indicative of a slower negative feedback compared to males [91, 180, 184]. Studies in humans also suggest that feedback is slower in women than men [191–193]. Sex differences in negative feedback may be established by ovarian hormones, because female rats treated with estrogen have a slower glucocorticoid feedback than ovariectomized controls [198, 199]. This suggests that estrogen can decrease negative feedback, resulting in a prolonged release of glucocorticoids in females.
Sex differences in glucocorticoid negative feedback may be explained by sex differences in glucocorticoid receptor number. In the pituitary, female rats have fewer MRs and GRs than males [200]. Similarly, compared to male rats, glucocorticoid binding is lower in the hypothalamus of females, likely reflecting fewer receptors [201]. Estrogen regulates the expression of receptors for glucocorticoids, again highlighting the role of estrogen in establishing this sex difference. Specifically, estrogen treatment decreased GR mRNA in breast cancer cells [202]. Also, estrogen treatment of ovariectomized females downregulated GR expression in the hippocampus, hypothalamus, and pituitary [200, 203]. Thus, direct downregulation of glucocorticoid receptors by estrogen is possible.
Sex differences in GR translocation and other feedback regulators
Another mechanism by which sex differences in glucocorticoid negative feedback can be established involves GR translocation [204]. The process of translocation is initiated by the binding of glucocorticoids to nuclear GRs. The GRs then are shuttled by multiple co-chaperones (e.g., PPID and FKBP52) into the nucleus to regulate gene transcription [205–207]. In addition to co-chaperones that promote GR translocation, other co-chaperones, such as Bag1 and Fkbp5, inhibit GR translocation, highlighting that this process is highly regulated (Figure 2) [205, 207–210]. Bourke and colleagues (2012) identified several co-chaperones that were regulated in a sex-specific manner, and often these sex differences were exacerbated by exposure to chronic stress. Alterations in co-chaperone expression observed in female rats with a history of chronic adolescent stress were linked to changes in glucocorticoid negative feedback [204]. Specifically, when these females were exposed as adults to an acute stress challenge, elevated levels of Bag1 and Fkbp5 were observed. Acute stressor exposure also attenuated GR translocation and negative feedback in this group when compared to females that were not stressed during adolescence. These findings suggest that stress-induced increases in co-chaperones that inhibit GR translocation result in impaired glucocorticoid negative feedback in females. Interestingly, a history of adolescent stress did not alter GR translocation or HPA responses to acute stressor exposure in males, indicating that females are more vulnerable to stress during the adolescent period.

This schematic depicts the role of GR translocation in glucocorticoid negative feedback. Glucocorticoids readily cross the plasma membrane to bind nuclear GRs. GRs then are shuttled into the nucleus by multiple co-chaperones (in green), such as PPID and FKBP52. Once inside, GRs bind to glucocorticoid response elements (GREs) on genes to repress transcription of CRF and precursors for ACTH. Other co-chaperones (in red), including Bag1 and Fkbp5, inhibit GR translocation, an effect which can attenuate negative feedback.
Sex differences in glucocorticoid negative feedback may also occur via mechanisms other than GR expression and translocation. Progesterone can compete with glucocorticoids for binding cites on GR itself, while estrogen receptors can compete with GRs for a binding site on the promoter region of genes, demonstrating that ovarian hormones can directly block glucocorticoid actions [211, 212]. Additionally, because estrogen receptors co-localize with GABA neurons in the area around the paraventricular of the hypothalamus nucleus, it has been proposed that estrogen can reduce GABA input into the hypothalamus, thereby indirectly inhibiting negative feedback in females [213]. Together these studies suggest that the regulation of glucocorticoid negative feedback by ovarian hormones is complex and not limited to effects solely at GR regulation. Nevertheless, the reduction in GR levels and the prevention of their translocation into the nucleus in stressed females would decrease the ability of glucocorticoids to repress CRF and ACTH gene transcription. Thus, sex differences at the level of GRs are likely important mechanisms that lead to increased stress hormones levels in women. Under typical conditions, slight increases in glucocorticoid levels due to a protracted negative feedback may not impact women. However, under conditions of chronic stress, the slower termination of the HPA axis response in women could result in high glucocorticoid levels for an extended period of time. As hypercortisolemia is thought to cause depression, sex differences in glucocorticoid negative feedback may predispose women to this disease [148, 149, 151].
Implications for pharmacotherapy
The aforementioned studies reveal sex differences at the level of two receptors that mediate responses to stress: CRF1 and GR. Not only can these sex differences contribute to the development of mood and anxiety disorders, but they have implications for pharmaceutical treatments. For example, CRF1 antagonists are currently being developed to treat stress-related disorders [214–216]. The fact that the CRF1 binds Gs and β-arrestin proteins differently in males versus females suggests sex differences in the conformation of this receptor. This conformational change could alter the binding of CRF1 antagonists, changing their efficacy in men versus women. Understanding the mechanisms that differentially regulate these receptors in males and females may also lead to novel treatments. Perhaps future pharmacotherapies could be developed to promote β-arrestin binding to CRF1, thereby stimulating internalization, a potentially longer lasting treatment than could be achieved with CRF1 antagonists. Other compounds targeting co-chaperones to increase GR translocation could accelerate negative feedback of the HPA response. Given the sex differences in CRF1internalization and GR translocation, both of these treatments are likely to be particularly effective in women.
These studies also underscore the importance of using female subjects in preclinical research. A review of the non-human animal studies published in 2009 found that single-sex studies of male animals outnumbered those of females 5.5 to 1 in neuroscience and 5 to 1 in pharmacology [217]. Given the evidence that sex differences occur at the receptor level, it is possible that receptor antagonists would work well in one sex but not the other. In fact, one interesting possibility is that the pharmacological treatments discussed above may only be effective for the treatment of stress-related disorders in females. Because compounds are typically screened exclusively in male animals, new drugs that appear to be ineffective for males would never get past the preclinical drug development stage, despite the fact that they may be very effective for females. This scenario could deny women with mood and anxiety disorders important treatment options.
Conclusions
The emerging literature on sex differences in stress-related receptors indicates that, although psychological factors may play a role, biological factors also contribute to sex differences in stress-related psychiatric disorders. Moreover, these studies are not only advancing our understanding of the neurobiological bases for this sex bias, but they also may guide the development of novel pharamotherapies. In addition, the fact that sex differences occur at the receptor level has even broader implications. These are the first reports, to my knowledge, of sex differences in G-protein coupled receptor signaling and steroid hormone receptor translocation. It is possible that similar sex differences occur in other G-protein coupled and steroid hormone receptors. If supported by future studies, sex differences in receptor function may also explain why many other psychiatric disorders (e.g., autism, attention-deficit disorder, etc.) occur at different rates in men and women [218, 219]. Thus, sex differences at the receptor level may be an important yet underexplored determinant of vulnerability to a wide range of psychiatric diseases.
Author’s contributions
DAB wrote and edited manuscript, and has read and approved the final version.
Abbreviations
- PTSD:
-
Post-traumatic stress disorder
- CRF:
-
Corticotropin releasing factor
- GRs:
-
Glucocorticoid receptors
- HPA:
-
Hypothalamic pituitary adrenal
- CRF1 :
-
CRF1 receptor type
- CRF2 :
-
CRF2 receptor type
- BNST:
-
Bed nucleus of the stria terminalis
- cAMP:
-
cyclic adenosine monophosphate
- PKA:
-
Protein kinase A
- LC:
-
Locus coeruleus
- ACTH:
-
Adrenocorticotropic hormone
- MR:
-
Mineralocorticoid receptor
- GREs:
-
Glucocorticoid response elements.
References
Kendler KS, Kessler RC, Walters EE, MacLean C, Neale MC, Heath AC, Eaves LJ: Stressful life events, genetic liability, and onset of an episode of major depression in women. Am J Psychiatry 1995, 152: 833–842.
Stein MB, Steckler T: Behavioral neurobiology of anxiety and its treatment. Preface. Curr Top Behav Neurosci 2010, 2: v-vii.
Melchior M, Caspi A, Milne BJ, Danese A, Poulton R, Moffitt TE: Work stress precipitates depression and anxiety in young, working women and men. Psychol Med 2007, 37: 1119–1129. 10.1017/S0033291707000414
Breslau N: The epidemiology of trauma, PTSD, and other posttrauma disorders. Trauma Violence Abuse 2009, 10: 198–210. 10.1177/1524838009334448
Breslau N, Kessler RC, Chilcoat HD, Schultz LR, Davis GC, Andreski P: Trauma and posttraumatic stress disorder in the community: the 1996 detroit area survey of trauma. Arch Gen Psychiatry 1998, 55: 626–632. 10.1001/archpsyc.55.7.626
Faravelli C: Life events preceding the onset of panic disorder. J Affect Disorders 1985, 9: 103–105. 10.1016/0165-0327(85)90016-3
Newman SC, Bland RC: Life events and the 1-year prevalence of major depressive episode, generalized anxiety disorder, and panic disorder in a community sample. Compr Psychiatry 1994, 35: 76–82. 10.1016/0010-440X(94)90173-2
Shabsigh R, Rowland D: The diagnostic and statistical manual of mental disorders, fourth edition, text revision as an appropriate diagnostic for premature ejaculation. J Sex Med 2007, 4: 1468–1478. 10.1111/j.1743-6109.2007.00557.x
Gutman DA, Nemeroff CB: Neurobiology of early life stress: rodent studies. Semin Clin Neuropsychiatry 2002, 7: 89–95. 10.1053/scnp.2002.31781
McCrory E, De Brito SA, Viding E: The link between child abuse and psychopathology: a review of neurobiological and genetic research. J R Soc Med 2012, 105: 151–156. 10.1258/jrsm.2011.110222
Bremne JD, Vermetten E: Stress and development: behavioral and biological consequences. Dev Psychopathol 2001, 13: 473–489. 10.1017/S0954579401003042
Kessler RC: Epidemiology of women and depression. J Affect Disord 2003, 74: 5–13. 10.1016/S0165-0327(02)00426-3
Kessler RC, McGonagle KA, Swartz M, Blazer DG, Nelson CB: Sex and depression in the national comorbidity survey. I: lifetime prevalence, chronicity and recurrence. J Affect Disord 1993, 29: 85–96. 10.1016/0165-0327(93)90026-G
Tolin DF, Foa EB: Sex differences in trauma and posttraumatic stress disorder: a quantitative review of 25 years of research. Psychol Bull 2006, 132: 959–992.
Breslau N: Gender differences in trauma and posttraumatic stress disorder. J Gend Specif Med 2002, 5: 34–40.
Tolin DF, Foa EB: Gender and PTSD: A cognitive model. In In Gender and PTSD. Edited by: Kimerling R, Ouimette P, Wolfe J. New York, NY US: Guilford Press; 2002:76–97.
Hankin BL, Abramson LY: Development of gender differences in depression: an elaborated cognitive vulnerability-transactional stress theory. Psychol Bull 2001, 127: 773–796.
Cyranowski JM, Frank E, Young E, Shear MK: Adolescent onset of the gender difference in lifetime rates of major depression: A theoretical model. In Annual progress in child psychiatry and child development: 2000–2001. Edited by: Hertzig ME, Farber EA. New York, NY US: Brunner-Routledge; 2003:383–398.
Kendler KS, Kessler RC, Neale MC, Heath AC: The prediction of major depression in women: toward an integrated etiologic model. Am J Psychiatry 1993, 150: 1139–1148.
Mezulis AH, Funasaki KS, Charbonneau AM, Hyde JS: Gender differences in the cognitive vulnerability-stress model of depression in the transition to adolescence. Cogn Ther Res 2010, 34: 501–513. 10.1007/s10608-009-9281-7
McLaughlin K, Nolen-Hoeksema S: The role of rumination in promoting and preventing depression in adolescent girls. In In Depression in adolescent girls: Science and prevention. Edited by: Strauman TJ, Costanzo PR, Garber J. New York, NY US: Guilford Press; 2011:112–129. Duke series in child development and public policy]
Wichstrøm L: The emergence of gender difference in depressed mood during adolescence: the role of intensified gender socialization. Dev Psychol 1999, 35: 232–245.
Hyde JS, Mezulis AH, Abramson LY: The ABCs of depression: integrating affective, biological, and cognitive models to explain the emergence of the gender difference in depression. Psychol Rev 2008, 115: 291–313.
Breslau N, Chilcoat HD, Kessler RC, Peterson EL, Lucia VC: Vulnerability to assaultive violence: further specification of the sex difference in post-traumatic stress disorder. Psychol Med 1999, 29: 813–821. 10.1017/S0033291799008612
Iteke O, Bakare MO, Agomoh AO, Uwakwe R, Onwukwe JU: Road traffic accidents and posttraumatic stress disorder in an orthopedic setting in south-eastern nigeria: a controlled study. Scand J Trauma Resusc Emerg Med 2011, 19: 39. 10.1186/1757-7241-19-39
Vale W, Spiess J, Rivier C, Rivier J: Characterization of a 41-residue ovine hypothalamic peptide that stimulates secretion of corticotropin and beta-endorphin. Science 1981, 213: 1394–1397. 10.1126/science.6267699
Dallman MF, Akana SF, Jacobson L, Levin N, Cascio CS, Shinsako J: Characterization of corticosterone feedback regulation of ACTH secretion. Ann N Y Acad Sci 1987, 512: 402–414. 10.1111/j.1749-6632.1987.tb24976.x
Sapolsky RM, Meaney MJ, McEwen BS: The development of the glucocorticoid receptor system in the rat limbic brain. III. Negative-feedback regulation. Brain research 1985, 350: 169–173.
McEwen BS, Gianaros PJ: Stress- and allostasis-induced brain plasticity. Annu Rev Med 2011, 62: 431–445. 10.1146/annurev-med-052209-100430
Merchenthaler I: Corticotropin releasing factor (CRF)-like immunoreactivity in the rat central nervous system. Extrahypothalamic distribution. Peptides 1984,5(Suppl 1):53–69.
Swanson LW, Sawchenko PE, Rivier J, Vale WW: Organization of ovine corticotropin-releasing factor immunoreactive cells and fibers in the rat brain: an immunohistochemical study. Neuroendocrinology 1983, 36: 165–186. 10.1159/000123454
Cummings S, Elde R, Ells J, Lindall A: Corticotropin-releasing factor immunoreactivity is widely distributed within the central nervous system of the rat: an immunohistochemical study. J Neurosci 1983, 3: 1355–1368.
Isogawa K, Bush DE, Ledoux JE: Contrasting effects of pretraining, posttraining, and pretesting infusions of corticotropin-releasing factor into the lateral amygdala: attenuation of fear memory formation but facilitation of its expression. Biol Psychiatry 2012. Epub ahead of print
Gafford GM, Guo JD, Flandreau EI, Hazra R, Rainnie DG, Ressler KJ: Cell-type specific deletion of GABA(a)alpha1 in corticotropin-releasing factor-containing neurons enhances anxiety and disrupts fear extinction. Proc Natl Acad Sci USA 2012, 109: 16330–16335. 10.1073/pnas.1119261109
Regev L, Tsoory M, Gil S, Chen A: Site-specific genetic manipulation of amygdala corticotropin-releasing factor reveals its imperative role in mediating behavioral response to challenge. Biol Psychiatry 2012, 71: 317–326. 10.1016/j.biopsych.2011.05.036
Pitts MW, Takahashi LK: The central amygdala nucleus via corticotropin-releasing factor is necessary for time-limited consolidation processing but not storage of contextual fear memory. Neurobiol Learn Mem 2011, 95: 86–91. 10.1016/j.nlm.2010.11.006
Bijlsma EY, van Leeuwen ML, Westphal KG, Olivier B, Groenink L: Local repeated corticotropin-releasing factor infusion exacerbates anxiety- and fear-related behavior: differential involvement of the basolateral amygdala and medial prefrontal cortex. Neuroscience 2011, 173: 82–92.
Jasnow AM, Davis M, Huhman KL: Involvement of central amygdalar and bed nucleus of the stria terminalis corticotropin-releasing factor in behavioral responses to social defeat. Behav Neurosci 2004, 118: 1052–1061.
Gray TS: Amygdaloid CRF pathways. Role in autonomic, neuroendocrine, and behavioral responses to stress. Ann N Y Acad Sci 1993, 697: 53–60. 10.1111/j.1749-6632.1993.tb49922.x
Rodaros D, Caruana DA, Amir S, Stewart J: Corticotropin-releasing factor projections from limbic forebrain and paraventricular nucleus of the hypothalamus to the region of the ventral tegmental area. Neuroscience 2007, 150: 8–13. 10.1016/j.neuroscience.2007.09.043
Wanat MJ, Hopf FW, Stuber GD, Phillips PE, Bonci A: Corticotropin-releasing factor increases mouse ventral tegmental area dopamine neuron firing through a protein kinase C-dependent enhancement of Ih. J Physiol 2008, 586: 2157–2170. 10.1113/jphysiol.2007.150078
Valentino RJ, Van Bockstaele E: Convergent regulation of locus coeruleus activity as an adaptive response to stress. Eur J Pharmacol 2008, 583: 194–203. 10.1016/j.ejphar.2007.11.062
Valentino RJ, Commons KG: Peptides that fine-tune the serotonin system. Neuropeptides 2005, 39: 1–8. 10.1016/j.npep.2004.09.005
Chang CP, Pearse RV 2nd, O′Connell S, Rosenfeld MG: Identification of a seven transmembrane helix receptor for corticotropin-releasing factor and sauvagine in mammalian brain. Neuron 1993, 11: 1187–1195. 10.1016/0896-6273(93)90230-O
Chen R, Lewis KA, Perrin MH, Vale WW: Expression cloning of a human corticotropin-releasing-factor receptor. Proc Natl Acad Sci USA 1993, 90: 8967–8971. 10.1073/pnas.90.19.8967
Lovenberg TW, Liaw CW, Grigoriadis DE, Clevenger W, Chalmers DT, De Souza EB, Oltersdorf T: Cloning and characterization of a functionally distinct corticotropin-releasing factor receptor subtype from rat brain. Proc Natl Acad Sci USA 1995, 92: 836–840. 10.1073/pnas.92.3.836
Chalmers DT, Lovenberg TW, De Souza EB: Localization of novel corticotropin-releasing factor receptor (CRF2) mRNA expression to specific subcortical nuclei in rat brain: comparison with CRF1 receptor mRNA expression. J Neurosci 1995, 15: 6340–6350.
Primus RJ, Yevich E, Baltazar C, Gallager DW: Autoradiographic localization of CRF1 and CRF2 binding sites in adult rat brain. Neuropsychopharmacology: official publication of the American College of Neuropsychopharmacology 1997, 17: 308–316. 10.1016/S0893-133X(97)00071-7
Van Pett K, Viau V, Bittencourt JC, Chan RK, Li HY, Arias C, Prins GS, Perrin M, Vale W, Sawchenko PE: Distribution of mRNAs encoding CRF receptors in brain and pituitary of rat and mouse. J Comp Neurol 2000, 428: 191–212. 10.1002/1096-9861(20001211)428:2<191::AID-CNE1>3.0.CO;2-U
Bale TL, Vale WW: CRF and CRF receptors: role in stress responsivity and other behaviors. Annu Rev Pharmacol Toxicol 2004, 44: 525–557. 10.1146/annurev.pharmtox.44.101802.121410
Hauger RL, Risbrough V, Oakley RH, Olivares-Reyes JA, Dautzenberg FM: Role of CRF receptor signaling in stress vulnerability, anxiety, and depression. Ann N Y Acad Sci 2009, 1179: 120–143. 10.1111/j.1749-6632.2009.05011.x
Takahashi LK: Role of CRF(1) and CRF(2) receptors in fear and anxiety. Neurosci Biobehav Rev 2001, 25: 627–636. 10.1016/S0149-7634(01)00046-X
Heinrichs SC, Lapsansky J, Lovenberg TW, De Souza EB, Chalmers DT: Corticotropin-releasing factor CRF1, but not CRF2, receptors mediate anxiogenic-like behavior. Regul Pept 1997, 71: 15–21. 10.1016/S0167-0115(97)01005-7
Contarino A, Dellu F, Koob GF, Smith GW, Lee KF, Vale W, Gold LH: Reduced anxiety-like and cognitive performance in mice lacking the corticotropin-releasing factor receptor 1. Brain Res 1999, 835: 1–9. 10.1016/S0006-8993(98)01158-5
Smith GW, Aubry JM, Dellu F, Contarino A, Bilezikjian LM, Gold LH, Chen R, Marchuk Y, Hauser C, Bentley CA, et al.: Corticotropin releasing factor receptor 1-deficient mice display decreased anxiety, impaired stress response, and aberrant neuroendocrine development. Neuron 1998, 20: 1093–1102. 10.1016/S0896-6273(00)80491-2
Timpl P, Spanagel R, Sillaber I, Kresse A, Reul JM, Stalla GK, Blanquet V, Steckler T, Holsboer F, Wurst W: Impaired stress response and reduced anxiety in mice lacking a functional corticotropin-releasing hormone receptor 1. Nat Genet 1998, 19: 162–166. 10.1038/520
Holsboer F, Ising M: Central CRH system in depression and anxiety–evidence from clinical studies with CRH1 receptor antagonists. Eur J Pharmacol 2008, 583: 350–357. 10.1016/j.ejphar.2007.12.032
Coste SC, Murray SE, Stenzel-Poore MP: Animal models of CRH excess and CRH receptor deficiency display altered adaptations to stress. Peptides 2001, 22: 733–741. 10.1016/S0196-9781(01)00386-2
Bale TL, Contarino A, Smith GW, Chan R, Gold LH, Sawchenko PE, Koob GF, Vale WW, Lee KF: Mice deficient for corticotropin-releasing hormone receptor-2 display anxiety-like behaviour and are hypersensitive to stress. Nat Genet 2000, 24: 410–414. 10.1038/74263
Muller MB, Holsboer F: Mice with mutations in the HPA-system as models for symptoms of depression. Biol Psychiatry 2006, 59: 1104–1115. 10.1016/j.biopsych.2006.02.008
Preil J, Muller MB, Gesing A, Reul JM, Sillaber I, van Gaalen MM, Landgrebe J, Holsboer F, Stenzel-Poore M, Wurst W: Regulation of the hypothalamic-pituitary-adrenocortical system in mice deficient for CRH receptors 1 and 2. Endocrinology 2001, 142: 4946–4955. 10.1210/en.142.11.4946
Valdez GR, Zorrilla EP, Rivier J, Vale WW, Koob GF: Locomotor suppressive and anxiolytic-like effects of urocortin 3, a highly selective type 2 corticotropin-releasing factor agonist. Brain Res 2003, 980: 206–212. 10.1016/S0006-8993(03)02971-8
Coste SC, Kesterson RA, Heldwein KA, Stevens SL, Heard AD, Hollis JH, Murray SE, Hill JK, Pantely GA, Hohimer AR, et al.: Abnormal adaptations to stress and impaired cardiovascular function in mice lacking corticotropin-releasing hormone receptor-2. Nat Genet 2000, 24: 403–409. 10.1038/74255
Zhao Y, Valdez GR, Fekete EM, Rivier JE, Vale WW, Rice KC, Weiss F, Zorrilla EP: Subtype-selective corticotropin-releasing factor receptor agonists exert contrasting, but not opposite, effects on anxiety-related behavior in rats. J Pharmacol Exp Ther 2007, 323: 846–854. 10.1124/jpet.107.123208
Henry B, Vale W, Markou A: The effect of lateral septum corticotropin-releasing factor receptor 2 activation on anxiety is modulated by stress. J Neurosci 2006, 26: 9142–9152. 10.1523/JNEUROSCI.1494-06.2006
Hammack SE, Schmid MJ, LoPresti ML, Der-Avakian A, Pellymounter MA, Foster AC, Watkins LR, Maier SF: Corticotropin releasing hormone type 2 receptors in the dorsal raphe nucleus mediate the behavioral consequences of uncontrollable stress. J Neurosci 2003, 23: 1019–1025.
Commons KG, Valentino RJ: Cellular basis for the effects of substance P in the periaqueductal gray and dorsal raphe nucleus. J Comp Neurol 2002, 447: 82–97. 10.1002/cne.10228
Gold PW, Chrousos GP: Clinical studies with corticotropin releasing factor: implications for the diagnosis and pathophysiology of depression, Cushing′s disease, and adrenal insufficiency. Psychoneuroendocrinology 1985, 10: 401–419. 10.1016/0306-4530(85)90080-0
Gold PW, Chrousos GP: Organization of the stress system and its dysregulation in melancholic and atypical depression: high vs low CRH/NE states. Mol Psychiatry 2002, 7: 254–275. 10.1038/sj.mp.4001032
Bremner JD, Licinio J, Darnell A, Krystal JH, Owens MJ, Southwick SM, Nemeroff CB, Charney DS: Elevated CSF corticotropin-releasing factor concentrations in posttraumatic stress disorder. Am J Psychiatry 1997, 154: 624–629.
Nemeroff CB, Widerlov E, Bissette G, Walleus H, Karlsson I, Eklund K, Kilts CD, Loosen PT, Vale W: Elevated concentrations of CSF corticotropin-releasing factor-like immunoreactivity in depressed patients. Science 1984, 226: 1342–1344. 10.1126/science.6334362
Baker DG, West SA, Nicholson WE, Ekhator NN, Kasckow JW, Hill KK, Bruce AB, Orth DN, Geracioti TD Jr: Serial CSF corticotropin-releasing hormone levels and adrenocortical activity in combat veterans with posttraumatic stress disorder. Am J Psychiatry 1999, 156: 585–588.
Sautter FJ, Bissette G, Wiley J, Manguno-Mire G, Schoenbachler B, Myers L, Johnson JE, Cerbone A, Malaspina D: Corticotropin-releasing factor in posttraumatic stress disorder (PTSD) with secondary psychotic symptoms, nonpsychotic PTSD, and healthy control subjects. Biol Psychiatry 2003, 54: 1382–1388. 10.1016/S0006-3223(03)00571-7
Banki CM, Karmacsi L, Bissette G, Nemeroff CB: Cerebrospinal fluid neuropeptides in mood disorder and dementia. J Affect Disord 1992, 25: 39–45. 10.1016/0165-0327(92)90091-J
Raadsheer FC, Hoogendijk WJ, Stam FC, Tilders FJ, Swaab DF: Increased numbers of corticotropin-releasing hormone expressing neurons in the hypothalamic paraventricular nucleus of depressed patients. Neuroendocrinology 1994, 60: 436–444. 10.1159/000126778
Wang SS, Kamphuis W, Huitinga I, Zhou JN, Swaab DF: Gene expression analysis in the human hypothalamus in depression by laser microdissection and real-time PCR: the presence of multiple receptor imbalances. Mol Psychiatry 2008, 13: 786–799. 741 10.1038/mp.2008.38
Austin MC, Janosky JE, Murphy HA: Increased corticotropin-releasing hormone immunoreactivity in monoamine-containing pontine nuclei of depressed suicide men. Mol Psychiatry 2003, 8: 324–332. 10.1038/sj.mp.4001250
Bissette G, Klimek V, Pan J, Stockmeier C, Ordway G: Elevated concentrations of CRF in the locus coeruleus of depressed subjects. Neuropsychopharmacology 2003, 28: 1328–1335. 10.1038/sj.npp.1300191
Owens MJ, Bissette G, Nemeroff CB: Acute effects of alprazolam and adinazolam on the concentrations of corticotropin-releasing factor in the rat brain. Synapse 1989, 4: 196–202. 10.1002/syn.890040304
Owens MJ, Vargas MA, Knight DL, Nemeroff CB: The effects of alprazolam on corticotropin-releasing factor neurons in the rat brain: acute time course, chronic treatment and abrupt withdrawal. J Pharmacol Exp Ther 1991, 258: 349–356.
Heuser I, Bissette G, Dettling M, Schweiger U, Gotthardt U, Schmider J, Lammers CH, Nemeroff CB, Holsboer F: Cerebrospinal fluid concentrations of corticotropin-releasing hormone, vasopressin, and somatostatin in depressed patients and healthy controls: response to amitriptyline treatment. Depress Anxiety 1998, 8: 71–79.
De Bellis MD, Gold PW, Geracioti TD Jr, Listwak SJ, Kling MA: Association of fluoxetine treatment with reductions in CSF concentrations of corticotropin-releasing hormone and arginine vasopressin in patients with major depression. Am J Psychiatry 1993, 150: 656–657.
Nemeroff CB, Bissette G, Akil H, Fink M: Neuropeptide concentrations in the cerebrospinal fluid of depressed patients treated with electroconvulsive therapy. Corticotrophin-releasing factor, beta-endorphin and Somatostatin. Br J Psychiatry 1991, 158: 59–63. 10.1192/bjp.158.1.59
Keck ME, Kern N, Erhardt A, Unschuld PG, Ising M, Salyakina D, Muller MB, Knorr CC, Lieb R, Hohoff C, et al.: Combined effects of exonic polymorphisms in CRHR1 and AVPR1B genes in a case/control study for panic disorder. Am J Med Genet B Neuropsychiatr Genet 2008, 147B: 1196–1204. 10.1002/ajmg.b.30750
Amstadter AB, Nugent NR, Yang BZ, Miller A, Siburian R, Moorjani P, Haddad S, Basu A, Fagerness J, Saxe G, et al.: Corticotrophin-releasing hormone type 1 receptor gene (CRHR1) variants predict posttraumatic stress disorder onset and course in pediatric injury patients. Dis Markers 2011, 30: 89–99.
Wasserman D, Sokolowski M, Rozanov V, Wasserman J: The CRHR1 gene: a marker for suicidality in depressed males exposed to low stress. Genes Brain Behav 2008, 7: 14–19.
Liu Z, Zhu F, Wang G, Xiao Z, Wang H, Tang J, Wang X, Qiu D, Liu W, Cao Z, Li W: Association of corticotropin-releasing hormone receptor1 gene SNP and haplotype with major depression. Neurosci Lett 2006, 404: 358–362. 10.1016/j.neulet.2006.06.016
Polanczyk G, Caspi A, Williams B, Price TS, Danese A, Sugden K, Uher R, Poulton R, Moffitt TE: Protective effect of CRHR1 gene variants on the development of adult depression following childhood maltreatment: replication and extension. Arch Gen Psychiatry 2009, 66: 978–985. 10.1001/archgenpsychiatry.2009.114
Ishitobi Y, Nakayama S, Yamaguchi K, Kanehisa M, Higuma H, Maruyama Y, Ninomiya T, Okamoto S, Tanaka Y, Tsuru J, et al.: Association of CRHR1 and CRHR2 with major depressive disorder and panic disorder in a japanese population. Am J Med Genet B Neuropsychiatr Genet 2012, 159B: 429–436. 10.1002/ajmg.b.32046
Zobel AW, Nickel T, Kunzel HE, Ackl N, Sonntag A, Ising M, Holsboer F: Effects of the high-affinity corticotropin-releasing hormone receptor 1 antagonist R121919 in major depression: the first 20 patients treated. J Psychiatr Res 2000, 34: 171–181. 10.1016/S0022-3956(00)00016-9
Iwasaki-Sekino A, Mano-Otagiri A, Ohata H, Yamauchi N, Shibasaki T: Gender differences in corticotropin and corticosterone secretion and corticotropin-releasing factor mRNA expression in the paraventricular nucleus of the hypothalamus and the central nucleus of the amygdala in response to footshock stress or psychological stress in rats. Psychoneuroendocrinology 2009, 34: 226–237. 10.1016/j.psyneuen.2008.09.003
Sterrenburg L, Gaszner B, Boerrigter J, Santbergen L, Bramini M, Roubos EW, Peeters BW, Kozicz T: Sex-dependent and differential responses to acute restraint stress of corticotropin-releasing factor-producing neurons in the rat paraventricular nucleus, central amygdala, and bed nucleus of the stria terminalis. J Neurosci Res 2012, 90: 179–192. 10.1002/jnr.22737
Viau V, Bingham B, Davis J, Lee P, Wong M: Gender and puberty interact on the stress-induced activation of parvocellular neurosecretory neurons and corticotropin-releasing hormone messenger ribonucleic acid expression in the rat. Endocrinology 2005, 146: 137–146.
Duncko R, Kiss A, Skultetyova I, Rusnak M, Jezova D: Corticotropin-releasing hormone mRNA levels in response to chronic mild stress rise in male but not in female rats while tyrosine hydroxylase mRNA levels decrease in both sexes. Psychoneuroendocrinology 2001, 26: 77–89. 10.1016/S0306-4530(00)00040-8
Bale TL, Vale WW: Increased depression-like behaviors in corticotropin-releasing factor receptor-2-deficient mice: sexually dichotomous responses. J Neurosci 2003, 23: 5295–5301.
Weathington JM, Cooke BM: Corticotropin-releasing factor receptor binding in the amygdala changes across puberty in a Sex-specific manner. Endocrinology 2012, 153: 5701–5705. 10.1210/en.2012-1815
Lim MM, Nair HP, Young LJ: Species and sex differences in brain distribution of corticotropin-releasing factor receptor subtypes 1 and 2 in monogamous and promiscuous vole species. J Comp Neurol 2005, 487: 75–92. 10.1002/cne.20532
Sztainberg Y, Kuperman Y, Tsoory M, Lebow M, Chen A: The anxiolytic effect of environmental enrichment is mediated via amygdalar CRF receptor type 1. Mol Psychiatry 2010, 15: 905–917. 10.1038/mp.2009.151
Sandi C, Cordero MI, Ugolini A, Varea E, Caberlotto L, Large CH: Chronic stress-induced alterations in amygdala responsiveness and behavior–modulation by trait anxiety and corticotropin-releasing factor systems. Eur J Neurosci 2008, 28: 1836–1848. 10.1111/j.1460-9568.2008.06451.x
Maejima Y, Aoyama M, Ookawara S, Hirao A, Sugita S: Distribution of the androgen receptor in the diencephalon and the pituitary gland in goats: Co-localisation with corticotrophin releasing hormone, arginine vasopressin and corticotrophs. Vet J 2009, 181: 193–199. 10.1016/j.tvjl.2008.02.021
Shughrue PJ, Lane MV, Merchenthaler I: Comparative distribution of estrogen receptor-alpha and -beta mRNA in the rat central nervous system. J Comp Neurol 1997, 388: 507–525. 10.1002/(SICI)1096-9861(19971201)388:4<507::AID-CNE1>3.0.CO;2-6
Auger CJ, De Vries GJ: Progestin receptor immunoreactivity within steroid-responsive vasopressin-immunoreactive cells in the male and female rat brain. J Neuroendocrinol 2002, 14: 561–567. 10.1046/j.1365-2826.2002.00809.x
Zuloaga DG, Yahn SL, Pang Y, Quihuis AM, Oyola MG, Reyna A, Thomas P, Handa RJ, Mani SK: Distribution and estrogen regulation of membrane progesterone receptor-beta in the female rat brain. Endocrinology 2012, 153: 4432–4443. 10.1210/en.2012-1469
Simerly RB, Chang C, Muramatsu M, Swanson LW: Distribution of androgen and estrogen receptor mRNA-containing cells in the rat brain: an in situ hybridization study. J Comp Neurol 1990, 294: 76–95. 10.1002/cne.902940107
Weiser MJ, Goel N, Sandau US, Bale TL, Handa RJ: Androgen regulation of corticotropin-releasing hormone receptor 2 (CRHR2) mRNA expression and receptor binding in the rat brain. Exp Neurol 2008, 214: 62–68. 10.1016/j.expneurol.2008.07.013
Karteris E, Markovic D, Chen J, Hillhouse EW, Grammatopoulos DK: Identification of a novel corticotropin-releasing hormone type 1beta-like receptor variant lacking exon 13 in human pregnant myometrium regulated by estradiol-17beta and progesterone. Endocrinology 2010, 151: 4959–4968. 10.1210/en.2010-0622
Parham KL, Zervou S, Karteris E, Catalano RD, Old RW, Hillhouse EW: Promoter analysis of human corticotropin-releasing factor (CRF) type 1 receptor and regulation by CRF and urocortin. Endocrinology 2004, 145: 3971–3983. 10.1210/en.2004-0194
Catalano RD, Kyriakou T, Chen J, Easton A, Hillhouse EW: Regulation of corticotropin-releasing hormone type 2 receptors by multiple promoters and alternative splicing: identification of multiple splice variants. Mol Endocrinol 2003, 17: 395–410. 10.1210/me.2002-0302
Bangasser DA, Curtis A, Reyes BA, Bethea TT, Parastatidis I, Ischiropoulos H, Van Bockstaele EJ, Valentino RJ: Sex differences in corticotropin-releasing factor receptor signaling and trafficking: potential role in female vulnerability to stress-related psychopathology. Mol Psychiatry 2010,15(877):896–904.
Valentino RJ, Bangasser D, Van Bockstaele E: Sex biased stress signaling. Mol Pharmacol 2012. Epub ahead of print
Bangasser DA, Valentino RJ: Sex differences in molecular and cellular substrates of stress. Cell Mol Neurobiol 2012, 32: 709–723. 10.1007/s10571-012-9824-4
Hillhouse EW, Grammatopoulos DK: The molecular mechanisms underlying the regulation of the biological activity of corticotropin-releasing hormone receptors: implications for physiology and pathophysiology. Endocr Rev 2006, 27: 260–286. 10.1210/er.2005-0034
Grammatopoulos DK, Randeva HS, Levine MA, Kanellopoulou KA, Hillhouse EW: Rat cerebral cortex corticotropin-releasing hormone receptors: evidence for receptor coupling to multiple G-proteins. J Neurochem 2001, 76: 509–519. 10.1046/j.1471-4159.2001.00067.x
Chen FM, Bilezikjian LM, Perrin MH, Rivier J, Vale W: Corticotropin releasing factor receptor-mediated stimulation of adenylate cyclase activity in the rat brain. Brain Res 1986, 381: 49–57. 10.1016/0006-8993(86)90688-8
Battaglia G, Webster EL, De Souza EB: Characterization of corticotropin-releasing factor receptor-mediated adenylate cyclase activity in the rat central nervous system. Synapse 1987, 1: 572–581. 10.1002/syn.890010610
Keil MF, Briassoulis G, Gokarn N, Nesterova M, Wu TJ, Stratakis CA: Anxiety phenotype in mice that overexpress protein kinase a. Psychoneuroendocrinology 2012, 37: 836–843. 10.1016/j.psyneuen.2011.09.016
Favilla C, Abel T, Kelly MP: Chronic galphas signaling in the striatum increases anxiety-related behaviors independent of developmental effects. J Neurosci 2008, 28: 13952–13956. 10.1523/JNEUROSCI.4986-08.2008
Page ME, Berridge CW, Foote SL, Valentino RJ: Corticotropin-releasing factor in the locus coeruleus mediates EEG activation associated with hypotensive stress. Neurosci Lett 1993, 164: 81–84. 10.1016/0304-3940(93)90862-F
Snyder K, Wang WW, Han R, McFadden K, Valentino RJ: Corticotropin-releasing factor in the norepinephrine nucleus, locus coeruleus. Neuropsychopharmacology: Facilitates Behavioral Flexibility; 2011.
Curtis AL, Lechner SM, Pavcovich LA, Valentino RJ: Activation of the locus coeruleus noradrenergic system by intracoerulear microinfusion of corticotropin-releasing factor: effects on discharge rate, cortical norepinephrine levels and cortical electroencephalographic activity. J Pharmacol Exp Ther 1997, 281: 163–172.
Curtis AL, Bethea T, Valentino RJ: Sexually dimorphic responses of the brain norepinephrine system to stress and corticotropin-releasing factor. Neuropsychopharmacology 2006, 31: 544–554. 10.1038/sj.npp.1300875
Jedema HP, Grace AA: Corticotropin-releasing hormone directly activates noradrenergic neurons of the locus ceruleus recorded in vitro. J Neurosci 2004, 24: 9703–9713. 10.1523/JNEUROSCI.2830-04.2004
Valentino RJ, Van Bockstaele E: Functional interactions between stress neuromediator and the locus coeruleur-noradrenaline system. In In Handbook of Stress and the Brain. Edited by: Steckler TK N. The Netherlands: Elsevier; 2005:465–486.
Hauger RL, Smith RD, Braun S, Dautzenberg FM, Catt KJ: Rapid agonist-induced phosphorylation of the human CRF receptor, type 1: a potential mechanism for homologous desensitization. Biochem Biophys Res Commun 2000, 268: 572–576. 10.1006/bbrc.2000.2183
Teli T, Markovic D, Levine MA, Hillhouse EW, Grammatopoulos DK: Regulation of corticotropin-releasing hormone receptor type 1alpha signaling: structural determinants for G protein-coupled receptor kinase-mediated phosphorylation and agonist-mediated desensitization. Mol Endocrinol 2005, 19: 474–490.
Reyes BA, Fox K, Valentino RJ, Van Bockstaele EJ: Agonist-induced internalization of corticotropin-releasing factor receptors in noradrenergic neurons of the rat locus coeruleus. Eur J Neurosci 2006, 23: 2991–2998. 10.1111/j.1460-9568.2006.04820.x
Reyes BA, Valentino RJ, Van Bockstaele EJ: Stress-induced intracellular trafficking of corticotropin-releasing factor receptors in rat locus coeruleus neurons. Endocrinology 2008, 149: 122–130.
Holmes KD, Babwah AV, Dale LB, Poulter MO, Ferguson SS: Differential regulation of corticotropin releasing factor 1alpha receptor endocytosis and trafficking by beta-arrestins and Rab GTPases. J Neurochem 2006, 96: 934–949. 10.1111/j.1471-4159.2005.03603.x
Oakley RH, Olivares-Reyes JA, Hudson CC, Flores-Vega F, Dautzenberg FM, Hauger RL: Carboxyl-terminal and intracellular loop sites for CRF1 receptor phosphorylation and beta-arrestin-2 recruitment: a mechanism regulating stress and anxiety responses. Am J Physiol Regul Integr Comp Physiol 2007, 293: R209–222. 10.1152/ajpregu.00099.2006
Stenzel-Poore MP, Cameron VA, Vaughan J, Sawchenko PE, Vale W: Development of Cushing′s syndrome in corticotropin-releasing factor transgenic mice. Endocrinology 1992, 130: 3378–3386. 10.1210/en.130.6.3378
Stenzel-Poore MP, Heinrichs SC, Rivest S, Koob GF, Vale WW: Overproduction of corticotropin-releasing factor in transgenic mice: a genetic model of anxiogenic behavior. J Neurosci 1994, 14: 2579–2584.
van Gaalen MM, Stenzel-Poore MP, Holsboer F, Steckler T: Effects of transgenic overproduction of CRH on anxiety-like behaviour. Eur J Neurosci 2002, 15: 2007–2015. 10.1046/j.1460-9568.2002.02040.x
Bangasser DA, Reyes BA, Piel D, Garachh V, Zhang XY, Plona ZM, Van Bockstaele EJ, Beck SG, Valentino RJ: Increased vulnerability of the brain norepinephrine system of females to corticotropin-releasing factor overexpression. Mol Psychiatry 2012. Epub ahead of print
O′Donnell T, Hegadoren KM, Coupland NC: Noradrenergic mechanisms in the pathophysiology of post-traumatic stress disorder. Neuropsychobiology 2004, 50: 273–283. 10.1159/000080952
Southwick SM, Bremner JD, Rasmusson A, Morgan CA 3rd, Arnsten A, Charney DS: Role of norepinephrine in the pathophysiology and treatment of posttraumatic stress disorder. Biol Psychiatry 1999, 46: 1192–1204. 10.1016/S0006-3223(99)00219-X
Gold PW, Chrousos GP: The endocrinology of melancholic and atypical depression: relation to neurocircuitry and somatic consequences. Proc Assoc Am Physicians 1999, 111: 22–34. 10.1046/j.1525-1381.1999.09423.x
Linton EA, Lowry PJ: Comparison of a specific two-site immunoradiometric assay with radioimmunoassay for rat/human CRF-41. Regul Pept 1986, 14: 69–84. 10.1016/0167-0115(86)90206-5
Orth DN, Mount CD: Specific high-affinity binding protein for human corticotropin-releasing hormone in normal human plasma. Biochem Biophys Res Commun 1987, 143: 411–417. 10.1016/0006-291X(87)91369-6
Westphal NJ, Seasholtz AF: CRH-BP: the regulation and function of a phylogenetically conserved binding protein. Front Biosci 2006, 11: 1878–1891. 10.2741/1931
Potter E, Behan DP, Linton EA, Lowry PJ, Sawchenko PE, Vale WW: The central distribution of a corticotropin-releasing factor (CRF)-binding protein predicts multiple sites and modes of interaction with CRF. Proc Natl Acad Sci USA 1992, 89: 4192–4196. 10.1073/pnas.89.9.4192
Potter E, Behan DP, Fischer WH, Linton EA, Lowry PJ, Vale WW: Cloning and characterization of the cDNAs for human and rat corticotropin releasing factor-binding proteins. Nature 1991, 349: 423–426. 10.1038/349423a0
Herringa RJ, Nanda SA, Hsu DT, Roseboom PH, Kalin NH: The effects of acute stress on the regulation of central and basolateral amygdala CRF-binding protein gene expression. Brain Res Mol Brain Res 2004, 131: 17–25. 10.1016/j.molbrainres.2004.08.005
Lombardo KA, Herringa RJ, Balachandran JS, Hsu DT, Bakshi VP, Roseboom PH, Kalin NH: Effects of acute and repeated restraint stress on corticotropin-releasing hormone binding protein mRNA in rat amygdala and dorsal hippocampus. Neurosci Lett 2001, 302: 81–84. 10.1016/S0304-3940(01)01680-9
Herringa RJ, Mackenrodt DB, Barlow JD, Roseboom PH, Nanda SA, Kalin NH: Corticotropin-releasing factor (CRF), but not corticosterone, increases basolateral amygdala CRF-binding protein. Brain Res 2006, 1083: 21–28. 10.1016/j.brainres.2006.01.122
Ungless MA, Singh V, Crowder TL, Yaka R, Ron D, Bonci A: Corticotropin-releasing factor requires CRF binding protein to potentiate NMDA receptors via CRF receptor 2 in dopamine neurons. Neuron 2003, 39: 401–407. 10.1016/S0896-6273(03)00461-6
Speert DB, SJ MC, Seasholtz AF: Sexually dimorphic expression of corticotropin-releasing hormone-binding protein in the mouse pituitary. Endocrinology 2002, 143: 4730–4741. 10.1210/en.2002-220556
Deuschle M, Schweiger U, Weber B, Gotthardt U, Korner A, Schmider J, Standhardt H, Lammers CH, Heuser I: Diurnal activity and pulsatility of the hypothalamus-pituitary-adrenal system in male depressed patients and healthy controls. J Clin Endocrinol Metab 1997, 82: 234–238. 10.1210/jc.82.1.234
Holsboer F: Stress, hypercortisolism and corticosteroid receptors in depression: implications for therapy. J Affect Disorders 2001, 62: 77–91. 10.1016/S0165-0327(00)00352-9
Barden N: Implication of the hypothalamic-pituitary-adrenal axis in the physiopathology of depression. J Psychiatry Neurosci 2004, 29: 185–193.
Board F, Persky H, Hamburg DA: Psychological stress and endocrine functions; blood levels of adrenocortical and thyroid hormones in acutely disturbed patients. Psychosom Med 1956, 18: 324–333.
Nestler EJ, Barrot M, DiLeone RJ, Eisch AJ, Gold SJ, Monteggia LM: Neurobiology of depression. Neuron 2002, 34: 13–25. 10.1016/S0896-6273(02)00653-0
Strohle A, Holsboer F: Stress responsive neurohormones in depression and anxiety. Pharmacopsychiatry 2003,36(Suppl 3):S207–214.
Pariante CM, Miller AH: Glucocorticoid receptors in major depression: relevance to pathophysiology and treatment. Biol Psychiatry 2001, 49: 391–404. 10.1016/S0006-3223(00)01088-X
Sapolsky RM, Romero LM, Munck AU: How do glucocorticoids influence stress responses? integrating permissive, suppressive, stimulatory, and preparative actions. Endocr Rev 2000, 21: 55–89. 10.1210/er.21.1.55
De Kloet ER, Vreugdenhil E, Oitzl MS, Joels M: Brain corticosteroid receptor balance in health and disease. Endocr Rev 1998, 19: 269–301. 10.1210/er.19.3.269
Reul JM, de Kloet ER: Two receptor systems for corticosterone in rat brain: microdistribution and differential occupation. Endocrinology 1985, 117: 2505–2511. 10.1210/endo-117-6-2505
Veldhuis HD, Van Koppen C, Van Ittersum M, De Kloet ER: Specificity of the adrenal steroid receptor system in rat hippocampus. Endocrinology 1982, 110: 2044–2051. 10.1210/endo-110-6-2044
Spencer RL, Young EA, Choo PH, McEwen BS: Adrenal steroid type I and type II receptor binding: estimates of in vivo receptor number, occupancy, and activation with varying level of steroid. Brain Res 1990, 514: 37–48. 10.1016/0006-8993(90)90433-C
De Kloet ER, Reul JM: Feedback action and tonic influence of corticosteroids on brain function: a concept arising from the heterogeneity of brain receptor systems. Psychoneuroendocrinology 1987, 12: 83–105. 10.1016/0306-4530(87)90040-0
Tasker JG, Herman JP: Mechanisms of rapid glucocorticoid feedback inhibition of the hypothalamic-pituitary-adrenal axis. Stress 2011, 14: 398–406.
Di S, Malcher-Lopes R, Halmos KC, Tasker JG: Nongenomic glucocorticoid inhibition via endocannabinoid release in the hypothalamus: a fast feedback mechanism. J Neurosci 2003, 23: 4850–4857.
Charron J, Drouin J: Glucocorticoid inhibition of transcription from episomal proopiomelanocortin gene promoter. Proc Natl Acad Sci USA 1986, 83: 8903–8907. 10.1073/pnas.83.23.8903
Newton R: Molecular mechanisms of glucocorticoid action: what is important? Thorax 2000, 55: 603–613. 10.1136/thorax.55.7.603
Jingami H, Matsukura S, Numa S, Imura H: Effects of adrenalectomy and dexamethasone administration on the level of prepro-corticotropin-releasing factor messenger ribonucleic acid (mRNA) in the hypothalamus and adrenocorticotropin/beta-lipotropin precursor mRNA in the pituitary in rats. Endocrinology 1985, 117: 1314–1320. 10.1210/endo-117-4-1314
Kageyama K, Suda T: Regulatory mechanisms underlying corticotropin-releasing factor gene expression in the hypothalamus. Endocr J 2009, 56: 335–344. 10.1507/endocrj.K09E-075
Malkoski SP, Dorin RI: Composite glucocorticoid regulation at a functionally defined negative glucocorticoid response element of the human corticotropin-releasing hormone gene. Mol Endocrinol 1999, 13: 1629–1644. 10.1210/me.13.10.1629
Pozzoli G, Bilezikjian LM, Perrin MH, Blount AL, Vale WW: Corticotropin-releasing factor (CRF) and glucocorticoids modulate the expression of type 1 CRF receptor messenger ribonucleic acid in rat anterior pituitary cell cultures. Endocrinology 1996, 137: 65–71. 10.1210/en.137.1.65
Arana GW: Dexamethasone suppression test in the diagnosis of depression. JAMA 1991, 265: 2253–2254.
Arana GW, Baldessarini RJ, Ornsteen M: The dexamethasone suppression test for diagnosis and prognosis in psychiatry. Commentary and review. Arch Gen Psychiatry 1985, 42: 1193–1204. 10.1001/archpsyc.1985.01790350067012
Carroll BJ, Feinberg M, Greden JF, Tarika J, Albala AA, Haskett RF, James NM, Kronfol Z, Lohr N, Steiner M, et al.: A specific laboratory test for the diagnosis of melancholia. Standardization, validation, and clinical utility. Arch Gen Psychiatry 1981, 38: 15–22. 10.1001/archpsyc.1981.01780260017001
Holsboer F: The corticosteroid receptor hypothesis of depression. Neuropsychopharmacology 2000, 23: 477–501. 10.1016/S0893-133X(00)00159-7
Reul JM, van den Bosch FR, de Kloet ER: Relative occupation of type-I and type-II corticosteroid receptors in rat brain following stress and dexamethasone treatment: functional implications. J Endocrinol 1987, 115: 459–467. 10.1677/joe.0.1150459
Watson S, Gallagher P, Smith MS, Ferrier IN, Young AH: The dex/CRH test–is it better than the DST? Psychoneuroendocrinology 2006, 31: 889–894. 10.1016/j.psyneuen.2006.03.001
Heuser I, Yassouridis A, Holsboer F: The combined dexamethasone/CRH test: a refined laboratory test for psychiatric disorders. J Psychiatr Res 1994, 28: 341–356. 10.1016/0022-3956(94)90017-5
Ising M, Kunzel HE, Binder EB, Nickel T, Modell S, Holsboer F: The combined dexamethasone/CRH test as a potential surrogate marker in depression. Prog Neuropsychopharmacol Biol Psychiatry 2005, 29: 1085–1093. 10.1016/j.pnpbp.2005.03.014
Arborelius L, Owens MJ, Plotsky PM, Nemeroff CB: The role of corticotropin-releasing factor in depression and anxiety disorders. J Endocrinol 1999, 160: 1–12. 10.1677/joe.0.1600001
Pereira AM, Tiemensma J, Romijn JA: Neuropsychiatric disorders in Cushing′s syndrome. Neuroendocrinology 2010,92(Suppl 1):65–70.
Cohen SI: Cushing′s Syndrome: a psychiatric study of 29 patients. Br J Psychiatry 1980, 136: 120–124. 10.1192/bjp.136.2.120
Kitay JI: Sex differences in adrenal cortical secretion in the rat. Endocrinology 1961, 68: 818–824. 10.1210/endo-68-5-818
Weinstock M, Razin M, Schorer-Apelbaum D, Men D, McCarty R: Gender differences in sympathoadrenal activity in rats at rest and in response to footshock stress. Int J Dev Neurosci 1998, 16: 289–295. 10.1016/S0736-5748(98)00021-5
Handa RJ, Burgess LH, Kerr JE, O′Keefe JA: Gonadal steroid hormone receptors and sex differences in the hypothalamo-pituitary-adrenal axis. Horm Behav 1994, 28: 464–476. 10.1006/hbeh.1994.1044
Rivier C: Gender, sex steroids, corticotropin-releasing factor, nitric oxide, and the HPA response to stress. Pharmacol Biochem Behav 1999, 64: 739–751.
Heinsbroek RP, Van Haaren F, Feenstra MG, Endert E, Van de Poll NE: Sex- and time-dependent changes in neurochemical and hormonal variables induced by predictable and unpredictable footshock. Physiol Behav 1991, 49: 1251–1256. 10.1016/0031-9384(91)90359-V
Seale JV, Wood SA, Atkinson HC, Harbuz MS, Lightman SL: Gonadal steroid replacement reverses gonadectomy-induced changes in the corticosterone pulse profile and stress-induced hypothalamic-pituitary-adrenal axis activity of male and female rats. J Neuroendocrinol 2004, 16: 989–998. 10.1111/j.1365-2826.2004.01258.x
Weathington JM, Arnold AR, Cooke BM: Juvenile social subjugation induces a sex-specific pattern of anxiety and depression-like behaviors in adult rats. Horm Behav 2012, 61: 91–99. 10.1016/j.yhbeh.2011.10.008
Uhart M, Chong RY, Oswald L, Lin PI, Wand GS: Gender differences in hypothalamic-pituitary-adrenal (HPA) axis reactivity. Psychoneuroendocrinology 2006, 31: 642–652. 10.1016/j.psyneuen.2006.02.003
Kirschbaum C, Kudielka BM, Gaab J, Schommer NC, Hellhammer DH: Impact of gender, menstrual cycle phase, and oral contraceptives on the activity of the hypothalamus-pituitary-adrenal axis. Psychosom Med 1999, 61: 154–162.
Kirschbaum C, Klauer T, Filipp SH, Hellhammer DH: Sex-specific effects of social support on cortisol and subjective responses to acute psychological stress. Psychosom Med 1995, 57: 23–31.
Seeman TE, Singer B, Wilkinson CW, McEwen B: Gender differences in age-related changes in HPA axis reactivity. Psychoneuroendocrinology 2001, 26: 225–240. 10.1016/S0306-4530(00)00043-3
Kirschbaum C, Wust S, Hellhammer D: Consistent sex differences in cortisol responses to psychological stress. Psychosom Med 1992, 54: 648–657.
Kudielka BM, Kirschbaum C: Sex differences in HPA axis responses to stress: a review. Biol Psychol 2005, 69: 113–132. 10.1016/j.biopsycho.2004.11.009
Gallucci WT, Baum A, Laue L, Rabin DS, Chrousos GP, Gold PW, Kling MA: Sex differences in sensitivity of the hypothalamic-pituitary-adrenal axis. Health Psychol 1993, 12: 420–425.
Heuser IJ, Gotthardt U, Schweiger U, Schmider J, Lammers CH, Dettling M, Holsboer F: Age-associated changes of pituitary-adrenocortical hormone regulation in humans: importance of gender. Neurobiol Aging 1994, 15: 227–231. 10.1016/0197-4580(94)90117-1
Friedmann B, Kindermann W: Energy metabolism and regulatory hormones in women and men during endurance exercise. Eur J Appl Physiol Occup Physiol 1989, 59: 1–9. 10.1007/BF02396572
Collins A, Frankenhaeuser M: Stress responses in male and female engineering students. J Human Stress 1978, 4: 43–48.
Paris JJ, Franco C, Sodano R, Freidenberg B, Gordis E, Anderson DA, Forsyth JP, Wulfert E, Frye CA: Sex differences in salivary cortisol in response to acute stressors among healthy participants, in recreational or pathological gamblers, and in those with posttraumatic stress disorder. Horm Behav 2010, 57: 35–45. 10.1016/j.yhbeh.2009.06.003
Desbonnet L, Garrett L, Daly E, McDermott KW, Dinan TG: Sexually dimorphic effects of maternal separation stress on corticotrophin-releasing factor and vasopressin systems in the adult rat brain. Int J Dev Neurosci 2008, 26: 259–268. 10.1016/j.ijdevneu.2008.02.004
Viau V, Meaney MJ: Variations in the hypothalamic-pituitary-adrenal response to stress during the estrous cycle in the rat. Endocrinology 1991, 129: 2503–2511. 10.1210/endo-129-5-2503
Burgess LH, Handa RJ: Chronic estrogen-induced alterations in adrenocorticotropin and corticosterone secretion, and glucocorticoid receptor-mediated functions in female rats. Endocrinology 1992, 131: 1261–1269. 10.1210/en.131.3.1261
Turner BB: Sex difference in glucocorticoid binding in rat pituitary is estrogen dependent. Life Sci 1990, 46: 1399–1406. 10.1016/0024-3205(90)90340-W
Turner BB, Weaver DA: Sexual dimorphism of glucocorticoid binding in rat brain. Brain Res 1985, 343: 16–23. 10.1016/0006-8993(85)91153-9
Krishnan AV, Swami S, Feldman D: Estradiol inhibits glucocorticoid receptor expression and induces glucocorticoid resistance in MCF-7 human breast cancer cells. J Steroid Biochem Mol Biol 2001, 77: 29–37. 10.1016/S0960-0760(01)00030-9
Burgess LH, Handa RJ: Estrogen-induced alterations in the regulation of mineralocorticoid and glucocorticoid receptor messenger RNA expression in the female rat anterior pituitary gland and brain. Mol Cell Neurosci 1993, 4: 191–198. 10.1006/mcne.1993.1023
Bourke CH, Raees MQ, Malviya S, Bradburn CA, Binder EB, Neigh GN: Glucocorticoid sensitizers Bag1 and ppid are regulated by adolescent stress in a sex-dependent manner. Psychoneuroendocrinology 2012, 38: 84–93.
Grad I, Picard D: The glucocorticoid responses are shaped by molecular chaperones. Mol Cell Endocrinol 2007, 275: 2–12. 10.1016/j.mce.2007.05.018
Pratt WB, Morishima Y, Murphy M, Harrell M: Chaperoning of glucocorticoid receptors. Handb Exp Pharmacol 2006, 172: 111–138. 10.1007/3-540-29717-0_5
Bourke CH, Harrell CS, Neigh GN: Stress-induced sex differences: adaptations mediated by the glucocorticoid receptor. Horm Behav 2012, 62: 210–218. 10.1016/j.yhbeh.2012.02.024
Binder EB: The role of FKBP5, a co-chaperone of the glucocorticoid receptor in the pathogenesis and therapy of affective and anxiety disorders. Psychoneuroendocrinology 2009,34(Suppl 1):S186–195.
Schmidt U, Wochnik GM, Rosenhagen MC, Young JC, Hartl FU, Holsboer F, Rein T: Essential role of the unusual DNA-binding motif of BAG-1 for inhibition of the glucocorticoid receptor. J Biol Chem 2003, 278: 4926–4931. 10.1074/jbc.M212000200
Kanelakis KC, Morishima Y, Dittmar KD, Galigniana MD, Takayama S, Reed JC, Pratt WB: Differential effects of the hsp70-binding protein BAG-1 on glucocorticoid receptor folding by the hsp90-based chaperone machinery. J Biol Chem 1999, 274: 34134–34140. 10.1074/jbc.274.48.34134
Uht RM, Anderson CM, Webb P, Kushner PJ: Transcriptional activities of estrogen and glucocorticoid receptors are functionally integrated at the AP-1 response element. Endocrinology 1997, 138: 2900–2908. 10.1210/en.138.7.2900
Kontula K, Paavonen T, Luukkainen T, Andersson LC: Binding of progestins to the glucocorticoid receptor. Correlation to their glucocorticoid-like effects on in vitro functions of human mononuclear leukocytes. Biochem Pharmacol 1983, 32: 1511–1518. 10.1016/0006-2952(83)90474-4
Weiser MJ, Handa RJ: Estrogen impairs glucocorticoid dependent negative feedback on the hypothalamic-pituitary-adrenal axis via estrogen receptor alpha within the hypothalamus. Neuroscience 2009, 159: 883–895. 10.1016/j.neuroscience.2008.12.058
Million M, Grigoriadis DE, Sullivan S, Crowe PD, McRoberts JA, Zhou H, Saunders PR, Maillot C, Mayer EA, Tache Y: A novel water-soluble selective CRF1 receptor antagonist, NBI 35965, blunts stress-induced visceral hyperalgesia and colonic motor function in rats. Brain Res 2003, 985: 32–42. 10.1016/S0006-8993(03)03027-0
Nemeroff CB, Vale WW: The neurobiology of depression: inroads to treatment and new drug discovery. J Clin Psychiatry 2005,66(Suppl 7):5–13.
Kehne JH: The CRF1 receptor, a novel target for the treatment of depression, anxiety, and stress-related disorders. CNS Neurol Disord Drug Targets 2007, 6: 163–182. 10.2174/187152707780619344
Beery AK, Zucker I: Sex bias in neuroscience and biomedical research. Neurosci Biobehav Rev 2011, 35: 565–572. 10.1016/j.neubiorev.2010.07.002
Gaub M, Carlson CL: Gender differences in ADHD: a meta-analysis and critical review. J Am Acad Child Adolesc Psychiatry 1997, 36: 1036–1045. 10.1097/00004583-199708000-00011
Ramtekkar UP, Reiersen AM, Todorov AA, Todd RD: Sex and age differences in attention-deficit/hyperactivity disorder symptoms and diagnoses: implications for DSM-V and ICD-11. J Am Acad Child Adolesc Psychiatry 2010, 49: 217–228. e211–213
Acknowledgements
The author acknowledges the support of PHS grant MH092438. Special thanks to D. E. Waxler for comments on the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The author has no competing interests.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Bangasser, D.A. Sex differences in stress-related receptors: "micro" differences with "macro" implications for mood and anxiety disorders. Biol Sex Differ 4, 2 (2013). https://doi.org/10.1186/2042-6410-4-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/2042-6410-4-2