
Abstract
Bloom Syndrome (BS) is an autosomal recessive disorder due to mutation in Bloom helicase (referred in literature either as BLM helicase or BLM). Patients with BS are predisposed to almost all forms of cancer. BS patients are even today diagnosed in the clinics by hyper-recombination phenotype that is manifested by high rates of Sister Chromatid Exchange. The function of BLM as a helicase and its role during the regulation of homologous recombination (HR) is well characterized. However in the last few years the role of BLM as a DNA damage sensor has been revealed. For example, it has been demonstrated that BLM can stimulate the ATPase and chromatin remodeling activities of RAD54 in vitro. This indicates that BLM may increase the accessibility of the sensor proteins that recognize the lesion. Over the years evidence has accumulated that BLM is one of the earliest proteins that accumulates at the site of the lesion. Finally BLM also acts like a "molecular node" by integrating the upstream signals and acting as a bridge between the transducer and effector proteins (which again includes BLM itself), which in turn repair the DNA damage. Hence BLM seems to be a protein involved in multiple functions - all of which may together contribute to its reported role as a "caretaker tumor suppressor". In this review the recent literature documenting the upstream BLM functions has been elucidated and future directions indicated.
Similar content being viewed by others


Role of protein phosphorylation in response to DNA damage
Signal transduction during DNA damage response is mediated by two proximal sensory kinases, ATM (ataxia telangiectasia-mutated) and ATR (ATM-Rad3-related) [1, 2]. ATM and ATR initiate the signaling cascade via phosphorylation of its downstream checkpoint effector kinases, Chk1 and Chk2 [3]. ATR and Chk1 predominantly sense the damage in response to the stalling and subsequent collapse of the replication forks (called stalled replication forks), leading to replication arrest. On other hand ATM and Chk2 are involved in response to double strand breaks (DSBs), typically generated in vivo by exposure of cells to ionizing radiation (IR) or drugs like neocarzinostatin or bleomycin. Both stalled replication forks and DSBs lead to the generation of nuclear chromatinized foci called stalled replication foci and ionizing radiation-induced foci (IRIF), respectively. Replication arrest can also lead to the generation of DSBs [4], thereby hinting at partial common mechanistic framework in response to two common forms of DNA damage. ATM/ATR along with Chk1/Chk2, which accumulate at the chromatinized structures, are known to phosphorylate extensive network of downstream substrates in response to DNA damage [5].
The protein that was initially demonstrated to accumulate at the site of IRIF was the phosphorylated form of histone variant H2AX (γH2AX) [6] (Figure 1B). However subsequently it was observed that H2AX phosphorylation was dispensable for the initial recognition of DNA breaks and was instead proposed to concentrate proteins in the vicinity of DNA lesions [7]. Since then a growing number of proteins, containing either or both the phospho-protein binding motifs BRCA1 C-terminal (BRCT) and forkhead associated (FHA) domains, have been identified to be present both at IRIF and sites of stalled replication.

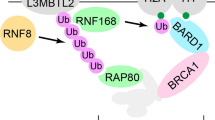
Proposed model for the functions of BLM helicase during DNA damage response. (A) DSBs (red line) are recognized after BLM and/or RAD51-stimulated RAD54-dependent chromatin remodeling. BLM affects chromatin organization by interacting with and regulating the function of CAF-1. On remodeled chromatin, BLM accumulates and helps in the optimal ATM activation and MRN complex accumulation. (B) MRN complex promotes H2AX phosphorylation (γH2AX, red dots) which recruits MDC1. MRN complex, stabilized on the DNA lesion by MDC1, promotes further accumulation of activated ATM. ATM phosphorylates MDC1 (purple dots), promoting the binding and recruitment of RNF8/Ubc13 complex, which catalyzes the Lys63-linked ubiquitylation of H2A and H2AX (yellow dots), causing a more accessible conformation of the chromatin. (C) RNF8/Ubc13 ubiquitylated histones recruits of RNF168. RNF168/Ubc13 attaches K63 linked polyubiquitin moieties to RNF8-ubiquitylated histones (yellow dots). (D) Poly-ubiquitylated histones recruits RAP80, which helps in the accumulation of Abraxas/BRCA1/BARD1 at DSBs. Constitutive methylation of histones H3 and H4 (blue dots) are probably exposed due to RNF168/Ubc13-dependent ubiquitylation. This results in the efficient recruitment of 53BP1 to the site of DNA damage. BLM again accumulates on the lesion in a 53BP1-dependent manner. Pro-recombinogenic proteins RAD51 and RAD54, interact with BLM, and accumulate at DSBs. (E) BLM functionally interacts with its partners like RAD51, RAD54, 53BP1 and p53 during HR. RAD51 binds to the single stranded DNA by displacing replication protein A (green dots). While BLM, 53BP1 and p53 have anti-recombinogenic property; BLM also has a pro-recombinogenic resection function in coordination with Exo1.
One such FHA-BRCT domain containing protein that accumulates at the sites of DNA damage is the mediator of DNA damage checkpoint 1 (MDC1) (Figure 1B). Recruitment of MDC1 occurs in a BRCT-dependent manner by binding to the C-terminal phosphorylated tail of H2AX [8]. MDC1 is required for intra-S phase DNA damage checkpoint [9–11]. At the IRIF, MDC1 acts like a molecular adaptor required for the localization of a number of other DNA damage response proteins including MRE11-RAD50-NBS1 (MRN) complex.
NBS1 (named for Nijmegen breakage syndrome; also called nibrin), a key member of the MRN complex, accumulates at the IRIF due to its own phospho-peptide binding FHA-BRCT domain [12] (Figure 1B). MDC1 stabilizes NBS1 at the sites of DNA damage, thereby promoting further accumulation of the MRN complex and activated ATM [13, 14]. Recent studies have revealed that phosphorylation of Ser-Asp-Thr-Asp (SDTD) repeats within MDC1 mediate its interaction with the FHA-BRCT domain of NBS1. This phospho-dependent interaction mediates the retention of MRN complex at the sites of DNA damage, thereby ensuring optimal S-phase checkpoint activation [15–18].
Role of protein ubiquitylation in response to DNA damage
The localization of conjugated ubiquitin at the sites of DNA damage had been demonstrated quite sometime back [19, 20]. Subsequently receptor associated protein 80 (RAP80) that contains tandem ubiquitin interacting motif (UIM) and binds to Lys63 and Lys6 polyubiquitin chains was identified at the IRIF [21, 22]. RAP80 targets Breast Cancer gene 1 (BRCA1) and BRCA1-associated Ring Domain 1 (BARD1) complex to the sites of DNA damage utilizing its association with Abraxas (ABRA1) [23–25]. The recruitment of BRCA1/BARD1 complex is required for its function in DNA damage resistance, intra-S and G2/M checkpoint control as well as DNA repair [25].
Interestingly around this time it was reported that the E3 ligase, Ring finger protein 8 (RNF8) assembles at the site of DSBs via the interaction of its FHA domain with the conserved Thr-Gln-any amino acid-Phe (TQXF) motif in MDC1. Phosphorylation of the MDC1 TQXF motifs by ATM and ATR is required for the interaction with RNF8 [26–28] (Figure 1B). Once recruited RNF8 and E2 conjugating enzyme Ubc13 catalyzes the Lys63-linked ubiquitylation of H2A and H2AX. This ubiquitylation promotes the transition of chromatin into a more accessible conformation leading to the recruitment of p53 binding protein 1 (53BP1) and RAP80/Abraxas/BRCA1 complex to the DNA damage foci [26–29].
A subsequent study reported that a patient with RIDDLE syndrome (radiosensitivity, immunodeficiency, dysmorphic features and learning difficulties) was defective in the recruitment of 53BP1 and BRCA1 to the DSBs [30], indicating the presence of another protein in the RNF8-dependent 53BP1 recruitment process. A siRNA screen using 53BP1 foci formation as the readout revealed an E3 ligase, Ring finger protein 168 (RNF168), as the gene mutated in RIDDLE syndrome [31, 32]. RNF168, which contains two motifs that interact with ubiquitin (MIU), is recruited to the to sites of DNA damage by binding to ubiquitinated H2A. The assembly of RNF168 at DSBs occurs in RNF8-dependent manner and leads to the amplification of RNF8-dependent substrate ubiquitylation (Figure 1C). RNF168/Ubc13 mediated histone poly-ubiquitylation recruits RAP80 to the sites of DNA damage, which in turn helps in the accumulation of Abraxas/BRCA1/BARD1 at DSBs (Figure 1D).
53BP1 accumulates at IRIF by interacting with methylated histones H3 and H4 via its Tudor domain [33, 34]. Histones H3 and H4 are constitutively methylated. However exposure to DNA damage causes a transition of histone H3 and H4 to a more accessible conformation due to RNF8/RNF168/Ubc13-dependent poly-ubiquitylation, which exposes the H4-K20 and/or K3-K79, methylated histones. This results in the recruitment of 53BP1 to the site of DNA damage (Figure 1D). Hence lack of both RNF8 and RNF168 leads to disruption in the recruitment of 53BP1.
Bloom (BLM) helicase and DNA damage response
BLM helicase and cancer
Bloom Syndrome (BS) is an autosomal recessive disorder that is associated with predisposition to cancer [35]. BS is characterized by proportional dwarfism, sun-induced chronic erythema, type II diabetes, male infertility and female subfertility and frequent infections due to immune deficiency. The BS afflicted individuals are predisposed to cancers. However unlike other cancer predisposition syndromes, BS patients suffer from almost all the major types of cancer [36]. This indicates that BLM is possibly involved at an early stage during neoplastic transformation - a step that maybe common for all forms of cancer. Hence understanding the cascades which regulates BLM functions and also deciphering the processes that the helicase itself regulates can give clues regarding the "common master regulatory step" which may precede the divergent epigenetic and genetic alterations that subsequently drive tumor formation.
The mean age of cancer diagnosis in BLM patients is 24 years and death is generally associated before the age of 30 [36, 37]. Germ line mutations in BLM give rise to BS. Though BS patients are rare, BLM heterozygotes that carry a BLM mutation may be faced with a higher probability of developing colorectal cancer [38]. The exact percentage of BLM heterozygous individuals in general population is unknown. However in Ashkenazi Jewish population the frequency of BS is approximately 1 in 48,000. This is due to a founder effect, approximately 1% of the Ashkenazi Jewish population being heterozygous carriers of the BLMAshmutation (a six nucleotide deletion and a seven nucleotide insertion at position 2281 of the cDNA) [39]. Transgenic mouse model studies also support the hypothesis that carriers of a single defective BLM allele are cancer prone [40]. Based on recent studies (described below) is quite possible that BLM is involved in the detection, transmission and finally the resolution of damaged DNA - in collaboration of other stage specific regulatory partners. Hence it can be hypothesized that the lack of BLM, may lead to a change in the stoichiometry of the proteins involved in DNA damage sensing and repair, which in turn may have an adverse effect during the neoplastic transformation process.
Multiple functions of BLM helicase during DNA damage response
Though the functions of BLM in the resolution of DNA damage are well characterized, the role of this helicase in the DNA damage response is yet to be fully deciphered. The response of BLM to DNA damage signal is a direct consequence of the changes in the intranuclear localization of the helicase. In asynchronous cells BLM is found to be in promyelocytic leukemia nuclear bodies (PML NBs) and nucleolus [41, 42]. Exposure to replication inhibitors (like hydroxyurea, HU) results in relocalization of BLM to the sites of stalled replication forks. It has been recently demonstrated that Chk1 constitutively phosphorylates BLM at Ser646, and this specific phosphorylation event rapidly decreases after exposure to DNA damage [43]. Lack of Ser646 phosphorylation post-DNA damage results in diminished interaction of BLM with nucleolin and PML isoforms and consequently leads to decrease in the accumulation of the helicase in nucleolus and PML NBs. Instead post-damage BLM colocalizes and associates with MRE11-Rad50-NBS1 complex [43–45], ATM [46, 47] and ATR [45, 48]. Both ATM and ATR phosphorylate BLM, indicating a possible role of the helicase in the recognition of DSBs and stalled replication [47, 48].
Multiple lines of evidence exist indicating that BLM may function very early in response to DNA damage (Figure 1A). BLM is induced by treatment of cells with γ-irradiation in an ATM independent manner. This induction depends on G2 delay because it fails to occur when G2 phase is prevented or bypassed [49]. Secondly, ATR and ATM-dependent intranuclear trafficking of BLM helicase also occurs during replication stress, which ensures optimal ATM activation and 53BP1 focus formation [50]. Hence cells from BS patients undergo delayed assembly of BRCA1 and NBS1 repair complexes at stalled replication forks [44]. Thirdly, both endogenous and overexpressed BLM accumulates at sites of laser-induced DSBs within 10 seconds and colocalizes with γ-H2AX and ATM. The early accumulation of BLM at DSBs is independent of ATM, RAD17 and NBS1 [51]. Finally, absence of BLM impairs the ability of Chromatin Accessibility Factor-1 (CAF-1) to be mobilized at the sites of DNA damage within the nucleus [52], thereby indicating that BLM may have an effect at the chromatin remodeling stage. Incidentally apart from its functions during the presynaptic, synaptic and post-synaptic phases of HR [53, 54], RAD54 also functions as a chromatin remodeller, both in vitro [55–57] and in vivo [58]. BLM stimulates the ATPase and chromatin remodeling activities of RAD54 in vitro [59], and is therefore potentially capable of enhancing the accessibility of the DNA damage sensor proteins to the DNA lesion in vivo. Using Fluorescence Recovery After Photobleaching (FRAP) the residence time of BLM in the HU-induced foci is only 7.2 seconds, providing evidence about the transitory nature of BLM during the sensing and recognition of DNA damage.
However it has been reported that BLM also accumulates at the site of stalled replication around 1 hour post-HU-treatment [60, 61]. This second wave of BLM accumulation depends on 53BP1 with which it physically interacts (Figure 1D). The accumulation of BLM/53BP1 foci and the physical interaction between them was dependent on phosphorylation-mediated interactions [62]. These observations have led to the hypothesis that BLM also plays a role at a relatively later stage in the hierarchy of proteins accumulating at the site of damage. According to this model BLM acts as a "molecular node" in response to replication stress during S-phase checkpoint. During this step BLM may integrate the signal(s) obtained from the upstream damage recognizing proteins and coordinate with repair and recombination proteins downstream to efficiently remove the deleterious lesion [60, 61].
Hence BLM seems to have roles in multiple phases of the DNA damage response pathway. In the immediate early phase BLM acts independently in the pathway, not even requiring other known early sensors of DNA damage like γH2AX, MDC1 and 53BP1. In the intermediate phase BLM acts as a molecular node, physically and functionally interacting with multiple proteins already associated at the site of damage and acting either in hierarchical or combinatorial manner so that the downstream repair proteins can receive and process the DNA damage signal. It is in this last step, the repair phase (Figure 1E), BLM acts in combination with its partners like RAD51 [60, 63–67], RAD54 [59, 68], 53BP1 [62, 66] and p53 [60] to remove the deleterious lesions so that the genome integrity can be maintained. While the predominant function of BLM is anti-recombinogenic [59], the helicase also interacts with human exonuclease 1 (Exo1) to resect DNA and initiate the process of DNA repair [69, 70]. This process indicates a pro-recombinogenic function of BLM. Detailed description of BLM functions in the repair phase can be obtained from several excellent recent reviews [71–75], and hence have not been described in detail here.
Conclusions
BLM helicase has attracted much attention due to the hyper-recombinogenic phenotype of BS patients and their predisposition to almost all forms of cancers known to human. Since BLM was first discovered to be a helicase [76], its in vitro biochemical role during HR had become the focus of intense research. Maybe due to the above reason and also possibly due to the non-availability of the desired tools and reagents, initial studies deciphering the in vivo cellular functions of BLM had been much fewer and less well characterized. However in the last half decade using a range of immortalized genetically engineered cell lines, specific antibodies that recognize endogenous BLM or its phosphorylated forms and with the widespread availability of robust microscopic techniques, the focus has shifted to towards deciphering how the helicase functions in a cellular context. This review had aimed to summarize all the important findings in this emerging field.
Perhaps the most important message that emerges is that BLM helicase has a fascinating "double life" apart from its well-characterized role as a helicase functioning during HR. Hence the functions of BLM are not confined to its role during HR but much earlier when the cells are exposed to the deleterious lesions. Hence BLM acts as a sensor, transmitter and finally the effector at different steps during the entire DNA damage signaling cascade - effectively acting as the "caretaker tumor suppressor" [36]. The fact that BS patients are predisposed to almost all forms of cancers can be interpreted to indicate that certain functions common to neoplastic transformation process is being regulated by BLM. Apart from HR, other repair pathways are also employed by eukaryotic cells [77, 78], on which BLM does not seem to exert much or any control. Hence regulation of repair pathways by BLM (essentially HR) may not be the only reason for the wide spectrum cancer phenotype observed in BS patience. It can be argued that the "caretaker tumor suppressor" function of BLM at least partially depends on its upstream DNA damage sensor and transmission functions.
It is perhaps important to point out the similarity of the proposed multiple functions of BLM with those of NBS1 and 53BP1. One of the first functions assigned to MRE11-RAD50-NBS1 (MRN) complex was a role in the repair of DNA double strand breaks [79]. Initially NBS1 was shown to be a substrate for ATM phosphorylation. Thus cells from NBS patients were defective in checkpoint response [79, 80]. However evidence is accumulating that MRN complex also functions upstream of ATM [81, 82]. Recent mouse models have confirmed that NBS1 is required for activation of ATM during DNA double strand breaks [83]. Similarly 53BP1 has been well characterized as a DNA damage sensor protein recruited to the site of damage by an ubiquitylation-dependent cascade [84, 85]. However recently a role of 53BP1 in DNA repair has been established. 53BP1 has an anti-recombinogenic function [86], which is dependent on both BLM [66] and BRCA1 [87]. 53BP1 also affects the classical and alternate end-joining pathway during class switch recombination [88, 89]. Hence it is quite possible that proteins involved in either sensing and transmission of DNA damage signal and those regulating DNA repair processes can have additional functions which are different than that presently ascribed to them.
Finally the question arises - what are the future directions of research on BLM especially in relation to its role as a damage sensor. It remains to be firmly established whether BLM can actually act as the universal damage sensor and transmitter as most of the studies until now have been done on cells containing either stalled forks or DSBs. The study of BLM post-translational modifications (PTMs) individually and in combination, and how the PTMs affect BLM functions during signal recognition and transmission is bound to be of much interest to researchers. But perhaps in the genomics era, the time has come not to look at BLM functions in isolation. With the help of high throughput technologies it is imperative that BLM functions are analyzed on a "global" scale perhaps in conjunctions with the dynamicity of its interaction with its chromatin (which is its actual in vivo substrate) but also with its stage specific protein partners, a few known but perhaps many unknown. The information available till date already indicates that the time has come for BLM helicase to bloom and show its true potential to the researchers.
Authors' information
ST received her Masters in Biotechnology from University of Jammu in 2007. She is a graduate student in SS's lab and her interests include the study of post-translational modifications in proteins involved in DNA damage response.
SS received his Ph.D. in 1997 from the Indian Institute of Science, India, for his research on the regulation of nitrogen assimilation in yeasts. During his postdoctoral studies at the Institut de Génétique et de Biologie Moléculaire et Cellulaire (IGBMC), France, he examined the functional interaction between the tumor suppressor p53 and the glucocorticoid receptor. Subsequently, in the Laboratory of Human Carcinogenesis, National Cancer Institute, Bethesda, USA, SS studied the inter-regulatory roles of p53 and RecQ helicases. His ongoing research interests involve the study of the interactions between the members of the eukaryotic signal-transduction cascade and the DNA-repair and -recombination machinery. He joined the National Institute of Immunology, India, as a group leader in September 2004.
References
Cimprich KA, Cortez D: ATR: an essential regulator of genome integrity. Nat Rev Mol Cell Biol. 2008, 9: 616-627. 10.1038/nrm2450.
Hurley PJ, Bunz F: ATM and ATR: components of an integrated circuit. Cell Cycle. 2007, 6: 414-417. 10.4161/cc.6.4.3886.
Bartek J, Lukas J: Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell. 2003, 3: 421-429. 10.1016/S1535-6108(03)00110-7.
Saintigny Y, Delacote F, Vares G, Petitot F, Lambert S, Averbeck D, Lopez BS: Characterization of homologous recombination induced by replication inhibition in mammalian cells. Embo J. 2001, 20: 3861-3870. 10.1093/emboj/20.14.3861.
Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER, Hurov KE, Luo J, Bakalarski CE, Zhao Z, Solimini N, Lerenthal Y, et al: ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. 2007, 316: 1160-1166. 10.1126/science.1140321.
Burma S, Chen BP, Murphy M, Kurimasa A, Chen DJ: ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J Biol Chem. 2001, 276: 42462-42467. 10.1074/jbc.C100466200.
Celeste A, Fernandez-Capetillo O, Kruhlak MJ, Pilch DR, Staudt DW, Lee A, Bonner RF, Bonner WM, Nussenzweig A: Histone H2AX phosphorylation is dispensable for the initial recognition of DNA breaks. Nat Cell Biol. 2003, 5: 675-679. 10.1038/ncb1004.
Stucki M, Clapperton JA, Mohammad D, Yaffe MB, Smerdon SJ, Jackson SP: MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell. 2005, 123: 1213-1226. 10.1016/j.cell.2005.09.038.
Goldberg M, Stucki M, Falck J, D'Amours D, Rahman D, Pappin D, Bartek J, Jackson SP: MDC1 is required for the intra-S-phase DNA damage checkpoint. Nature. 2003, 421: 952-956. 10.1038/nature01445.
Lou Z, Minter-Dykhouse K, Wu X, Chen J: MDC1 is coupled to activated CHK2 in mammalian DNA damage response pathways. Nature. 2003, 421: 957-961. 10.1038/nature01447.
Stewart GS, Wang B, Bignell CR, Taylor AM, Elledge SJ: MDC1 is a mediator of the mammalian DNA damage checkpoint. Nature. 2003, 421: 961-966. 10.1038/nature01446.
Zhao S, Renthal W, Lee EY: Functional analysis of FHA and BRCT domains of NBS1 in chromatin association and DNA damage responses. Nucleic Acids Res. 2002, 30: 4815-4822. 10.1093/nar/gkf612.
Lou Z, Minter-Dykhouse K, Franco S, Gostissa M, Rivera MA, Celeste A, Manis JP, van Deursen J, Nussenzweig A, Paull TT, et al: MDC1 maintains genomic stability by participating in the amplification of ATM-dependent DNA damage signals. Mol Cell. 2006, 21: 187-200. 10.1016/j.molcel.2005.11.025.
Lukas C, Melander F, Stucki M, Falck J, Bekker-Jensen S, Goldberg M, Lerenthal Y, Jackson SP, Bartek J, Lukas J: Mdc1 couples DNA double-strand break recognition by Nbs1 with its H2AX-dependent chromatin retention. Embo J. 2004, 23: 2674-2683. 10.1038/sj.emboj.7600269.
Chapman JR, Jackson SP: Phospho-dependent interactions between NBS1 and MDC1 mediate chromatin retention of the MRN complex at sites of DNA damage. EMBO Rep. 2008, 9: 795-801. 10.1038/embor.2008.103.
Melander F, Bekker-Jensen S, Falck J, Bartek J, Mailand N, Lukas J: Phosphorylation of SDT repeats in the MDC1 N terminus triggers retention of NBS1 at the DNA damage-modified chromatin. J Cell Biol. 2008, 181: 213-226. 10.1083/jcb.200708210.
Spycher C, Miller ES, Townsend K, Pavic L, Morrice NA, Janscak P, Stewart GS, Stucki M: Constitutive phosphorylation of MDC1 physically links the MRE11-RAD50-NBS1 complex to damaged chromatin. J Cell Biol. 2008, 181: 227-240. 10.1083/jcb.200709008.
Wu L, Luo K, Lou Z, Chen J: MDC1 regulates intra-S-phase checkpoint by targeting NBS1 to DNA double-strand breaks. Proc Natl Acad Sci USA. 2008, 105: 11200-11205. 10.1073/pnas.0802885105.
Morris JR, Solomon E: BRCA1: BARD1 induces the formation of conjugated ubiquitin structures, dependent on K6 of ubiquitin, in cells during DNA replication and repair. Hum Mol Genet. 2004, 13: 807-817. 10.1093/hmg/ddh095.
Polanowska J, Martin JS, Garcia-Muse T, Petalcorin MI, Boulton SJ: A conserved pathway to activate BRCA1-dependent ubiquitylation at DNA damage sites. EMBO J. 2006, 25: 2178-2188. 10.1038/sj.emboj.7601102.
Yan J, Kim YS, Yang XP, Li LP, Liao G, Xia F, Jetten AM: The ubiquitin-interacting motif containing protein RAP80 interacts with BRCA1 and functions in DNA damage repair response. Cancer Res. 2007, 67: 6647-6656. 10.1158/0008-5472.CAN-07-0924.
Yan J, Yang XP, Kim YS, Jetten AM: RAP80 responds to DNA damage induced by both ionizing radiation and UV irradiation and is phosphorylated at Ser 205. Cancer Res. 2008, 68: 4269-4276. 10.1158/0008-5472.CAN-07-5950.
Kim H, Chen J, Yu X: Ubiquitin-binding protein RAP80 mediates BRCA1-dependent DNA damage response. Science. 2007, 316: 1202-1205. 10.1126/science.1139621.
Sobhian B, Shao G, Lilli DR, Culhane AC, Moreau LA, Xia B, Livingston DM, Greenberg RA: RAP80 targets BRCA1 to specific ubiquitin structures at DNA damage sites. Science. 2007, 316: 1198-1202. 10.1126/science.1139516.
Wang B, Matsuoka S, Ballif BA, Zhang D, Smogorzewska A, Gygi SP, Elledge SJ: Abraxas and RAP80 form a BRCA1 protein complex required for the DNA damage response. Science. 2007, 316: 1194-1198. 10.1126/science.1139476.
Huen MS, Grant R, Manke I, Minn K, Yu X, Yaffe MB, Chen J: RNF8 transduces the DNA-damage signal via histone ubiquitylation and checkpoint protein assembly. Cell. 2007, 131: 901-914. 10.1016/j.cell.2007.09.041.
Kolas NK, Chapman JR, Nakada S, Ylanko J, Chahwan R, Sweeney FD, Panier S, Mendez M, Wildenhain J, Thomson TM, et al: Orchestration of the DNA-damage response by the RNF8 ubiquitin ligase. Science. 2007, 318: 1637-1640. 10.1126/science.1150034.
Mailand N, Bekker-Jensen S, Faustrup H, Melander F, Bartek J, Lukas C, Lukas J: RNF8 ubiquitylates histones at DNA double-strand breaks and promotes assembly of repair proteins. Cell. 2007, 131: 887-900. 10.1016/j.cell.2007.09.040.
Wang B, Elledge SJ: Ubc13/Rnf8 ubiquitin ligases control foci formation of the Rap80/Abraxas/Brca1/Brcc36 complex in response to DNA damage. Proc Natl Acad Sci USA. 2007, 104: 20759-20763. 10.1073/pnas.0710061104.
Stewart GS, Stankovic T, Byrd PJ, Wechsler T, Miller ES, Huissoon A, Drayson MT, West SC, Elledge SJ, Taylor AM: RIDDLE immunodeficiency syndrome is linked to defects in 53BP1-mediated DNA damage signaling. Proc Natl Acad Sci USA. 2007, 104: 16910-16915. 10.1073/pnas.0708408104.
Doil C, Mailand N, Bekker-Jensen S, Menard P, Larsen DH, Pepperkok R, Ellenberg J, Panier S, Durocher D, Bartek J, et al: RNF168 binds and amplifies ubiquitin conjugates on damaged chromosomes to allow accumulation of repair proteins. Cell. 2009, 136: 435-446. 10.1016/j.cell.2008.12.041.
Stewart GS, Panier S, Townsend K, Al-Hakim AK, Kolas NK, Miller ES, Nakada S, Ylanko J, Olivarius S, Mendez M, et al: The RIDDLE syndrome protein mediates a ubiquitin-dependent signaling cascade at sites of DNA damage. Cell. 2009, 136: 420-434. 10.1016/j.cell.2008.12.042.
Botuyan MV, Lee J, Ward IM, Kim JE, Thompson JR, Chen J, Mer G: Structural basis for the methylation state-specific recognition of histone H4-K20 by 53BP1 and Crb2 in DNA repair. Cell. 2006, 127: 1361-1373. 10.1016/j.cell.2006.10.043.
Huyen Y, Zgheib O, Ditullio RA, Gorgoulis VG, Zacharatos P, Petty TJ, Sheston EA, Mellert HS, Stavridi ES, Halazonetis TD: Methylated lysine 79 of histone H3 targets 53BP1 to DNA double-strand breaks. Nature. 2004, 432: 406-411. 10.1038/nature03114.
Ellis NA, Groden J, Ye TZ, Straughen J, Lennon DJ, Ciocci S, Proytcheva M, German J: The Bloom's syndrome gene product is homologous to RecQ helicases. Cell. 1995, 83: 655-666. 10.1016/0092-8674(95)90105-1.
Hickson ID: RecQ helicases: caretakers of the genome. Nat Rev Cancer. 2003, 3: 169-178. 10.1038/nrc1012.
Bachrati CZ, Hickson ID: RecQ helicases: suppressors of tumorigenesis and premature aging. Biochem J. 2003, 374: 577-606. 10.1042/BJ20030491.
Gruber SB, Ellis NA, Scott KK, Almog R, Kolachana P, Bonner JD, Kirchhoff T, Tomsho LP, Nafa K, Pierce H, et al: BLM heterozygosity and the risk of colorectal cancer. Science. 2002, 297: 2013-10.1126/science.1074399.
Shahrabani-Gargir L, Shomrat R, Yaron Y, Orr-Urtreger A, Groden J, Legum C: High frequency of a common Bloom syndrome Ashkenazi mutation among Jews of Polish origin. Genet Test. 1998, 2: 293-296. 10.1089/gte.1998.2.293.
Goss KH, Risinger MA, Kordich JJ, Sanz MM, Straughen JE, Slovek LE, Capobianco AJ, German J, Boivin GP, Groden J: Enhanced tumor formation in mice heterozygous for Blm mutation. Science. 2002, 297: 2051-2053. 10.1126/science.1074340.
Sanz MM, Proytcheva M, Ellis NA, Holloman WK, German J: BLM, the Bloom's syndrome protein, varies during the cell cycle in its amount, distribution, and co-localization with other nuclear proteins. Cytogenet Cell Genet. 2000, 91: 217-223. 10.1159/000056848.
Yankiwski V, Marciniak RA, Guarente L, Neff NF: Nuclear structure in normal and Bloom syndrome cells. Proc Natl Acad Sci USA. 2000, 97: 5214-5219. 10.1073/pnas.090525897.
Kaur S, Modi P, Srivastava V, Mudgal R, Tikoo S, Arora P, Mohanty D, Sengupta S: Chk1-Dependent Constitutive Phosphorylation of BLM Helicase at Serine 646 Decreases after DNA Damage. Mol Cancer Res. 2010, 8: 1234-1247. 10.1158/1541-7786.MCR-10-0233.
Davalos AR, Campisi J: Bloom syndrome cells undergo p53-dependent apoptosis and delayed assembly of BRCA1 and NBS1 repair complexes at stalled replication forks. J Cell Biol. 2003, 162: 1197-1209. 10.1083/jcb.200304016.
Franchitto A, Pichierri P: Bloom's syndrome protein is required for correct relocalization of RAD50/MRE11/NBS1 complex after replication fork arrest. J Cell Biol. 2002, 157: 19-30. 10.1083/jcb.200110009.
Ababou M, Dutertre S, Lecluse Y, Onclercq R, Chatton B, Amor-Gueret M: ATM-dependent phosphorylation and accumulation of endogenous BLM protein in response to ionizing radiation. Oncogene. 2000, 19: 5955-5963. 10.1038/sj.onc.1204003.
Beamish H, Kedar P, Kaneko H, Chen P, Fukao T, Peng C, Beresten S, Gueven N, Purdie D, Lees-Miller S, et al: Functional link between BLM defective in Bloom's syndrome and the ataxia-telangiectasia-mutated protein, ATM. J Biol Chem. 2002, 277: 30515-30523. 10.1074/jbc.M203801200.
Davies SL, North PS, Dart A, Lakin ND, Hickson ID: Phosphorylation of the Bloom's syndrome helicase and its role in recovery from S-phase arrest. Mol Cell Biol. 2004, 24: 1279-1291. 10.1128/MCB.24.3.1279-1291.2004.
Bischof O, Kim SH, Irving J, Beresten S, Ellis NA, Campisi J: Regulation and localization of the Bloom syndrome protein in response to DNA damage. J Cell Biol. 2001, 153: 367-380. 10.1083/jcb.153.2.367.
Davalos AR, Kaminker P, Hansen RK, Campisi J: ATR and ATM-dependent movement of BLM helicase during replication stress ensures optimal ATM activation and 53BP1 focus formation. Cell Cycle. 2004, 3: 1579-1586.
Karmakar P, Seki M, Kanamori M, Hashiguchi K, Ohtsuki M, Murata E, Inoue E, Tada S, Lan L, Yasui A, Enomoto T: BLM is an early responder to DNA double-strand breaks. Biochem Biophys Res Commun. 2006, 348: 62-69. 10.1016/j.bbrc.2006.07.037.
Jiao R, Bachrati CZ, Pedrazzi G, Kuster P, Petkovic M, Li JL, Egli D, Hickson ID, Stagljar I: Physical and functional interaction between the Bloom's syndrome gene product and the largest subunit of chromatin assembly factor 1. Mol Cell Biol. 2004, 24: 4710-4719. 10.1128/MCB.24.11.4710-4719.2004.
Heyer WD, Li X, Rolfsmeier M, Zhang XP: Rad54: the Swiss Army knife of homologous recombination?. Nucleic Acids Res. 2006, 34: 4115-4125. 10.1093/nar/gkl481.
Sung P, Krejci L, Van Komen S, Sehorn MG: Rad51 recombinase and recombination mediators. J Biol Chem. 2003, 278: 42729-42732. 10.1074/jbc.R300027200.
Alexeev A, Mazin A, Kowalczykowski SC: Rad54 protein possesses chromatin-remodeling activity stimulated by the Rad51-ssDNA nucleoprotein filament. Nat Struct Biol. 2003, 10: 182-186. 10.1038/nsb901.
Alexiadis V, Lusser A, Kadonaga JT: A conserved N-terminal motif in Rad54 is important for chromatin remodeling and homologous strand pairing. J Biol Chem. 2004, 279: 27824-27829. 10.1074/jbc.M402648200.
Jaskelioff M, Van Komen S, Krebs JE, Sung P, Peterson CL: Rad54p is a chromatin remodeling enzyme required for heteroduplex DNA joint formation with chromatin. J Biol Chem. 2003, 278: 9212-9218. 10.1074/jbc.M211545200.
Wolner B, Peterson CL: ATP-dependent and ATP-independent roles for the Rad54 chromatin remodeling enzyme during recombinational repair of a DNA double strand break. J Biol Chem. 2005, 280: 10855-10860. 10.1074/jbc.M414388200.
Srivastava V, Modi P, Tripathi V, Mudgal R, De S, Sengupta S: BLM helicase stimulates the ATPase and chromatin-remodeling activities of RAD54. J Cell Sci. 2009, 122: 3093-3103. 10.1242/jcs.051813.
Sengupta S, Linke SP, Pedeux R, Yang Q, Farnsworth J, Garfield SH, Valerie K, Shay JW, Ellis NA, Wasylyk B, Harris CC: BLM helicase-dependent transport of p53 to sites of stalled DNA replication forks modulates homologous recombination. Embo J. 2003, 22: 1210-1222. 10.1093/emboj/cdg114.
Sengupta S, Robles AI, Linke SP, Sinogeeva NI, Zhang R, Pedeux R, Ward IM, Celeste A, Nussenzweig A, Chen J, et al: Functional interaction between BLM helicase and 53BP1 in a Chk1-mediated pathway during S-phase arrest. J Cell Biol. 2004, 166: 801-813. 10.1083/jcb.200405128.
Tripathi V, Kaur S, Sengupta S: Phosphorylation-dependent interactions of BLM and 53BP1 are required for their anti-recombinogenic roles during homologous recombination. Carcinogenesis. 2008, 29: 52-61. 10.1093/carcin/bgm238.
Bugreev DV, Mazina OM, Mazin AV: Bloom syndrome helicase stimulates RAD51 DNA strand exchange activity through a novel mechanism. J Biol Chem. 2009, 284: 26349-26359. 10.1074/jbc.M109.029371.
Bugreev DV, Yu X, Egelman EH, Mazin AV: Novel pro- and anti-recombination activities of the Bloom's syndrome helicase. Genes Dev. 2007, 21: 3085-3094. 10.1101/gad.1609007.
Ouyang KJ, Woo LL, Zhu J, Huo D, Matunis MJ, Ellis NA: SUMO modification regulates BLM and RAD51 interaction at damaged replication forks. PLoS Biol. 2009, 7: e1000252-10.1371/journal.pbio.1000252.
Tripathi V, Nagarjuna T, Sengupta S: BLM helicase-dependent and -independent roles of 53BP1 during replication stress-mediated homologous recombination. J Cell Biol. 2007, 178: 9-14. 10.1083/jcb.200610051.
Wu L, Davies SL, Levitt NC, Hickson ID: Potential role for the BLM helicase in recombinational repair via a conserved interaction with RAD51. J Biol Chem. 2001, 276: 19375-19381. 10.1074/jbc.M009471200.
Chu WK, Hanada K, Kanaar R, Hickson ID: BLM has early and late functions in homologous recombination repair in mouse embryonic stem cells. Oncogene. 2010, 29: 4705-4714. 10.1038/onc.2010.214.
Gravel S, Chapman JR, Magill C, Jackson SP: DNA helicases Sgs1 and BLM promote DNA double-strand break resection. Genes Dev. 2008, 22: 2767-2772. 10.1101/gad.503108.
Nimonkar AV, Ozsoy AZ, Genschel J, Modrich P, Kowalczykowski SC: Human exonuclease 1 and BLM helicase interact to resect DNA and initiate DNA repair. Proc Natl Acad Sci USA. 2008, 105: 16906-16911. 10.1073/pnas.0809380105.
Bachrati CZ, Hickson ID: RecQ helicases: guardian angels of the DNA replication fork. Chromosoma. 2008, 117: 219-233. 10.1007/s00412-007-0142-4.
Cheok CF, Bachrati CZ, Chan KL, Ralf C, Wu L, Hickson ID: Roles of the Bloom's syndrome helicase in the maintenance of genome stability. Biochem Soc Trans. 2005, 33: 1456-1459. 10.1042/BST20051456.
Chu WK, Hickson ID: RecQ helicases: multifunctional genome caretakers. Nat Rev Cancer. 2009, 9: 644-654. 10.1038/nrc2682.
Ouyang KJ, Woo LL, Ellis NA: Homologous recombination and maintenance of genome integrity: cancer and aging through the prism of human RecQ helicases. Mech Ageing Dev. 2008, 129: 425-440. 10.1016/j.mad.2008.03.003.
Singh DK, Ahn B, Bohr VA: Roles of RECQ helicases in recombination based DNA repair, genomic stability and aging. Biogerontology. 2009, 10: 235-252. 10.1007/s10522-008-9205-z.
Karow JK, Chakraverty RK, Hickson ID: The Bloom's syndrome gene product is a 3'-5' DNA helicase. J Biol Chem. 1997, 272: 30611-30614. 10.1074/jbc.272.49.30611.
Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K, Linn S: Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem. 2004, 73: 39-85. 10.1146/annurev.biochem.73.011303.073723.
Wood RD: DNA repair in eukaryotes. Annu Rev Biochem. 1996, 65: 135-167. 10.1146/annurev.bi.65.070196.001031.
Stracker TH, Theunissen JW, Morales M, Petrini JH: The Mre11 complex and the metabolism of chromosome breaks: the importance of communicating and holding things together. DNA Repair (Amst). 2004, 3: 845-854. 10.1016/j.dnarep.2004.03.014.
Lim DS, Kim ST, Xu B, Maser RS, Lin J, Petrini JH, Kastan MB: ATM phosphorylates p95/nbs1 in an S-phase checkpoint pathway. Nature. 2000, 404: 613-617. 10.1038/35007091.
Carson CT, Schwartz RA, Stracker TH, Lilley CE, Lee DV, Weitzman MD: The Mre11 complex is required for ATM activation and the G2/M checkpoint. Embo J. 2003, 22: 6610-6620. 10.1093/emboj/cdg630.
Uziel T, Lerenthal Y, Moyal L, Andegeko Y, Mittelman L, Shiloh Y: Requirement of the MRN complex for ATM activation by DNA damage. Embo J. 2003, 22: 5612-5621. 10.1093/emboj/cdg541.
Difilippantonio S, Celeste A, Fernandez-Capetillo O, Chen HT, Martin BR, Laethem FV, Yang YP, Petukhova GV, Eckhaus M, Feigenbaum L, et al: Role of Nbs1 in the activation of the Atm kinase revealed in humanized mouse models. Nat Cell Biol. 2005, 7: 675-685. 10.1038/ncb1270.
FitzGerald JE, Grenon M, Lowndes NF: 53BP1: function and mechanisms of focal recruitment. Biochem Soc Trans. 2009, 37: 897-904. 10.1042/BST0370897.
Stewart GS: Solving the RIDDLE of 53BP1 recruitment to sites of damage. Cell Cycle. 2009, 8: 1532-1538. 10.4161/cc.8.10.8351.
Adams MM, Wang B, Xia Z, Morales JC, Lu X, Donehower LA, Bochar DA, Elledge SJ, Carpenter PB: 53BP1 oligomerization is independent of its methylation by PRMT1. Cell Cycle. 2005, 4: 1854-1861.
Bunting SF, Callen E, Wong N, Chen HT, Polato F, Gunn A, Bothmer A, Feldhahn N, Fernandez-Capetillo O, Cao L, et al: 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell. 2010, 141: 243-254. 10.1016/j.cell.2010.03.012.
Bothmer A, Robbiani DF, Feldhahn N, Gazumyan A, Nussenzweig A, Nussenzweig MC: 53BP1 regulates DNA resection and the choice between classical and alternative end joining during class switch recombination. J Exp Med. 2010, 207: 855-865. 10.1084/jem.20100244.
Ward IM, Reina-San-Martin B, Olaru A, Minn K, Tamada K, Lau JS, Cascalho M, Chen L, Nussenzweig A, Livak F, et al: 53BP1 is required for class switch recombination. J Cell Biol. 2004, 165: 459-464. 10.1083/jcb.200403021.
Acknowledgements
The work in SS's lab is supported by National Institute of Immunology core funds, Department of Biotechnology, India (BT/PR9598/Med/30/33/2007, BT/PR11258/BRB/10/645/2008), Council of Scientific and Industrial Research [37(1348)/08/EMR-II], India and National Institutes of Health, USA (1 R01 TW007302-05).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
Both ST and SS have been involved in conceptualizing and writing the review. Both the authors read and approved the final version of the manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Tikoo, S., Sengupta, S. Time to Bloom. Genome Integrity 1, 14 (2010). https://doi.org/10.1186/2041-9414-1-14
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/2041-9414-1-14