
Abstract
White matter damage is a clinically important part of stroke. However, compared to the mechanisms of neuronal injury in gray matter, white matter pathophysiology remains relatively understudied and poorly understood. This mini-review aims at summarizing current knowledge on experimental systems for analyzing the role of white matter injury relevant to stroke. In vitro platforms comprise primary cultures of both mature oligodendrocytes (OLGs) as well as oligodendrocyte precursor cells (OPCs). Tissue platforms involve preparations of optic nerve systems. Whole-animal platforms comprise in vivo models of cerebral ischemia that attempt to target white matter brain areas. While there is no single perfect model system, the collection of these experimental approaches have recently allowed a better understanding of the molecular and cellular pathways underlying OLG/OPC damage and demyelination. A systematic utilization of these cell, tissue and whole-animal platforms may eventually lead us to discover new targets for treating white matter injury in stroke and other CNS disorders.
Similar content being viewed by others
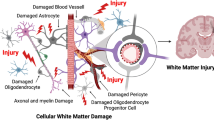


Introduction
Over the past decade, impressive advances have been made in understanding the basic molecular mechanisms underlying neuronal death. However, clinically effective neuroprotectants have not yet been discovered. This has been especially true in stroke, where many drugs targeting excitotoxicity, oxidative stress, and inflammation have all failed [1–3]. Although there are many difficult reasons for these translational problems [4–6], one potential issue worth examining further is the lack of emphasis on white matter.
In the CNS, white matter is primarily comprised of axonal bundles ensheathed with myelin. The cells forming these sheaths are the oligodendrocytes (OLGs), which tend to be arranged in rows parallel to axonal tracts. Just before and after birth, oligodendrocyte precursor cells (OPCs) multiply rapidly, mature into OLGs, and develop processes, which are then involved in the formation of myelin. Damage to OPCs and OLGs causes loss of myelin synthesis and interruption of proper axonal function. Hence, even if we protect neurons in gray matter, loss of myelin and axonal integrity would interfere with neuronal connectivity and function. Neuroprotection cannot be truly attained without oligoprotection.
White matter ischemia is different from gray matter in many ways. For instance, white matter ischemia is typically more severe than gray matter because white matter blood flow is lower than gray matter and there is little collateral blood supply in deep white matter [6]. Moreover, mechanisms responsible for ischemic cell death may be different between white matter and gray matter. Although an increase in intracellular Ca2+ is involved in both white matter and gray matter ischemia, the routes of Ca2+ entry might differ. In white matter, pathological Ca2+ entry occurs in part due to increased intracellular Na+ and membrane depolarization [7–9]. Therefore, Na+ channel blockade has been proposed as a protective strategy in white matter [7–10]. In contrast, voltage dependent Ca2+ channels and glutamate-activated ionic receptors in gray matter have been traditionally viewed as the primary routes of pathological Ca2+ entry [11, 12]. Of course, beyond these differences in calcium handling, there are potentially many other aspects of gray versus white matter function that differ. Thus far, white matter pathophysiology remains relatively poorly understood compared to gray matter. Because white matter damage is a clinically important part of stroke, we might need to take white matter ischemia more into account to develop effective stroke therapy.
In this mini-review, we summarize the current knowledge on experimental systems for assessing white matter damage by ischemic stress. A systematic utilization of these cell, tissue and whole-animal platforms should enable us to better dissect mechanisms of white matter pathophysiology and help our search for oligoprotectants in stroke and other CNS disorders.
In vitro white matter ischemia models
In pathological conditions, excitotoxic cell death is a critical part of neuronal injury, and it has been implicated in acute injury to the CNS and in chronic neurodegenerative disorders [13–15]. Numerous studies have now revealed that, in addition to neurons, glial cells can also be damaged by excitotoxicity. Among the glial cell types, the OLG lineage may be the most vulnerable to excitotoxicity.
In the gray matter of the brain, neuronal cell death is often caused by a rise of extracellular glutamate concentration, which activates NMDA receptors and leads an excessive rise of intracellular Ca2+ concentrations. Glutamate can also damage white matter OLGs, in both acute and chronic diseases [16]. Since excessive glutamate release can be seen under brain ischemic conditions, glutamate-induced OLG death has been used as a model for in vitro white matter ischemia to pursue the mechanism of OLG death by excitotoxicity. So far, there are at least three different mechanisms of glutamate-induced OLG death. Using cultured OLGs prepared from new born rat brain, Oka et al. reported that OLGs in cultures are highly vulnerable to glutamate. They showed that 24-h exposure of glutamate caused OLG death by reversing cystine-glutamate exchange, which induces glutathione depletion [17]. AMPA/kainate receptors have also indicated to be involved in glutamate-induced OLG death. Sanchez-Gomez et al. used primary cultures of OLGs derived from the optic nerves of young-adult rat or mouse to examine the involvement AMPA/kainite receptors in OLG death. In the report, they revealed that excessive activation of AMPA/kainate receptors causes Na+ and Ca2+ influx through the receptor channel complex leading to OLG death [18]. Until recently, it had been thought that NMDA receptor was not involved in OLG death by excitotoxicity. However, three recent studies have demonstrated that OLGs also express NMDA receptors as neurons [19–21]. NMDA receptors of OLGs are activated by glutamate in white matter ischemia [19], and activation of these receptors leads to rise of intracellular Ca2+ concentration [21]. Hence, NMDA receptors might be also participate in glutamate-induced OLG damage.
In addition to these primary excitotoxic mechanisms, glutamate can also kill OLGs via immune-system-related pathways. Alberdi et al. showed that brief incubation with glutamate followed by exposure to complement is lethal to OLGs in vitro [22]. Thus, even glutamate at non-toxic concentrations can kill OLGs by sensitizing these cells to complement attack. Further, inflammatory cytokines such as TNF-alpha and IL-1beta are also known to be involved in glutamate-induced OLG death. Those cytokines are released by reactive microglia and can impair glutamate uptake and trigger excitotoxic OLG death [23]. Since glutamate is reported to release TNF-alpha from microglia through AMPA/kainate activation, glutamate can also induce OLG death in indirect mechanisms [24].
Although elevations in glutamate certainly occur in stroke, alterations in other extracellular mediators may also be important. In the CNS, extracellular ATP can act as an excitatory neurotransmitter. ATP activates ionotropic P2X receptor and metabotropic P2Y receptor [25, 26]. Both P2X and P2Y receptors are expressed in OLGs. James and Butt used isolated optic nerves to show that ATP increased intracellular Ca2+ concentrations in OLGs through P2Y receptors [27]. In addition, they also demonstrated that a P2X receptor agonist evoked a smaller but significant OLG Ca2+ signal. In brain ischemia and spinal cord injury models, P2X receptors were reported to mediate signaling cascades leading to neurodegeneration [28, 29]. Also in OLGs, P2X was shown to be involved in cell death. Matute et al. have demonstrated that ATP or P2X agonists, but not P2Y agonists, was toxic to differentiated OLGs in vitro [16].
No matter what the upstream trigger might be, energetic stress should be a common denominator for white matter injury. In this regard, oxygen-glucose deprivation (OGD) is a useful tool for mimicking in vitro ischemia. OGD can induce both OPC as well as OLG death in vitro [30, 31]. Indeed, it appeared that OPCs were more susceptible than OLGs [30]. More recently, using an immortalized mouse OLG cell line, Zhang et al showed that OGD-induced OLG death was induced by apoptosis via p75 and caspase-3 [32]. Thus, these cell systems can be productively used to dissect the molecular pathways of OGD-induced white matter injury.
Compared to astrocytes, both OLGs and OPCs are relatively weak. To maintain those cells in culture, a wide spectrum of culture supplements such as growth factors are needed. Hence, removing those factors from culture media causes OLG or OPC death. Although this starvation stress does not perfectly reflect in vivo ischemic conditions, the stress can be thought as one of the in vitro ischemia model and is now well used to induce OLG/OPC damage, especially for OPC cultures. For instance, Cui et al. reported two studies whereby IGF-1 promoted the proliferation of OPCs via PI3K/Akt, MEK/ERK, and src-like tyrosine kinase pathways [33, 34]. Rubio et al. showed that neurotrophin-3 is also important factor for OPC survival [35]. GGF/neuregulin has been also reported to be OPC protective [36]. Taken together, these serum deprivation paradigms provide evidence for the essential nature of trophic coupling for oligoprotection. Indeed, these types of growth factor gain-and-loss experiments may help us test drugs that can be used to salvage OPC and OLG health in a wide range of disorders.
The experimental systems described above are all useful for examining the mechanisms of OLG/OPC death by ischemic stress. The main read-outs comprise either cell survival or myelin synthesis in isolated cultures, or compound action potentials in optic nerve preparations. The ease of quantitation and the reproducibility of culture platforms might allow for relatively high-throughput screening. However, it must be acknowledged that in vitro systems have some limitations. Cell and tissue systems cannot truly replicate the inter-cellular interactions and anatomic geometry of in vivo white matter.
In vivo rodent white matter ischemia models
White matter lesions are observed frequently in stroke patients and experimental animal models of cerebral ischemia, and have been thought to contribute to cognitive impairment [37–39]. Since nonhuman primates have well-developed white matter and vascular architectures which are similar to those of human brains, it seems to be reasonable to use them for studying the mechanisms of white matter injury. However, from ethical and practical standpoints, there remains an important imperative for developing rodent models. Compared to in vitro studies of white matter injury, basic research of white matter injury via in vivo models is not as well developed.
Some research groups have shown that OLG damage occur in response to ischemia in rodent middle cerebral artery occlusion (MCAO) models. Irving et al. observed structural changes of the OLG cytoskeleton by 40 min occlusion of MCA [40]. They assessed these changes by detecting increase of tau immunoreactivity within OLGs. Tau is the microtubule associated protein, and the increase in immunoreactivity has been found to be a sensitive marker for OLG damage. The MCAO-induced OLG damage assessed by tau immunoreactivity has been confirmed by other groups [41, 42]. Axons and myelin structure have also shown to be damaged in rodent MCAO models [43]. Another report by Irving et al. carefully examined the methods of quantifying white matter injury following prolonged focal cerebral ischemia in the rat. This study showed that myelin basic protein, Tau 1, and amyloid precursor protein staining can be utilized to assess myelin and axonal integrity in rat MCAO model [44]. Schabitz et al. have also shown the white matter injury in rat stroke model [45]. The use of Luxol-fast blue-periodic acid-Schiff and Bielschowsky's silver impregnation allowed the detection of myelin and axons, which could then be semi-quantified as read-outs for specific experiments.
The two-vessel occlusion model by permanent bilateral occlusion of the common carotid arteries has also been used to induce white matter ischemia. This model produces chronic cerebral hypoperfusion in rat and gerbil [46, 47]. These models are characterized by pathological changes in white matter, which appear similar to those in human cerebrovascular white matter lesions [48, 49]. As seen in human white matter ischemia, the rat chronic cerebral hypoperfusion model by the ligation of the bilateral common carotid arteries is accompanied by cognitive impairment [50]. In this model, however, the visual pathway is also injured by the occlusion of the ophthalmic arteries, and thus may affect behavioral assessment [47]. Although rats and gerbils are mostly used for these chronic cerebral hypoperfusion models, mice have also been used recently. With newly designed micro-coils, Shibata et al. developed a new mouse model of chronic cerebral hypoperfusion with relative preservation of the visual pathway [51]. In this model, white matter lesions occurred after 14 days without any gray mater involvement. Another report from Shibata et al. has demonstrated that the mouse hypoperfusion model showed impairment of working memory using the 8-arm radial maze [52]. The importance of the mouse model centers on the ability of future studies to utilize specific knockouts or transgenics for dissecting molecular mechanisms and mediators. For example, the use of knockout mice for matrix metalloproteinases (MMPs) could be anticipated. From a biochemical basis, MMPs can directly attack myelin components [53]. In fact, Nakaji et al. used the mouse chronic cerebral hypoperfusion model to show that gene knockout of MMP-2 reduced the severity of the white matter lesions [54]. Furthermore, MMP-9 knockout mice showed the resistance to MCAO-induced degradation of myelin-basic protein [55]. Depending on the proposed mechanisms, the combination of specific knockout mice with experimental induction of stroke or brain injury should be useful for exploring the underlying mechanisms involved in white matter pathophysiology.
In addition to the methods of occluding blood vessels, the direct injection of endothelin-1 (ET-1) into the neural parenchyma has also been used to induce white matter ischemia [56]. ET-1 is a potent vasoconstrictor peptide, and acts through different receptors called types A and B, which are distributed throughout the CNS [57–59]. Hughes et al. reported that microinjection of ET-1 into the striatum and cerebral cortex induces focal ischemia with a reduction of 40% of local blood flow in rats [60]. The damage in this model is localized in both gray and white matter without blood brain barrier breakdown. Frost et al. also used ET-1 microinjection model to induce white matter ischemia [61]. They injected ET-1 into the internal capsule, and tissue necrosis and demyelination in the infarcted white matter were found after a 14-day survival period. Further, infarcts resulted in measurable sensorimotor deficits. Regarding inflammatory response in the ET-1-injection white matter ischemia model, two studies recently came out. They showed that inflammatory response and white matter damage are closely related in the rat models of ET-1 microinjection into the striatum [62, 63].
The optic nerve has been thought as one of the most useful regions to study OLG functions. As cited in the previous section, numerous groups use OLG cultures from rodent optic nerve to investigate the OLG death by ischemia. Also in vivo system, the mechanisms of OLG damage have been examined in optic nerve. Retinal ganglion cell (RGC) axons comprise the optic nerve. So far, several in vivo models have been created to identify the RGC response to optic nerve damage [64–67]. A recent effort used photochemically-induced thrombosis to evoke anterior ischemic optic neuropathy (AION) in rats and mice [68, 69]. AION may mimic optic nerve strokes, which are among the most common causes of sudden optic nerve-related vision loss. Discrete histopathological changes of OLGs were reported in human AION [70]. Correspondingly, OLG dysfunction was observed in the mouse AION model [69].
Taken together, the emergence of various mechanical, thrombotic and chemical models now provides reasonable ways to move forward in vivo. But it must be acknowledged that white matter volumes in rodents are extremely small compared to human brains. Thus, methods for quantifying outcomes are challenging. Ultimately, comparisons of these rodent outcomes with human stroke patients must be performed carefully since white matter connectivity may be different between these vastly differing levels of brain evolution.
Remyelination in recovery phase in rodent stroke models
To date, the majority of studies using cell, tissue and whole-animal models of white matter injury have mostly focused on mechanisms and targets for acute injury. However, it may not be easy to block all multifactorial pathways of brain cell death in stroke patients. Therefore, an emerging emphasis on promoting recovery after stroke is beginning to take shape in our field. In the context of white matter mechanisms, it will be important to carefully define how each of the model platforms can be applied in this regard.
To support normal brain function, neuronal connectivity must be maintained. Hence, remyelination and regenerating axons in the border zone of cerebral infarcts and in secondarily lesioned areas are essential for stroke recovery. Fundamental research into these mechanisms of remyelination after ischemia remains somewhat limited, but a few studies are beginning to lead the way. Mandai et al. reported that myelin repair can occur in peri-infarct areas in mouse MCAO model, as judged by upregulated gene expression of proteolipid protein, one of the major protein components of CNS myelin [71]. Gregersen et al. studied the expression of myelin basic protein (MBP), another major component of CNS myelin, in peri-infarct areas using rat MCAO model [72]. They revealed that peri-infarct OLGs increased their expression of MBP mRNA from 24 h to maximal levels at day 7. These changes corresponded to the appearance of process-bearing MBP and occasional myelin oligodendrocyte glycoprotein-immunoreactive OLGs in parallel sections.
As described before, OLGs originate from their precursors; OPCs. OPCs are now found not only during development but pockets might also exist throughout the adult brain [73–76]. Therefore, OPCs may contribute to myelin maintenance and repair by generating new OLGs as a source of remyelination and repair. Experimental evidence is starting to be collected showing that OPCs in adult brain contribute to replenishment of OLGs after ischemic insults. Tanaka et al. examined the alteration of OLGs, OPCs and myelination in rat MCAO model [77]. They showed that a rapid and progressive decrease in the number of OLGs, OPCs, and myelin density after 2-day in the infarct core. In contrast, the peri-infarct area exhibited a moderate reduction in the number of OLGs and the myelin density with a slight increase in OPCs at 2 days after MCAO. Subsequently, a steady increase in the number of OPCs and a gradual recovery of OLGs were found in the peri-infarct area at 2 weeks of MCAO.
If OPCs provide a source of potential white matter repair, what signals and substrates are involved? A recent interesting study by Komitova et al. suggested that enriched environments may enhance the generation of OPCs after focal cortical ischemia. In this study, newly born OPCs were found to be immunoreactive for brain-derived neurotrophic factor (BDNF) [78]. Thus, it is tempting to speculate that BDNF may be related to remyelination by OPC proliferation/differentiation. What remains to be clarified is how this signal may interact with a multitude of other growth factors involved in OPC proliferation/differentiation. Since growth factor expression is upregulated after ischemia in peri-infarct area, a network response of growth factors might have essential roles in remyelination after stroke. These studies have mostly utilized in vivo models. As we dig down deeper mechanistically, a systems biology approach may ultimately be required as one asks broadly what gene profiles mediate cell-cell interactions in white matter during and after injury.
Potential therapeutic targets for white matter ischemia
The combined use of cell, tissue and whole-animal platforms discussed here may provide powerful tools for dissecting the pathophysiologic mechanisms of white matter injury in CNS disorders (see Figs. 1 and 2). Of course, no validated drugs yet exit. But several promising leads should be discussed.

Schematic for experimental systems and their endopoints. All systems have both advantages and disadvantages. A systematic utilization of these systems should enable us to better dissect mechanisms of white matter pathophysiology and help our search for oligoprotectants in stroke and other CNS disorders.

Summary for experimental systems for analyzing the role of white matter injury relevant to stroke.
Memantine, an uncompetitive NMDA receptor antagonist, has been well examined for the efficacy for preventing white matter from several insults including ischemic stress. Memantine is now licensed for moderate-to-severe Alzheimer's disease in US and EU [79]. Very recently, two studies suggested that memantine may be protective to white matter. Bakiri et al. reported that memantine reduced ischemic damage to mature and precursor OLGs in brain slices assessed by patch-clamp system [80]. Manning et al. showed that memantine attenuated white matter injury in a rat model of periventricular leukomalacia [81]. Therefore, NMDA receptor antagonists might be a good target for white matter injury after stroke.
Although the radical spin-trap NXY-059 failed in clinical stroke trial recently [82], antioxidant drugs are still potent therapeutic candidates for white matter. Imai et al. used rat transient ischemia models to evaluate the efficacy of ebselen, an anti-oxidant drug [41]. In this study, they showed that ebselen reduced axonal damage, and that OLG pathology was also reduced. Using rat MCAO model, Irving et al. have demonstrated that a free radical scavenger phenyl-N-tert-butyl-nitrone (PBN) reduced the number of tau-positive OLGs in the subcortical white matter of the ischemic hemisphere [40]. Lin et al. also examined the efficacy of PBN on white matter injury by hypoxia-ischemia in the neonatal rat brain [83]. In the study, the PBN treatment protected both OLGs and axons from ischemic insults. Subsequently, the same group has demonstrated that PBN also inhibited up-regulation of inflammatory cytokines such as IL-1beta, TNF-alpha and iNOS mRNA expression in the same model [84].
Finally, an ongoing NIH-funded trial is assessing minocycline for acute stroke. Minocycline is a second-generation tetracycline, which can cross the blood-brain barrier [85, 86]. Minocycline has been shown to be beneficial in a wide range of acute neurological injuries. In rodent brain ischemic models, this drug showed anti-inflammatory effects, based on its ability to inhibit immune mediators such as microglia [87, 88]. Although there is no report that minocycline directly protects OLGs against ischemic stress in adult rodent stroke model, this drug was shown to attenuate hypoxia/ischemia-induced white matter injury in the neonatal rat [89, 90]. Further, Hewlett and Corbett have shown that delayed minocycline treatment reduced long-term functional deficits as well as white matter injury in ET-1-induced rat ischemia model [91]. In spite of these experimental findings, it must be noted that a recent clinical trail using minocycline in ALS patients failed to show efficacy [92]. A potential caveat with this study is the long-term use of minocycline. Among its many actions, minocycline is a powerful metalloproteinase inhibitor. It has been recently suggested that long-term suppression of metalloproteinases may be detrimental for neurovascular homeostasis [93]. In stroke, short term applications of minocycline may still be possible. Ultimately, however, whether minocycline will be useful for white matter injury in stroke patients will have to be answered in a carefully analyzed randomized trial.
Conclusion
OLGs, myelin-forming glial cells in the CNS, are very vulnerable to ischemic stress, resulting in early loss of myelin and white matter dysfunction. Inhibiting axonal damage and/or OLG death, and accelerating the remyelination via OPC proliferation and differentiation may turn out to be critical for preventing acute neuronal disconnections as well as promoting repair and remodeling after stroke. Although there are no clinically validated treatments to date, several promising leads are beginning to be dissected in experimental systems. In this mini-review, we tried to provide a broad but brief survey of existing models at the cell, tissue and whole-animal levels. Many studies have productively used these model systems to dissect pathophysiology as well as assess treatment strategies. As we seek to cross the difficult translational hurdles between basic science and clinical challenges, the combined use of multiple model systems should help.
References
Ikonomidou C, Turski L: Why did NMDA receptor antagonists fail clinical trials for stroke and traumatic brain injury? Lancet Neurol 2002, 1: 383–386. 10.1016/S1474-4422(02)00164-3
Wang CX, Shuaib A: Neuroprotective effects of free radical scavengers in stroke. Drugs Aging 2007, 24: 537–546. 10.2165/00002512-200724070-00002
Kennedy TP, Vinten-Johansen J: A review of the clinical use of anti-inflammatory therapies for reperfusion injury in myocardial infarction and stroke: where do we go from here? Curr Opin Investig Drugs 2006, 7: 229–242.
Gladstone DJ, Black SE, Hakim AM: Toward wisdom from failure: lessons from neuroprotective stroke trials and new therapeutic directions. Stroke 2002, 33: 2123–2136. 10.1161/01.STR.0000025518.34157.51
Wahlgren NG, Ahmed N: Neuroprotection in cerebral ischaemia: facts and fancies--the need for new approaches. Cerebrovasc Dis 2004, 17(Suppl 1):153–166. 10.1159/000074808
Lo EH, Dalkara T, Moskowitz MA: Mechanisms, challenges and opportunities in stroke. Nat Rev Neurosci 2003, 4: 399–415. 10.1038/nrn1106
Imaizumi T, Kocsis JD, Waxman SG: Anoxic injury in the rat spinal cord: pharmacological evidence for multiple steps in Ca(2+)-dependent injury of the dorsal columns. J Neurotrauma 1997, 14: 299–311. 10.1089/neu.1997.14.299
Stys PK, Ransom BR, Waxman SG: Tertiary and quaternary local anesthetics protect CNS white matter from anoxic injury at concentrations that do not block excitability. J Neurophysiol 1992, 67: 236–240.
Stys PK: White matter injury mechanisms. Curr Mol Med 2004, 4: 113–130. 10.2174/1566524043479220
Hewitt KE, Stys PK, Lesiuk HJ: The use-dependent sodium channel blocker mexiletine is neuroprotective against global ischemic injury. Brain Res 2001, 898: 281–287. 10.1016/S0006-8993(01)02195-3
Choi DW: Ionic dependence of glutamate neurotoxicity. J Neurosci 1987, 7: 369–379.
Choi DW: Cerebral hypoxia: some new approaches and unanswered questions. J Neurosci 1990, 10: 2493–2501.
Choi DW: Glutamate neurotoxicity and diseases of the nervous system. Neuron 1988, 1: 623–634. 10.1016/0896-6273(88)90162-6
Lipton SA, Rosenberg PA: Excitatory amino acids as a final common pathway for neurologic disorders. N Engl J Med 1994, 330: 613–622. 10.1056/NEJM199403033300907
Lee JM, Zipfel GJ, Choi DW: The changing landscape of ischaemic brain injury mechanisms. Nature 1999, 399: A7–14.
Matute C, Alberdi E, Domercq M, Sanchez-Gomez MV, Perez-Samartin A, Rodriguez-Antiguedad A, Perez-Cerda F: Excitotoxic damage to white matter. J Anat 2007, 210: 693–702. 10.1111/j.1469-7580.2007.00733.x
Oka A, Belliveau MJ, Rosenberg PA, Volpe JJ: Vulnerability of oligodendroglia to glutamate: pharmacology, mechanisms, and prevention. J Neurosci 1993, 13: 1441–1453.
Sanchez-Gomez MV, Alberdi E, Ibarretxe G, Torre I, Matute C: Caspase-dependent and caspase-independent oligodendrocyte death mediated by AMPA and kainate receptors. J Neurosci 2003, 23: 9519–9528.
Karadottir R, Cavelier P, Bergersen LH, Attwell D: NMDA receptors are expressed in oligodendrocytes and activated in ischaemia. Nature 2005, 438: 1162–1166. 10.1038/nature04302
Salter MG, Fern R: NMDA receptors are expressed in developing oligodendrocyte processes and mediate injury. Nature 2005, 438: 1167–1171. 10.1038/nature04301
Micu I, Jiang Q, Coderre E, Ridsdale A, Zhang L, Woulfe J, Yin X, Trapp BD, McRory JE, Rehak R, et al.: NMDA receptors mediate calcium accumulation in myelin during chemical ischaemia. Nature 2006, 439: 988–992.
Alberdi E, Sanchez-Gomez MV, Torre I, Domercq M, Perez-Samartin A, Perez-Cerda F, Matute C: Activation of kainate receptors sensitizes oligodendrocytes to complement attack. J Neurosci 2006, 26: 3220–3228. 10.1523/JNEUROSCI.3780-05.2006
Takahashi JL, Giuliani F, Power C, Imai Y, Yong VW: Interleukin-1beta promotes oligodendrocyte death through glutamate excitotoxicity. Ann Neurol 2003, 53: 588–595. 10.1002/ana.10519
Merrill JE, Benveniste EN: Cytokines in inflammatory brain lesions: helpful and harmful. Trends Neurosci 1996, 19: 331–338. 10.1016/0166-2236(96)10047-3
Ralevic V, Burnstock G: Receptors for purines and pyrimidines. Pharmacol Rev 1998, 50: 413–492.
North RA: Molecular physiology of P2X receptors. Physiol Rev 2002, 82: 1013–1067.
James G, Butt AM: P2X and P2Y purinoreceptors mediate ATP-evoked calcium signalling in optic nerve glia in situ. Cell Calcium 2001, 30: 251–259. 10.1054/ceca.2001.0232
Le Feuvre RA, Brough D, Touzani O, Rothwell NJ: Role of P2X7 receptors in ischemic and excitotoxic brain injury in vivo. J Cereb Blood Flow Metab 2003, 23: 381–384. 10.1097/00004647-200303000-00013
Wang X, Arcuino G, Takano T, Lin J, Peng WG, Wan P, Li P, Xu Q, Liu QS, Goldman SA, Nedergaard M: P2X7 receptor inhibition improves recovery after spinal cord injury. Nat Med 2004, 10: 821–827. 10.1038/nm1082
Deng W, Rosenberg PA, Volpe JJ, Jensen FE: Calcium-permeable AMPA/kainate receptors mediate toxicity and preconditioning by oxygen-glucose deprivation in oligodendrocyte precursors. Proc Natl Acad Sci USA 2003, 100: 6801–6806. 10.1073/pnas.1136624100
Arai K, Lo EH: An oligovascular niche: cerebral endothelial cells promote the survival and proliferation of oligodendrocyte precursor cells. J Neurosci 2009, 29: 4351–4355. 10.1523/JNEUROSCI.0035-09.2009
Zhang J, Li Y, Zheng X, Gao Q, Liu Z, Qu R, Borneman J, Elias SB, Chopp M: Bone marrow stromal cells protect oligodendrocytes from oxygen-glucose deprivation injury. J Neurosci Res 2008, 86: 1501–1510. 10.1002/jnr.21617
Cui QL, Zheng WH, Quirion R, Almazan G: Inhibition of Src-like kinases reveals Akt-dependent and -independent pathways in insulin-like growth factor I-mediated oligodendrocyte progenitor survival. J Biol Chem 2005, 280: 8918–8928. 10.1074/jbc.M414267200
Cui QL, Almazan G: IGF-I-induced oligodendrocyte progenitor proliferation requires PI3K/Akt, MEK/ERK, and Src-like tyrosine kinases. J Neurochem 2007, 100: 1480–1493.
Rubio N, Rodriguez R, Arevalo MA: In vitro myelination by oligodendrocyte precursor cells transfected with the neurotrophin-3 gene. Glia 2004, 47: 78–87. 10.1002/glia.20035
Flores AI, Mallon BS, Matsui T, Ogawa W, Rosenzweig A, Okamoto T, Macklin WB: Akt-mediated survival of oligodendrocytes induced by neuregulins. J Neurosci 2000, 20: 7622–7630.
Pantoni L, Garcia JH: Pathogenesis of leukoaraiosis: a review. Stroke 1997, 28: 652–659.
Esiri MM: The interplay between inflammation and neurodegeneration in CNS disease. J Neuroimmunol 2007, 184: 4–16. 10.1016/j.jneuroim.2006.11.013
Medana IM, Esiri MM: Axonal damage: a key predictor of outcome in human CNS diseases. Brain 2003, 126: 515–530. 10.1093/brain/awg061
Irving EA, Yatsushiro K, McCulloch J, Dewar D: Rapid alteration of tau in oligodendrocytes after focal ischemic injury in the rat: involvement of free radicals. J Cereb Blood Flow Metab 1997, 17: 612–622. 10.1097/00004647-199706000-00003
Imai H, Masayasu H, Dewar D, Graham DI, Macrae IM: Ebselen protects both gray and white matter in a rodent model of focal cerebral ischemia. Stroke 2001, 32: 2149–2154. 10.1161/hs0901.095725
Valeriani V, Dewar D, McCulloch J: Quantitative assessment of ischemic pathology in axons, oligodendrocytes, and neurons: attenuation of damage after transient ischemia. J Cereb Blood Flow Metab 2000, 20: 765–771. 10.1097/00004647-200005000-00002
Dewar D, Dawson DA: Changes of cytoskeletal protein immunostaining in myelinated fibre tracts after focal cerebral ischaemia in the rat. Acta Neuropathol 1997, 93: 71–77. 10.1007/s004010050584
Irving EA, Bentley DL, Parsons AA: Assessment of white matter injury following prolonged focal cerebral ischaemia in the rat. Acta Neuropathol 2001, 102: 627–635.
Schabitz WR, Li F, Fisher M: The N-methyl-D-aspartate antagonist CNS 1102 protects cerebral gray and white matter from ischemic injury following temporary focal ischemia in rats. Stroke 2000, 31: 1709–1714.
Sarti C, Pantoni L, Bartolini L, Inzitari D: Cognitive impairment and chronic cerebral hypoperfusion: what can be learned from experimental models. J Neurol Sci 2002, 203–204: 263–266. 10.1016/S0022-510X(02)00302-7
Farkas E, Luiten PG, Bari F: Permanent, bilateral common carotid artery occlusion in the rat: a model for chronic cerebral hypoperfusion-related neurodegenerative diseases. Brain Res Rev 2007, 54: 162–180. 10.1016/j.brainresrev.2007.01.003
Farkas E, Donka G, de Vos RA, Mihaly A, Bari F, Luiten PG: Experimental cerebral hypoperfusion induces white matter injury and microglial activation in the rat brain. Acta Neuropathol 2004, 108: 57–64. 10.1007/s00401-004-0864-9
Wakita H, Tomimoto H, Akiguchi I, Matsuo A, Lin JX, Ihara M, McGeer PL: Axonal damage and demyelination in the white matter after chronic cerebral hypoperfusion in the rat. Brain Res 2002, 924: 63–70. 10.1016/S0006-8993(01)03223-1
Farkas E, Luiten PG: Cerebral microvascular pathology in aging and Alzheimer's disease. Prog Neurobiol 2001, 64: 575–611. 10.1016/S0301-0082(00)00068-X
Shibata M, Ohtani R, Ihara M, Tomimoto H: White matter lesions and glial activation in a novel mouse model of chronic cerebral hypoperfusion. Stroke 2004, 35: 2598–2603. 10.1161/01.STR.0000143725.19053.60
Shibata M, Yamasaki N, Miyakawa T, Kalaria RN, Fujita Y, Ohtani R, Ihara M, Takahashi R, Tomimoto H: Selective impairment of working memory in a mouse model of chronic cerebral hypoperfusion. Stroke 2007, 38: 2826–2832. 10.1161/STROKEAHA.107.490151
Chandler S, Coates R, Gearing A, Lury J, Wells G, Bone E: Matrix metalloproteinases degrade myelin basic protein. Neurosci Lett 1995, 201: 223–226. 10.1016/0304-3940(95)12173-0
Nakaji K, Ihara M, Takahashi C, Itohara S, Noda M, Takahashi R, Tomimoto H: Matrix metalloproteinase-2 plays a critical role in the pathogenesis of white matter lesions after chronic cerebral hypoperfusion in rodents. Stroke 2006, 37: 2816–2823. 10.1161/01.STR.0000244808.17972.55
Asahi M, Wang X, Mori T, Sumii T, Jung JC, Moskowitz MA, Fini ME, Lo EH: Effects of matrix metalloproteinase-9 gene knock-out on the proteolysis of blood-brain barrier and white matter components after cerebral ischemia. J Neurosci 2001, 21: 7724–7732.
Sozmen EG, Kolekar A, Havton LA, Carmichael ST: A white matter stroke model in the mouse: axonal damage, progenitor responses and MRI correlates. J Neurosci Methods 2009, 180: 261–272. 10.1016/j.jneumeth.2009.03.017
Masaki T, Yanagisawa M: Endothelins. Essays Biochem 1992, 27: 79–89.
Motte S, McEntee K, Naeije R: Endothelin receptor antagonists. Pharmacol Ther 2006, 110: 386–414. 10.1016/j.pharmthera.2005.08.012
Rubanyi GM, Polokoff MA: Endothelins: molecular biology, biochemistry, pharmacology, physiology, and pathophysiology. Pharmacol Rev 1994, 46: 325–415.
Hughes PM, Anthony DC, Ruddin M, Botham MS, Rankine EL, Sablone M, Baumann D, Mir AK, Perry VH: Focal lesions in the rat central nervous system induced by endothelin-1. J Neuropathol Exp Neurol 2003, 62: 1276–1286.
Frost SB, Barbay S, Mumert ML, Stowe AM, Nudo RJ: An animal model of capsular infarct: endothelin-1 injections in the rat. Behav Brain Res 2006, 169: 206–211. 10.1016/j.bbr.2006.01.014
Souza-Rodrigues RD, Costa AM, Lima RR, Dos Santos CD, Picanco-Diniz CW, Gomes-Leal W: Inflammatory response and white matter damage after microinjections of endothelin-1 into the rat striatum. Brain Res 2008, 1200: 78–88. 10.1016/j.brainres.2007.11.025
Dos Santos CD, Picanco-Diniz CW, Gomes-Leal W: Differential patterns of inflammatory response, axonal damage and myelin impairment following excitotoxic or ischemic damage to the trigeminal spinal nucleus of adult rats. Brain Res 2007, 1172: 130–144. 10.1016/j.brainres.2007.07.037
Selles-Navarro I, Ellezam B, Fajardo R, Latour M, McKerracher L: Retinal ganglion cell and nonneuronal cell responses to a microcrush lesion of adult rat optic nerve. Exp Neurol 2001, 167: 282–289. 10.1006/exnr.2000.7573
Krueger-Naug AM, Emsley JG, Myers TL, Currie RW, Clarke DB: Injury to retinal ganglion cells induces expression of the small heat shock protein Hsp27 in the rat visual system. Neuroscience 2002, 110: 653–665. 10.1016/S0306-4522(01)00453-5
Johnson EC, Deppmeier LM, Wentzien SK, Hsu I, Morrison JC: Chronology of optic nerve head and retinal responses to elevated intraocular pressure. Invest Ophthalmol Vis Sci 2000, 41: 431–442.
Cioffi GA, Orgul S, Onda E, Bacon DR, Van Buskirk EM: An in vivo model of chronic optic nerve ischemia: the dose-dependent effects of endothelin-1 on the optic nerve microvasculature. Curr Eye Res 1995, 14: 1147–1153. 10.3109/02713689508995821
Bernstein SL, Guo Y, Kelman SE, Flower RW, Johnson MA: Functional and cellular responses in a novel rodent model of anterior ischemic optic neuropathy. Invest Ophthalmol Vis Sci 2003, 44: 4153–4162. 10.1167/iovs.03-0274
Goldenberg-Cohen N, Guo Y, Margolis F, Cohen Y, Miller NR, Bernstein SL: Oligodendrocyte dysfunction after induction of experimental anterior optic nerve ischemia. Invest Ophthalmol Vis Sci 2005, 46: 2716–2725. 10.1167/iovs.04-0547
Knox DL, Kerrison JB, Green WR: Histopathologic studies of ischemic optic neuropathy. Trans Am Ophthalmol Soc 2000, 98: 203–220. discussion 221–202
Mandai K, Matsumoto M, Kitagawa K, Matsushita K, Ohtsuki T, Mabuchi T, Colman DR, Kamada T, Yanagihara T: Ischemic damage and subsequent proliferation of oligodendrocytes in focal cerebral ischemia. Neuroscience 1997, 77: 849–861. 10.1016/S0306-4522(97)00517-4
Gregersen R, Christensen T, Lehrmann E, Diemer NH, Finsen B: Focal cerebral ischemia induces increased myelin basic protein and growth-associated protein-43 gene transcription in peri-infarct areas in the rat brain. Exp Brain Res 2001, 138: 384–392. 10.1007/s002210100715
Levine JM, Reynolds R, Fawcett JW: The oligodendrocyte precursor cell in health and disease. Trends Neurosci 2001, 24: 39–47. 10.1016/S0166-2236(00)01691-X
Nishiyama A, Chang A, Trapp BD: NG2+ glial cells: a novel glial cell population in the adult brain. J Neuropathol Exp Neurol 1999, 58: 1113–1124. 10.1097/00005072-199911000-00001
Nishiyama A: NG2 cells in the brain: a novel glial cell population. Hum Cell 2001, 14: 77–82.
Chang A, Nishiyama A, Peterson J, Prineas J, Trapp BD: NG2-positive oligodendrocyte progenitor cells in adult human brain and multiple sclerosis lesions. J Neurosci 2000, 20: 6404–6412.
Tanaka K, Nogawa S, Suzuki S, Dembo T, Kosakai A: Upregulation of oligodendrocyte progenitor cells associated with restoration of mature oligodendrocytes and myelination in peri-infarct area in the rat brain. Brain Res 2003, 989: 172–179. 10.1016/S0006-8993(03)03317-1
Komitova M, Perfilieva E, Mattsson B, Eriksson PS, Johansson BB: Enriched environment after focal cortical ischemia enhances the generation of astroglia and NG2 positive polydendrocytes in adult rat neocortex. Exp Neurol 2006, 199: 113–121. 10.1016/j.expneurol.2005.12.007
Lipton SA: NMDA receptors, glial cells, and clinical medicine. Neuron 2006, 50: 9–11. 10.1016/j.neuron.2006.03.026
Bakiri Y, Hamilton NB, Karadottir R, Attwell D: Testing NMDA receptor block as a therapeutic strategy for reducing ischaemic damage to CNS white matter. Glia 2008, 56: 233–240. 10.1002/glia.20608
Manning SM, Talos DM, Zhou C, Selip DB, Park HK, Park CJ, Volpe JJ, Jensen FE: NMDA receptor blockade with memantine attenuates white matter injury in a rat model of periventricular leukomalacia. J Neurosci 2008, 28: 6670–6678. 10.1523/JNEUROSCI.1702-08.2008
Proctor PH, Tamborello LP: SAINT-I worked, but the neuroprotectant is not NXY-059. Stroke 2007, 38: e109. author reply e110 10.1161/STROKEAHA.107.489161
Lin S, Rhodes PG, Lei M, Zhang F, Cai Z: alpha-Phenyl-n-tert-butyl-nitrone attenuates hypoxic-ischemic white matter injury in the neonatal rat brain. Brain Res 2004, 1007: 132–141. 10.1016/j.brainres.2004.01.074
Lin S, Cox HJ, Rhodes PG, Cai Z: Neuroprotection of alpha-phenyl-n-tert-butyl-nitrone on the neonatal white matter is associated with anti-inflammation. Neurosci Lett 2006, 405: 52–56. 10.1016/j.neulet.2006.06.063
Macdonald H, Kelly RG, Allen ES, Noble JF, Kanegis LA: Pharmacokinetic studies on minocycline in man. Clin Pharmacol Ther 1973, 14: 852–861.
Saivin S, Houin G: Clinical pharmacokinetics of doxycycline and minocycline. Clin Pharmacokinet 1988, 15: 355–366. 10.2165/00003088-198815060-00001
Yrjanheikki J, Keinanen R, Pellikka M, Hokfelt T, Koistinaho J: Tetracyclines inhibit microglial activation and are neuroprotective in global brain ischemia. Proc Natl Acad Sci USA 1998, 95: 15769–15774. 10.1073/pnas.95.26.15769
Yrjanheikki J, Tikka T, Keinanen R, Goldsteins G, Chan PH, Koistinaho J: A tetracycline derivative, minocycline, reduces inflammation and protects against focal cerebral ischemia with a wide therapeutic window. Proc Natl Acad Sci USA 1999, 96: 13496–13500. 10.1073/pnas.96.23.13496
Carty ML, Wixey JA, Colditz PB, Buller KM: Post-insult minocycline treatment attenuates hypoxia-ischemia-induced neuroinflammation and white matter injury in the neonatal rat: a comparison of two different dose regimens. Int J Dev Neurosci 2008, 26: 477–485. 10.1016/j.ijdevneu.2008.02.005
Stolp HB, Ek CJ, Johansson PA, Dziegielewska KM, Potter AM, Habgood MD, Saunders NR: Effect of minocycline on inflammation-induced damage to the blood-brain barrier and white matter during development. Eur J Neurosci 2007, 26: 3465–3474. 10.1111/j.1460-9568.2007.05973.x
Hewlett KA, Corbett D: Delayed minocycline treatment reduces long-term functional deficits and histological injury in a rodent model of focal ischemia. Neuroscience 2006, 141: 27–33. 10.1016/j.neuroscience.2006.03.071
Gordon PH, Moore DH, Miller RG, Florence JM, Verheijde JL, Doorish C, Hilton JF, Spitalny GM, MacArthur RB, Mitsumoto H, et al.: Efficacy of minocycline in patients with amyotrophic lateral sclerosis: a phase III randomised trial. Lancet Neurol 2007, 6: 1045–1053. 10.1016/S1474-4422(07)70270-3
Zhao BQ, Wang S, Kim HY, Storrie H, Rosen BR, Mooney DJ, Wang X, Lo EH: Role of matrix metalloproteinases in delayed cortical responses after stroke. Nat Med 2006, 12: 441–445. 10.1038/nm1387
Acknowledgements and Funding
Supported in part by P01-NS55104, P50-NS10828, R01-NS37074, R01-NS48422, R01-NS53560, the American Heart Association and the Deane Institute.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
Study concept and design: KA and EHL; Drafting of the manuscript: KA and EHL; Critical revision of the manuscript for important intellectual content: KA and EHL; Obtained funding: KA and EHL. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Arai, K., Lo, E.H. Experimental models for analysis of oligodendrocyte pathophysiology in stroke. Exp & Trans Stroke Med 1, 6 (2009). https://doi.org/10.1186/2040-7378-1-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/2040-7378-1-6