
Abstract
Multiple sclerosis (MS) is the most common neurologic disease of young adults. In the recent years, our understanding on disease pathomechanisms has considerably improved and new therapies have emerged. Yet a cure for this devastating disorder is still a far cry away and human resources on ex vivo specimens are limited. More than 70 years after its first description, experimental autoimmune encephalomyelitis (EAE) remains an important tool to understand concepts of T cell mediated autoimmunity as well as the roles of the innate and the humoral immune systems. Some EAE models also well reflect mechanisms of tissue damage including demyelination, axonal injury and also cortical changes. A limitation of the classical EAE model is a neglect of CD8 T cell mediated immune mechanisms. Moreover, well characterized models for primary progressive MS or demyelination patterns involving primary oligodendrocyte dystrophy are still not available. Yet many current therapeutic concepts including glatiramer acetate or natalizumab stem from their successful first application in EAE models. New strategies include the widespread use of conditional knockout mice to understand the cell-type specific function of single genes, innovative approaches to establish models on the roles of B cells and CD8 T cells as well as on the relation of inflammation to primary degeneration. In summary, EAE models continue to play an important role in neuroimmunology thereby also stimulating research in other fields of the neurosciences and immunobiology.
Similar content being viewed by others

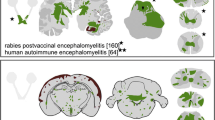
Classical EAE models: an overview
The model of experimental autoimmune encephalomyelitis (EAE) stems back from an attempt to understand the pathogenesis of post vaccinal encephalomyelitis, e.g., after rabies vaccination during the 1920s and 1930s (for overview see Gold et al., 2006). At that time, first immunization experiments with repeated inoculations of human spinal cord homogenate in rabbits and rhesus monkeys resulted in the first models of experimentally induced encephalomyelitis. Over the next 70-80 years, EAE was studied in many different species, including primates and rodents. Improved protocols included the application of a mineral oil-based adjuvant (Freund's adjuvant), and later on the addition of heat-inactivated mycobacteria (complete Freund's adjuvant, CFA) that facilitate disease induction after a single inoculation. Some models, especially in mice, also require the intraperitoneal injection of pertussis toxin which may deplete regulatory T cells and thus enhance autoimmune reactions [1].
After World War II, EAE models in rats and guinea pigs dominated neuroimmunological research for many decades. In these species, active immunization used either spinal cord homogenate, myelin or, as first purified antigen in the 1960s, myelin basic protein (MBP) in CFA. Such protocols resulted in a very reproducible disease with an onset 9-12 days after immunization and an acute monophasic or chronic relapsing-remitting/progressive disease course depending on the species or rodent strain [2]. Moreover, EAE can be induced not only by active immunization, but also passively by adoptive transfer of antigen specific T cells (AT-EAE). This was shown after in vitro propagation of MBP specific T cell lines and subsequent transfer of cells in mice [3]. In the Lewis rat strain, this regimen results in disease onset 3-4 days after cell transfer, followed by disease maximum 1-2 days later and a subsequent complete remission within the next few days. The histopathology of this model is characterized by a purely inflammatory pattern with minimal central nervous system (CNS) demyelination, thus rather reflecting acute disseminated encephalomyelitis (ADEM). Yet MBP-EAE provides an excellent paradigm to investigate T cell function and regulation in autoimmune neuroinflammation and nicely underpins the autoimmune origin of MS.
The induction of EAE is not restricted to myelin antigens, as the disease can also be elicited by astroglial antigens like S-100 or glial fibrillary acidic protein (for review see [4]) and neuronal antigens including Ma [5] or amyloid precursor protein (APP, [6]). Similarly, AT-EAE can also be induced by adoptive transfer of T cells specific for astroglial and neuronal antigens (reviewed in [7]).
Over the last 20-30 years, further encephalitogenic myelin antigens were identified, the first of which was proteolipid protein (PLP). Other myelin autoantigens in rodents include myelin oligodendrocyte glycoprotein (MOG; [8], myelin associated glycoprotein (MAG), 2', 3'-cyclic nucleotide 3'-phosphodiesterase (CNPase), oligodendrocyte (OL)-specific protein, and myelin-associated oligodendrocyte basic protein. The most recent studies were performed in inbred mouse strains in which immunodominant and encephalitogenic T cell epitopes, like PLP aa139-151 or MOG aa35-55, were also identified. Some of the mentioned myelin proteins are components not only of CNS, but also peripheral nervous system myelin. Thus immunization with e.g. MPB or PLP theoretically can also result in radiculitis or neuritis [9]. Importantly, not only the susceptibility to disease but also the pathology and clinical course are determined by both genetic factors and the specific immunogen/adjuvant used, resulting in established combinations of autoantigen and inbred mouse strain, e.g. PLP139-151 EAE in SJL mice. In some cases, tolerance to single autoantigens in non-susceptible strains can be overcome by specific protocols including booster immunization [10].
A milestone in multiple sclerosis (MS) research was marked by the characterization of MOG as an autoantigen. Although only a minor component of CNS myelin, MOG is part of the outer myelin layer thus being of special interest as target of a first autoimmune attack before antigen spreading may occur. Immunization with MOG induces not only encephalitogenic T cells, but also a demyelinating autoantibody response in susceptible species. Moreover, demyelinating anti-MOG antibodies enhance disease severity causing extensive demyelination after T cell-mediated CNS inflammation in rat and primate EAE models [11, 12]. Such antibodies to native MOG are also found in MS patients and mediate demyelination after transfer into EAE rats [13]. At first, immunization with MOG in rodents was mainly investigated in different rat strains [14]. Studies in the DA rat were able to reproduce a range of pathological and clinical phenotypes mimicking the spectrum of MS [15]. Induction of MOG-EAE in Brown Norway rats led to a Devic like disease (at least in clinical terms) with eosinophilic infiltrates also suggesting the involvement of T helper (Th2) cells [16]. In congenic Lewis rats, the contribution of genetic and environmental factors to disease susceptibility and modulation of target tissue responses were characterized. In contrast, immunization of marmoset monkeys with CNS tissue homogenates or recombinant MOG provided a disease model closely mimicking humoral disease patterns of MS in a species that is phylogenetically very close to man [17, 18]. Like in rats, pathogenic mechanisms include the contributions of encephalitogenic T cells and demyelinating autoantibodies [19]. While the investigation of marmoset EAE is somewhat limited by ethical issues as well as a fulminant disease course and lack of suitable markers in this species, this model may be of particular interest for pre-clinical treatment studies and imaging approaches.
In mice, MOG protein or MOG35-55 peptide can induce EAE in C57BL/6 mice or Biozzi ABH mice resulting in a relapsing-remitting or chronic disease course. In the C57BL/6 mouse strain, the H-2b MHC haplotype governs the ability to induce encephalitogenicity and to develop demyelinating autoantibodies in response to mouse or rat MOG. Transfer of these demyelinating anti-MOG antibodies also enhances demyelination and exacerbates disease severity in mouse models of EAE [8]. Over the last 15 years, the avenue of genetically engineered mice has greatly promoted the investigation of EAE pathomechanisms, particularly after backcrossing strains on the C57BL/6 background. Today, MOG-EAE in the C57BL/6 mouse is one of the most widely used models in neuroimmunological research with over 100 publications in recent years.
In summary, EAE models mimic many of the clinical, neuropathological and immunological aspects of MS but none is able to reproduce the whole spectrum of the human disease. In particular, many of the classical EAE models mainly affect the spinal cord (and not the brain) and are especially based on CD4 positive T cells, while the recently recognized importance of CD8 positive T cells in autoimmune inflammation of the CNS remains largely scotomized. So far, a model for the primary progressive course of the disease is still lacking and induction of EAE results in extensive tissue damage (including axons and neurons) rather than in primary demyelination. Nevertheless, the data from EAE studies still provide the most important cue for the notion of MS as an autoimmune disease where myelin specific cells initiate an inflammatory reaction in the CNS which ultimately leads to demyelination, gliosis and axonal injury.
EAE models contribute to unveiling the immunopathogenesis of MS
Research in EAE first focused on the adaptive immune system including T and B cell responses. Recently, components of the innate immune system, in particular macrophages, microglia, Toll-like receptors and even mast cells, have also been recognized as important parts of disease pathogenesis [20, 21]. In particular, the roles of vessel-associated dendritic cells for the re-activation of T cells in situ and of microglia for the development and maintenance of inflammatory CNS lesions have been highlighted [22, 23]. Another recent focus is on B lymphocytes. Besides production of demyelinating antibodies after differentiation into plasma cells (see also above), B cells may have a co-stimulatory function for T cell effectors, but can also play a regulatory role [24, 25].
The pivotal role of T lymphocytes in disease induction was confirmed by adoptive transfer of autoantigen specific T cells [26]. Later on, the importance of antigen-presenting cells (APC) governing T cell responses was recognized. Professional APC, like dendritic cells (DC), are characterized by a complete repertoire of co-stimulatory molecules like CD28/B7, inducible T cell co-stimulator (ICOS)/ICOS-L, and programmed death (PD)1/PD-1/2L thus enabling them to fully activate naive T cells, but also to regulate this activation (for overview see [27]. In contrast, non-professional APC, like macrophages, microglia or astrocytes, can upregulate the expression of immune molecules upon stimulation that facilitate activation only of memory, but not naive T cells. Recently, the special importance of perivascular DC for the reactivation of antigen specific T cells and their entry into the target organ was highlighted and DC may also present a valuable target for therapies as shown after intracerebral injection [28].
During T cell activation, the production of cytokines and chemokines influences the local microenvironment and finally determines the functional outcome of the immune response. In a classical paradigm, IFN-γ results in the differentiation of naive CD4 positive T cells into a "proinflammatory Th1" effector T cell subset which may initiate autoimmunity in the CNS [29]. In contrast, the "Th2" cytokines interleukin (IL)-4, IL-5, or IL-13 induce the differentiation of naive T cells into "Th2" T cells which are able to counterbalance the inflammatory reaction in the CNS. Only recently, the importance of IL-23 and not IL-12 for perpetuation of the immune response in EAE was recognized. This concept was further refined by the characterization of IL-17 producing "Th17" cells which seem to play a role for EAE induction in addition to "Th1" cells. While this concept was first established in rodents, a role for Th17 was recently also described in humans in vitro and in situ [30, 31]. The generation of Th17 cells requires the presence of the "proinflammatory" cytokine IL-6 in concert with the "regulatory" cytokine transforming growth factor (TGF)-β, whereas IL-23 is involved in their maintenance [32]. An alternative pathway for the differentiation of Th17 cells may be induced by IL-21 [33]. In fact, cytokine regulation may involve complex networks rather than linear pathways. This notion is further corroborated by the involvement of the IL-18 receptor pathway in the generation of IL-17 responses [34]. Moreover, some T cell clones may be capable of producing both interferon gamma and IL-17, while IL-22 seems to be more specific for Th17 cells [35]. Some studies point at the importance of Th17 cells as part of the first wave of T cell infiltration into the CNS which may be mediated by specific chemokines and their receptors like CCR6 and CCL20 [36]. While IL-17 promotes autoimmunity by triggering a positive-feedback loop via interleukin-6 induction [37], other reports also found that Th17 cells are not in all cases necessary for the propagation of EAE [38]. Some recent data rather point at the importance of the transcription factor T-bet for governing encephalitogenicity of myelin-specific T cells rather than pathway specific cytokines such as IFN-γ or IL-17 [39]. Finally, Th9 cells entered the stage as most recent effector T cells. In vitro, this population is generated by a combination of IL-4 and TGF-β [40]. Besides IL-9, these cells also produce IL-10 and contribute to tissue inflammation like colitis or, as shown recently, also autoimmune inflammation of the CNS [41]. A further level of complexity was introduced by the identification of further leukocyte subtypes which can be involved in disease pathogenesis. In some models, pathogenic neuroantigen-specific Th2 T cell responses or neutrophilic as well as eosinophilic granulocytes as effector cells may be involved in tissue damage [42, 43]. Moreover, mast cells produce IL-9 and thus may play a role in regulating T cell tolerance and in modulating the course of EAE [21, 44].
The invasion of all these immune cells into the CNS requires transmigration over the blood brain barrier. Here, a complex molecular interaction takes place between adhesion molecules on the surface of immune cells and the cerebral endothelial cells forming the blood brain barrier [45, 46]. These molecular interactions have been extensively studied in the 1990s. In particular, the interaction between the integrins very late antigen (VLA) 4 and leukocyte function associated antigen (LFA) 1 with their immunoglobulin-like counter-ligands vascular cell adhesion molecule (VCAM) 1 and intercellular adhesion molecule (ICAM) 1 was shown to be essential for T cell adhesion and migration into the central nervous system [47, 48]. Recently, proteomic based approaches defined an important function for the activated leukocyte cell adhesion molecule (ALCAM) in the recruitment of leukocytes into the brain [49]. Meanwhile, many studies point to a role of the brain endothelium not only as barrier, but also as active immunomodulator involved in DC polarization and Th17 signaling pathways [50].
While Th17 cells promote autoimmune tissue inflammation, several regulatory cell types contribute to the delicate balance between autoimmunity and immunoregulation. In EAE, a regulatory role was first established for CD8 positive effector cells [51] and later also natural killer (NK)/NK-T cells [52]. Most recent work indicated that particularly regulatory T cells (Treg) characterized by the presence of the forkhead/winged helix transcription factor family member FoxP3 are necessary and sufficient to prevent autoimmunity both in rodents and in man. Interestingly, Th17 cells and FoxP3 positive Treg seem to be dichotomously regulated by TGF-β, which induces FoxP3 in naive T cells, but together with IL-6 drives the generation of Th17 cells [32]. In rat EAE, polyclonal expansion of Treg prevents transmigration of encephalitogenic T cells into the CNS [53]. In a murine model, naturally occurring FoxP3 positive Treg expand in the peripheral immune organs and also accumulate in the central nervous system (CNS), but then do not prevent the onset of EAE [54]. Thus, therapeutic approaches strengthening regulatory mechanisms may be more promising when additional regimens control tissue inflammation in parallel.
Histopathology of EAE reflects many aspects of MS
Classical histopathological changes in MS lesions comprise perivascular inflammation, demyelination and gliosis in the white matter. Over the past 10 years, this concept has been refined and axonal injury, cortical demyelination as well as neuronal changes have come into the focus of interest. In acute demyelinating MS lesions, distinct patterns have been described pointing to patient-specific mechanisms of demyelination. In this classification, pattern I is characterized by the presence of T cells and macrophages and pattern II by antibody and complement deposition. In contrast, patterns III and IV are mediated by primary OL damage. In pattern III, an ischemic mechanism leading to loss of MAG, but not of MOG immunoreactivity, was postulated [55]. So far, EAE models predominantly reflect patterns I and II. In particular, antibody and complement deposition are detected in MOG-EAE lesions of marmosets and rats which are reminiscent of pattern II pathology [56]. In contrast, MOG-EAE in the C57BL/6 mouse results in less well demarcated demyelination which, after immunization with the T cell epitope MOG 35-55, is independent of humoral immune responses as revealed in μMT mice lacking mature B cells [57] and in models with deficiency for the complement component C3 or the C5a receptor [58, 59]. Although the exact mode of demyelination in this model remains to be determined, myelin damage seems to be critically dependent on the presence of macrophages and possibly also tumor necrosis factor (TNF)-α. To evoke a significant B cell response in C57BL/6 mice, immunization with whole recombinant MOG protein is preferred. Studies on the role of antibodies may also involve co-transfer approaches. Here, a role for complement activation, but not FcRγ receptors on phagocytes, could be delineated for antibody mediated demyelination [60].
To a large extent, inflammatory demyelination in MOG-EAE is paralleled by apoptotic loss of OL in the lesions [61]. Thus, oligodendroglial repopulation may constitute an important prerequisite for myelin repair. Indeed, OL precursor cells have been identified in the rodent and human adult CNS (for review see [62]). In different experimental models of demyelination including EAE, oligodendroglial cells are recruited, proliferate and can differentiate into mature OL [63]. Here, glial growth factors as well as the re-expression of developmentally regulated oligodendroglial pathways, including expression of potassium channels like Kv1.4, may be relevant [64]. Yet, in murine MOG-EAE lesions, repair is largely incomplete resulting in an accumulation of lesions over time which correlates with persistent disability of mice. Thus, mechanisms of remyelination are preferably studied in toxic models of demyelination including the application of cuprizone, lysolecithine or ethidium bromide (for review see [65]).
In recent years, further research focused on the presence of grey matter demyelination in the cortex. Although this specific feature is not predominant in murine MOG-EAE, cortical demyelination was well characterized in a focal model of rat MOG-EAE [66] and in some Lewis rat strains (LEW.1AR1 and LEW.1W strains, [67]).
With the avenue of confocal and magnetic resonance imaging techniques, the importance of axonal damage re-entered the stage in neuroimmunological research [68]. In rat MOG-EAE models, patterns of axonal damage and, in some settings, also neuronal injury, were characterized [69, 70]. After immunization with recombinant MOG, axonal damage was observed in the white matter of marmoset monkeys [17] and, in focal EAE models, also in the cortex of susceptible rat strains [66]. Investigation of axonal damage in chronic rat EAE and murine PLP-EAE underscored the concept that loss of axons may in particular correlate with permanent disability [71]. In murine MOG-EAE, axonal loss occurred early in lesions of white matter, but also in the grey matter of the spinal cord. At the later disease stages, disseminated axonal loss including the normal appearing white matter correlated with persistent disability [72]. Further studies in murine models were mainly performed in genetically engineered mice. Mice with a deficiency of the axonoprotective neurotrophic cytokine ciliary neurotrophic factor (CNTF) exhibited a more severe course of MOG-EAE with enhanced myelin and axonal pathology [61]. In a Thy1-GFP transgenic mouse model, patterns of axonal damage were directly characterized by fluorescent labelling of axon tracts in the spinal cord [73]. A focal EAE model in rats also allows the investigation of axonal remodelling in defined tracts [74]. In the near future, an elegant combination of these approaches in combination with 2-photon microscopy will be feasible in living animals [75]. Different mechanisms contribute to axonal damage during autoimmune demyelination (for overview see [76]). Among others, candidates include TNF-α mediated cytotoxicity, Fas-FasL interaction or glutamate excitotoxicity. Moreover, interaction of cytotoxic CD8 positive T cells with up-regulated major histocompatibility complex (MHC)-I on axons may play a role [77] although mice lacking MHC-I exhibit significant amounts of APP positive axons [78]. In several recent studies, altered expression of ion channels were implicated in axonal injury which may involve the sodoim channels Nav 1.2 or Nav1.6 [72] or the Ca2+ and Na+ permeable acid-sensing ion channel-1 [79]. Finally, EAE models allow investigation not only of axonal injury, but also of recovery and axonoprotection [80]. Recent studies focused on the role of the wlds fusion protein as an endogenous axonal protection mechanism [81] while some earlier studies in rodents characterized neuroprotective treatment approaches, like blocking of glutamate receptors [82]. Moreover, blocking of sodium channels may provide an attractive new therapeutic option (among others, see [83]), although expression of these channels in the immune system may somewhat limit these approaches and first studies in MS were reportedly not successful.
The value of experimental models for imaging studies and innovative therapies
EAE models may not only serve for the characterization of immunological and neurobiological mechanisms, but can also be valuable for the pre-clinical testing of new diagnostic or treatment modalities. Different magnetic resonance imaging (MRI) techniques have been employed to depict EAE lesions in rat brains, including contrast enhancement with gadolinium or ultra small iron particles [84], which allow macrophage tracking [85]. While most investigations focused on MBP-EAE models, some recent studies also investigated MOG-EAE in rats [86], marmosets [87] and also murine EAE models [88]. Yet imaging of spinal cord is still not satisfactory with routine protocols, and is only feasible with high-resolution techniques [89]. Advanced MR protocols may also include diffusion tensor imaging which is able to detect selective vulnerability of cerebral white matter in murine EAE [90]. More sophisticated protocols even allow imaging on a molecular level. These techniques include ultrasound based approaches which are able to depict and quantify adhesion molecules in living EAE rats with an axial resolution in the μm range [91]. Similarly, antibody-conjugated microparticles carrying iron oxide can provide quantifiable MRI contrast effects that delineate the architecture of activated cerebral blood vessels [92].
Regarding treatment approaches, many established present-day MS therapies like glatiramer acetate as well as natalizumab stem from their first successful application in EAE models. Yet a plethora of new therapeutic approaches are effective in animal models, but have failed in human trials. Well known failures include anti-TNF therapies in MS patients [93] as well as the application of superagonistic anti-CD28 antibodies in healthy volunteers [94]. In case of the anti-TNF studies, negative results in patients were better understood when data from knockout mouse models became available which revealed a beneficial role for TNF-α in apoptotic clearance of T cells from inflammatory lesions [95]. Even though a thorough characterization of CD28 superagonistic antibody therapy in animal models of both peripheral and CNS inflammation [96, 97] as well as later on also with live imaging of immune organs [98] was performed, this did not foresee the multiple cytokine release syndrome leading to severe side effects in a human phase I study. These failures call for the highest possible awareness with new therapies in human beings. EAE experiments cannot substitute for, but are rather complementary to, human clinical studies, and maximum safety standards during the first application of new compounds in patients must always apply. Yet, if interpreted correctly and chosen with care, EAE models can provide useful information, and sometimes may avoid complications which delay drug development or pose a threat to patients. In particular, EAE models may help to elucidate mechanisms of action for new MS therapies. One example is fumarate (FAE) therapy in MS, currently being tested in a human phase III trial. In the human skin disease psoriasis, FAE was shown to be effective via a Th1/Th2 shift. Yet, in EAE, FAE seems to exert effects on macrophages [99] and possibly also to harbour neuroprotective effects. Since other regenerative treatment approaches for MS are still a long way ahead, the best neuroprotective approach still consists in effective immunotherapy. This can be convincingly shown in treatment of rat MOG-EAE with liposomal steroids which effectively deplete phagocytes, modulate T cells and at the same time preserve axons in inflammatory lesions [100].
Future developments and new strategies
Future directions include EAE studies in conditional knockout mice to carve out the contribution of single molecules to the induction or effector phase of EAE. First studies used the Cre loxP system to conditionally delete genes involved in nuclear factor-κB (NF-κB) signalling pathways (EAE in CNS-restricted NEMO knockout mice, [101]). Together with the generation of bone marrow chimeras, these techniques provide powerful tools to dissect mechanisms during autoimmune inflammation of the CNS. Other innovative approaches include the immunization of Lewis rats with alpha synuclein leading to grey matter pathology which recently came into the focus of interest [102]. In the grey matter, also contactin2/TAG-1 mediated autoimmunity was recently characterized as contributing to cortical damage [103].
Moreover, the characterization of neurofascin as a novel autoantigen on axons at the node of Ranvier [104] and the identification of a protective role for the candidate astroglial autoantigen αβ crystallin in autoimmune demyelination [105] shed further light on non-myelin proteins as targets in disease pathogenesis. Further strategies are aiming at the implementation of models for aspects of MS which are not well mimicked in EAE paradigms so far. These efforts include studies on CD8 positive T cells which are a major component of the inflammatory infiltrate in chronic MS lesions [106]. In fact, several approaches in mice demonstrated that CD8 positive T lymphocytes also harbor an encephalitogenic potential [107–109] and are able to attack and transect MHC class I expressing axons in an antigen-specific manner. Recently, an elegant viral model studying cytotoxic T cells in lymphocytic choriomeningitis virus (LCMV) infection demonstrated that two related, but independently encountered, viral infections can lead to organ-specific immune disease without molecular mimicry or breaking immune tolerance [110].
CD8 T cell restricted autoimmune reactions can also be studied after conditional expression of MHC-I as shown in the muscle [111] or in double transgenic mice with simultaneous an OL sequestered antigen like ovalbumin or influenca hemagglutinin in the context of a respective MHC-I restricted T cell receptor [112, 113]. In such settings, monoclonal antibodies specific for MHC class I molecules presenting a dominant autoantigen peptide may allow selective immunotherapy [114]. Such transgenic approaches were also used to study CD4 T cell and B cell mediated immune reactions. In genetically engineered mice exclusively harbouring an MBP specific T cell receptor (TCR), EAE develops spontaneously with a high incidence [115]. In a similar approach, MOG-TCR transgenic mice suffer from spontaneous autoimmune optic neuritis to some degree [116]. In contrast, double transgenic mice with a MOG specific TCR and MOG-specific B cells spontaneously develop a Devic like disease pattern with lesions exclusively in the optic nerve and spinal cord, thus nicely underscoring the cooperation of B cells and T cells in the development of autoimmune inflammation [117, 118]. These spontaneously occurring models do not require artificial immunization protocols with adjuvant or pertussis toxin and are therefore of special value for neuroimmunological studies. In this MOG TCR transgenic model, autoreactive T cells harbor a bi-specificity and recognize their MOG epitope, but also neurofilament M as self-antigen. Such cumulative responses against several autoantigens by one autoreactive T cell clone may also contribute to pathogenicity in MS [119]. Whilst most of these spontaneously occurring models lead to a chronic disease course, a recently developed model with a transgenic MOG92-106 specific TCR in the context of an I-A(s) background was also able to spontaneously mimic relapsing remitting disease course [120]. Further elegant tools are humanized transgenic mice which can be used as tools to study human T cell receptors (TCR) in experimental models. This was first shown in a transgenic mouse model for an MHC-II restricted TCR specific for MBP84-102 [121]. While the incidence of spontaneous EAE in this model is low, immunization with a microbial peptide results in induction of disease, which may be due to structural mimicry of a binding hotspot shared by self and microbial antigens rather than to a degenerate TCR recognition [122]. Recently, such concepts were also analyzed in a transgenic model harboring a myelin specific T cell receptor of a CD8 T cell clone from a MS patient and the respective MHC class I allele. In this model, CD 8 positive T cells can exert disease promoting or protective functions, depending on the MHC class I context [123].
So far, EAE models cover the spectrum of demyelination patterns I and II, but they do not well reflect patterns III and IV with primary OL dystrophy. Yet, this obstacle may be overcome by the analysis of mutants with primary demyelination due to a genetic defect. This was recently suggested in a study of mice with peroxisome-deficient OL. Here, a metabolic defect in myelin producing cells led to demyelination and subsequently also CNS inflammation with infiltration of B cells and activated CD8 positive T cells [124], similar to previous investigations in knockout models of PNS myelin [125].
Finally, all these data from EAE models may also promote knowledge on immune reactions in other neurologic diseases, like infectious disorders of the CNS [126] or post-stroke immunodepression [127].
References
Cassan C, Piaggio E, Zappulla JP, Mars LT, Couturier N, Bucciarelli F, Desbois S, Bauer J, Gonzalez-Dunia D, Liblau RS: Pertussis toxin reduces the number of splenic Foxp3+ regulatory T cells. J Immunol 2006, 177: 1552–1560.
Traugott U, Raine CS: Acute experimental allergic encephalomyelitis. Myelin basic protein-reactive T cells in the circulation and in meningeal infiltrates. J Neurol Sci 1979, 42: 331–336. 10.1016/0022-510X(79)90166-7
Ben-Nun A, Lando Z: Detection of autoimmune cells proliferating to myelin basic protein and selection of T cell lines that mediate experimental autoimmune encephalomyelitis (EAE) in mice. J Immunol 1983, 130: 1205–1209.
Wekerle H, Kojima K, Lannes-Vieira J, Lassmann H, Linington C: Animal models. Ann Neurol 1994, 36(Suppl):S47-S53. 10.1002/ana.410360714
Pellkofer H, Schubart AS, Hoftberger R, Schutze N, Pagany M, Schuller M, Lassmann H, Hohlfeld R, Voltz R, Linington C: Modelling paraneoplastic CNS disease: T-cells specific for the onconeuronal antigen PNMA1 mediate autoimmune encephalomyelitis in the rat. Brain 2004, 127: 1822–1830. 10.1093/brain/awh205
Furlan R, Brambilla E, Sanvito F, Roccatagliata L, Olivieri S, Bergami A, Pluchino S, Uccelli A, Comi G, Martino G: Vaccination with amyloid-beta peptide induces autoimmune encephalomyelitis in C57/BL6 mice. Brain 2003, 126: 285–291. 10.1093/brain/awg031
Sospedra M, Martin R: Immunology of multiple sclerosis. Annu Rev Immunol 2005, 23: 683–747. 10.1146/annurev.immunol.23.021704.115707
Linington C, Bradl M, Lassmann H, Brunner C, Vass K: Augmentation of demyelination in rat acute allergic encephalomyelitis by circulating mouse monoclonal antibodies directed against a myelin/oligodendrocyte glycoprotein. Am J Pathol 1988, 130: 443–454.
Pender MP: The pathophysiology of myelin basic protein-induced acute experimental allergic encephalomyelitis in the Lewis rat. J Neurol Sci 1988, 86: 277–289. 10.1016/0022-510X(88)90105-0
Linker RA, Gold R: MBP-induced experimental autoimmune encephalomyelitis in C57BL/6 mice. J Immunol 2004, 173: 2896.
Linington C, Lassmann H: Antibody responses in chronic relapsing experimental allergic encephalomyelitis: correlation of serum demyelinating activity with antibody titre to the myelin/oligodendrocyte glycoprotein (MOG). J Neuroimmunol 1987, 17: 61–69. 10.1016/0165-5728(87)90031-2
Massacesi L, Genain CP, Lee-Parritz D, Letvin NL, Canfield D, Hauser SL: Active and passively induced experimental autoimmune encephalomyelitis in common marmosets: a new model for multiple sclerosis. Ann Neurol 1995, 37: 519–530. 10.1002/ana.410370415
Zhou D, Srivastava R, Nessler S, Grummel V, Sommer N, Bruck W, Hartung HP, Stadelmann C, Hemmer B: Identification of a pathogenic antibody response to native myelin oligodendrocyte glycoprotein in multiple sclerosis. Proc Natl Acad Sci USA 2006, 103: 19057–19062. 10.1073/pnas.0607242103
Weissert R, Wallstrom E, Storch MK, Stefferl A, Lorentzen J, Lassmann H, Linington C, Olsson T: MHC haplotype-dependent regulation of MOG-induced EAE in rats. J Clin Invest 1998, 102: 1265–1273. 10.1172/JCI3022
Storch MK, Stefferl A, Brehm U, Weissert R, Wallstrom E, Kerschensteiner M, Olsson T, Linington C, Lassmann H: Autoimmunity to myelin oligodendrocyte glycoprotein in rats mimics the spectrum of multiple sclerosis pathology. Brain Pathol 1998, 8: 681–694.
Stefferl A, Brehm U, Storch M, Lambracht-Washington D, Bourquin C, Wonigeit K, Lassmann H, Linington C: Myelin oligodendrocyte glycoprotein induces experimental autoimmune encephalomyelitis in the "resistant" Brown Norway rat: disease susceptibility is determined by MHC and MHC-linked effects on the B cell response. J Immunol 1999, 163: 40–49.
Mancardi G, Hart B, Roccatagliata L, Brok H, Giunti D, Bontrop R, Massacesi L, Capello E, Uccelli A: Demyelination and axonal damage in a non-human primate model of multiple sclerosis. J Neurol Sci 2001, 184: 41–49. 10.1016/S0022-510X(00)00490-1
Merkler D, Boscke R, Schmelting B, Czeh B, Fuchs E, Bruck W, Stadelmann C: Differential macrophage/microglia activation in neocortical EAE lesions in the marmoset monkey. Brain Pathol 2006, 16: 117–123. 10.1111/j.1750-3639.2006.00004.x
von Budingen HC, Hauser SL, Ouallet JC, Tanuma N, Menge T, Genain CP: Frontline: Epitope recognition on the myelin/oligodendrocyte glycoprotein differentially influences disease phenotype and antibody effector functions in autoimmune demyelination. Eur J Immunol 2004, 34: 2072–2083. 10.1002/eji.200425050
Prinz M, Garbe F, Schmidt H, Mildner A, Gutcher I, Wolter K, Piesche M, Schroers R, Weiss E, Kirschning CJ, et al.: Innate immunity mediated by TLR9 modulates pathogenicity in an animal model of multiple sclerosis. J Clin Invest 2006, 116: 456–464. 10.1172/JCI26078
Secor VH, Secor WE, Gutekunst CA, Brown MA: Mast cells are essential for early onset and severe disease in a murine model of multiple sclerosis. J Exp Med 2000, 191: 813–822. 10.1084/jem.191.5.813
Greter M, Heppner FL, Lemos MP, Odermatt BM, Goebels N, Laufer T, Noelle RJ, Becher B: Dendritic cells permit immune invasion of the CNS in an animal model of multiple sclerosis. Nat Med 2005, 11: 328–334. 10.1038/nm1197
Heppner FL, Greter M, Marino D, Falsig J, Raivich G, Hovelmeyer N, Waisman A, Rulicke T, Prinz M, Priller J, et al.: Experimental autoimmune encephalomyelitis repressed by microglial paralysis. Nat Med 2005, 11: 146–152. 10.1038/nm1177
Litzenburger T, Fassler R, Bauer J, Lassmann H, Linington C, Wekerle H, Iglesias A: B lymphocytes producing demyelinating autoantibodies: development and function in gene-targeted transgenic mice. J Exp Med 1998, 188: 169–180. 10.1084/jem.188.1.169
Matsushita T, Yanaba K, Bouaziz JD, Fujimoto M, Tedder TF: Regulatory B cells inhibit EAE initiation in mice while other B cells promote disease progression. J Clin Invest 2008, 118: 3420–3430.
Ben-Nun A, Liblau RS, Cohen L, Lehmann D, Tournier-Lasserve E, Rosenzweig A, Zhang JW, Raus JC, Bach MA: Restricted T-cell receptor V beta gene usage by myelin basic protein-specific T-cell clones in multiple sclerosis: predominant genes vary in individuals. Proc Natl Acad Sci USA 1991, 88: 2466–2470. 10.1073/pnas.88.6.2466
Racke MK, Ratts RB, Arredondo L, Perrin PJ, Lovett-Racke A: The role of costimulation in autoimmune demyelination. J Neuroimmunol 2000, 107: 205–215. 10.1016/S0165-5728(00)00230-7
Zozulya AL, Ortler S, Lee J, Weidenfeller C, Sandor M, Wiendl H, Fabry Z: Intracerebral dendritic cells critically modulate encephalitogenic versus regulatory immune responses in the CNS. J Neurosci 2009, 29: 140–152. 10.1523/JNEUROSCI.2199-08.2009
Gran B, Zhang GX, Rostami A: Role of the IL-12/IL-23 system in the regulation of T-cell responses in central nervous system inflammatory demyelination. Crit Rev Immunol 2004, 24: 111–128. 10.1615/CritRevImmunol.v24.i2.20
Kebir H, Kreymborg K, Ifergan I, Dodelet-Devillers A, Cayrol R, Bernard M, Giuliani F, Arbour N, Becher B, Prat A: Human TH17 lymphocytes promote blood-brain barrier disruption and central nervous system inflammation. Nat Med 2007, 13: 1173–1175. 10.1038/nm1651
Tzartos JS, Friese MA, Craner MJ, Palace J, Newcombe J, Esiri MM, Fugger L: Interleukin-17 Production in Central Nervous System-Infiltrating T Cells and Glial Cells Is Associated with Active Disease in Multiple Sclerosis. Am J Pathol 2007, 172: 146–155. 10.2353/ajpath.2008.070690
Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK: Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 2006, 441: 235–238. 10.1038/nature04753
Korn T, Bettelli E, Gao W, Awasthi A, Jager A, Strom TB, Oukka M, Kuchroo VK: IL-21 initiates an alternative pathway to induce proinflammatory T(H)17 cells. Nature 2007, 448: 484–487. 10.1038/nature05970
Gutcher I, Urich E, Wolter K, Prinz M, Becher B: Interleukin 18-independent engagement of interleukin 18 receptor-alpha is required for autoimmune inflammation. Nat Immunol 2006, 7: 946–953. 10.1038/ni1377
Kreymborg K, Etzensperger R, Dumoutier L, Haak S, Rebollo A, Buch T, Heppner FL, Renauld JC, Becher B: IL-22 Is Expressed by Th17 Cells in an IL-23-Dependent Fashion, but Not Required for the Development of Autoimmune Encephalomyelitis. J Immunol 2007, 179: 8098–8104.
Reboldi A, Coisne C, Baumjohann D, Benvenuto F, Bottinelli D, Lira S, Uccelli A, Lanzavecchia A, Engelhardt B, Sallusto F: C-C chemokine receptor 6-regulated entry of TH-17 cells into the CNS through the choroid plexus is required for the initiation of EAE. Nat Immunol 2009, 10: 514–523. 10.1038/ni.1716
Ogura H, Murakami M, Okuyama Y, Tsuruoka M, Kitabayashi C, Kanamoto M, Nishihara M, Iwakura Y, Hirano T: Interleukin-17 promotes autoimmunity by triggering a positive-feedback loop via interleukin-6 induction. Immunity 2008, 29: 628–636. 10.1016/j.immuni.2008.07.018
Haak S, Croxford AL, Kreymborg K, Heppner FL, Pouly S, Becher B, Waisman A: IL-17A and IL-17F do not contribute vitally to autoimmune neuro-inflammation in mice. J Clin Invest 2009, 119: 61–69.
Yang Y, Weiner J, Liu Y, Smith AJ, Huss DJ, Winger R, Peng H, Cravens PD, Racke MK, Lovett-Racke AE: T-bet is essential for encephalitogenicity of both Th1 and Th17 cells. J Exp Med 2009, 206: 1549–1564. 10.1084/jem.20082584
Dardalhon V, Awasthi A, Kwon H, Galileos G, Gao W, Sobel RA, Mitsdoerffer M, Strom TB, Elyaman W, Ho IC, et al.: IL-4 inhibits TGF-beta-induced Foxp3+ T cells and, together with TGF-beta, generates IL-9+ IL-10+ Foxp3(-) effector T cells. Nat Immunol 2008, 9: 1347–1355. 10.1038/ni.1677
Nowak EC, Weaver CT, Turner H, Begum-Haque S, Becher B, Schreiner B, Coyle AJ, Kasper LH, Noelle RJ: IL-9 as a mediator of Th17-driven inflammatory disease. J Exp Med 2009, 206: 1653–1660. 10.1084/jem.20090246
Lafaille JJ, Keere FV, Hsu AL, Baron JL, Haas W, Raine CS, Tonegawa S: Myelin basic protein-specific T helper 2 (Th2) cells cause experimental autoimmune encephalomyelitis in immunodeficient hosts rather than protect them from the disease. J Exp Med 1997, 186: 307–312. 10.1084/jem.186.2.307
Maatta JA, Sjoholm UR, Nygardas PT, Salmi AA, Hinkkanen AE: Neutrophils secreting tumor necrosis factor alpha infiltrate the central nervous system of BALB/c mice with experimental autoimmune encephalomyelitis. J Neuroimmunol 1998, 90: 162–175. 10.1016/S0165-5728(98)00135-0
Lu LF, Lind EF, Gondek DC, Bennett KA, Gleeson MW, Pino-Lagos K, Scott ZA, Coyle AJ, Reed JL, Van Snick J, et al.: Mast cells are essential intermediaries in regulatory T-cell tolerance. Nature 2006, 442: 997–1002. 10.1038/nature05010
Archelos JJ, Hartung HP: The role of adhesion molecules in multiple sclerosis: biology, pathogenesis and therapeutic implications. Mol Med Today 1997, 3: 310–321. 10.1016/S1357-4310(97)01066-6
Engelhardt B, Conley FK, Butcher EC: Cell adhesion molecules on vessels during inflammation in the mouse central nervous system. J Neuroimmunol 1994, 51: 199–208. 10.1016/0165-5728(94)90082-5
Archelos JJ, Previtali SC, Hartung HP: The role of integrins in immune-mediated diseases of the nervous system. Trends Neurosci 1999, 22: 30–38. 10.1016/S0166-2236(98)01287-9
Engelhardt B, Vajkoczy P, Laschinger M: Detection of endothelial/lymphocyte interaction in spinal cord microvasculature by intravital videomicroscopy. Methods Mol Med 2003, 89: 83–93.
Cayrol R, Wosik K, Berard JL, Dodelet-Devillers A, Ifergan I, Kebir H, Haqqani AS, Kreymborg K, Krug S, Moumdjian R, et al.: Activated leukocyte cell adhesion molecule promotes leukocyte trafficking into the central nervous system. Nat Immunol 2007, 9: 137–145. 10.1038/ni1551
Ifergan I, Kebir H, Bernard M, Wosik K, Dodelet-Devillers A, Cayrol R, Arbour N, Prat A: The blood brain barrier induces differentiation of migrating monocytes into Th17-polarizing dendritic cells. Brain 2007, 131: 785–799. 10.1093/brain/awm295
Koh DR, Fung-Leung WP, Ho A, Gray D, Acha-Orbea H, Mak TW: Less mortality but more relapses in experimental allergic encephalomyelitis in CD8-/- mice. Science 1992, 256: 1210–1213. 10.1126/science.256.5060.1210
Zhang B, Yamamura T, Kondo T, Fujiwara M, Tabira T: Regulation of experimental autoimmune encephalomyelitis by natural killer (NK) cells. J Exp Med 1997, 186: 1677–1687. 10.1084/jem.186.10.1677
Tischner D, Weishaupt A, van den Brandt J, Muller N, Beyersdorf N, Ip CW, Toyka KV, Hunig T, Gold R, Kerkau T, et al.: Polyclonal expansion of regulatory T cells interferes with effector cell migration in a model of multiple sclerosis. Brain 2006, 129: 2635–2647. 10.1093/brain/awl213
Korn T, Reddy J, Gao W, Bettelli E, Awasthi A, Petersen TR, Backstrom BT, Sobel RA, Wucherpfennig KW, Strom TB, et al.: Myelin-specific regulatory T cells accumulate in the CNS but fail to control autoimmune inflammation. Nat Med 2007, 13: 423–431. 10.1038/nm1564
Lassmann H, Bruck W, Lucchinetti CF: The immunopathology of multiple sclerosis: an overview. Brain Pathol 2007, 17: 210–218. 10.1111/j.1750-3639.2007.00064.x
Merkler D, Schmelting B, Czeh B, Fuchs E, Stadelmann C, Bruck W: Myelin oligodendrocyte glycoprotein-induced experimental autoimmune encephalomyelitis in the common marmoset reflects the immunopathology of pattern II multiple sclerosis lesions. Mult Scler 2006, 12: 369–374. 10.1191/1352458506ms1290oa
Dittel BN, Urbania TH, Janeway CA Jr: Relapsing and remitting experimental autoimmune encephalomyelitis in B cell deficient mice. J Autoimmun 2000, 14: 311–318. 10.1006/jaut.2000.0371
Calida DM, Constantinescu C, Purev E, Zhang GX, Ventura ES, Lavi E, Rostami A: Cutting edge: C3, a key component of complement activation, is not required for the development of myelin oligodendrocyte glycoprotein peptide-induced experimental autoimmune encephalomyelitis in mice. J Immunol 2001, 166: 723–726.
Reiman R, Gerard C, Campbell IL, Barnum SR: Disruption of the C5a receptor gene fails to protect against experimental allergic encephalomyelitis. Eur J Immunol 2002, 32: 1157–1163. 10.1002/1521-4141(200204)32:4<1157::AID-IMMU1157>3.0.CO;2-M
Urich E, Gutcher I, Prinz M, Becher B: Autoantibody-mediated demyelination depends on complement activation but not activatory Fc-receptors. Proc Natl Acad Sci USA 2006, 103: 18697–18702. 10.1073/pnas.0607283103
Linker RA, Maurer M, Gaupp S, Martini R, Holtmann B, Giess R, Rieckmann P, Lassmann H, Toyka KV, Sendtner M, et al.: CNTF is a major protective factor in demyelinating CNS disease: a neurotrophic cytokine as modulator in neuroinflammation. Nat Med 2002, 8: 620–624. 10.1038/nm0602-620
Levine JM, Reynolds R, Fawcett JW: The oligodendrocyte precursor cell in health and disease. Trends Neurosci 2001, 24: 39–47. 10.1016/S0166-2236(00)01691-X
Reynolds R, Dawson M, Papadopoulos D, Polito A, Di BI, Pham-Dinh D, Levine J: The response of NG2-expressing oligodendrocyte progenitors to demyelination in MOG-EAE and MS. J Neurocytol 2002, 31: 523–536. 10.1023/A:1025747832215
Herrero-Herranz E, Pardo LA, Bunt G, Gold R, Stuhmer W, Linker RA: Re-expression of a developmentally restricted potassium channel in autoimmune demyelination: Kv1.4 is implicated in oligodendroglial proliferation. Am J Pathol 2007, 171: 589–598. 10.2353/ajpath.2007.061241
Zawadzka M, Franklin RJ: Myelin regeneration in demyelinating disorders: new developments in biology and clinical pathology. Curr Opin Neurol 2007, 20: 294–298. 10.1097/WCO.0b013e32813aee7f
Merkler D, Ernsting T, Kerschensteiner M, Bruck W, Stadelmann C: A new focal EAE model of cortical demyelination: multiple sclerosis-like lesions with rapid resolution of inflammation and extensive remyelination. Brain 2006, 129: 1972–1983. 10.1093/brain/awl135
Storch MK, Bauer J, Linington C, Olsson T, Weissert R, Lassmann H: Cortical demyelination can be modeled in specific rat models of autoimmune encephalomyelitis and is major histocompatability complex (MHC) haplotype-related. J Neuropathol Exp Neurol 2006, 65: 1137–1142. 10.1097/01.jnen.0000248547.13176.9d
Trapp BD, Peterson J, Ransohoff RM, Rudick R, Mork S, Bo L: Axonal transection in the lesions of multiple sclerosis. N Engl J Med 1998, 338: 278–285. 10.1056/NEJM199801293380502
Hobom M, Storch MK, Weissert R, Maier K, Radhakrishnan A, Kramer B, Bahr M, Diem R: Mechanisms and time course of neuronal degeneration in experimental autoimmune encephalomyelitis. Brain Pathol 2004, 14: 148–157.
Kornek B, Storch MK, Weissert R, Wallstroem E, Stefferl A, Olsson T, Linington C, Schmidbauer M, Lassmann H: Multiple sclerosis and chronic autoimmune encephalomyelitis: a comparative quantitative study of axonal injury in active, inactive, and remyelinated lesions. Am J Pathol 2000, 157: 267–276.
Wujek JR, Bjartmar C, Richer E, Ransohoff RM, Yu M, Tuohy VK, Trapp BD: Axon loss in the spinal cord determines permanent neurological disability in an animal model of multiple sclerosis. J Neuropathol Exp Neurol 2002, 61: 23–32.
Herrero-Herranz E, Pardo LA, Gold R, Linker RA: Pattern of axonal injury in murine myelin oligodendrocyte glycoprotein induced experimental autoimmune encephalomyelitis: implications for multiple sclerosis. Neurobiol Dis 2008, 30: 162–173. 10.1016/j.nbd.2008.01.001
Bannerman PG, Hahn A: Enhanced visualization of axonopathy in EAE using thy1-YFP transgenic mice. J Neurol Sci 2007, 260: 23–32. 10.1016/j.jns.2007.03.020
Kerschensteiner M, Bareyre FM, Buddeberg BS, Merkler D, Stadelmann C, Bruck W, Misgeld T, Schwab ME: Remodeling of axonal connections contributes to recovery in an animal model of multiple sclerosis. J Exp Med 2004, 200: 1027–1038. 10.1084/jem.20040452
Kerschensteiner M, Schwab ME, Lichtman JW, Misgeld T: In vivo imaging of axonal degeneration and regeneration in the injured spinal cord. Nat Med 2005, 11: 572–577. 10.1038/nm1229
Linker RA, Sendtner M, Gold R: Mechanisms of axonal degeneration in EAE--lessons from CNTF and MHC I knockout mice. J Neurol Sci 2005, 233: 167–172. 10.1016/j.jns.2005.03.021
Medana I, Martinic MA, Wekerle H, Neumann H: Transection of major histocompatibility complex class I-induced neurites by cytotoxic T lymphocytes. Am J Pathol 2001, 159: 809–815.
Linker RA, Rott E, Hofstetter HH, Hanke T, Toyka KV, Gold R: EAE in beta-2 microglobulin-deficient mice: axonal damage is not dependent on MHC-I restricted immune responses. Neurobiol Dis 2005, 19: 218–228. 10.1016/j.nbd.2004.12.017
Friese MA, Craner MJ, Etzensperger R, Vergo S, Wemmie JA, Welsh MJ, Vincent A, Fugger L: Acid-sensing ion channel-1 contributes to axonal degeneration in autoimmune inflammation of the central nervous system. Nat Med 2007, 13: 1483–1489. 10.1038/nm1668
Diem R, Sattler MB, Bahr M: Neurodegeneration and -protection in autoimmune CNS inflammation. J Neuroimmunol 2007, 184: 27–36. 10.1016/j.jneuroim.2006.11.025
Kaneko S, Wang J, Kaneko M, Yiu G, Hurrell JM, Chitnis T, Khoury SJ, He Z: Protecting axonal degeneration by increasing nicotinamide adenine dinucleotide levels in experimental autoimmune encephalomyelitis models. J Neurosci 2006, 26: 9794–9804. 10.1523/JNEUROSCI.2116-06.2006
Pitt D, Werner P, Raine CS: Glutamate excitotoxicity in a model of multiple sclerosis. Nat Med 2000, 6: 67–70. 10.1038/71555
Black JA, Liu S, Hains BC, Saab CY, Waxman SG: Long-term protection of central axons with phenytoin in monophasic and chronic-relapsing EAE. Brain 2006, 129: 3196–3208. 10.1093/brain/awl216
Baeten K, Hendriks JJ, Hellings N, Theunissen E, Vanderlocht J, Ryck LD, Gelan J, Stinissen P, Adriaensens P: Visualisation of the kinetics of macrophage infiltration during experimental autoimmune encephalomyelitis by magnetic resonance imaging. J Neuroimmunol 2008, 195: 1–6. 10.1016/j.jneuroim.2007.11.008
Bendszus M, Stoll G: Caught in the act: in vivo mapping of macrophage infiltration in nerve injury by magnetic resonance imaging. J Neurosci 2003, 23: 10892–10896.
Linker RA, Kroner A, Horn T, Gold R, Maurer M, Bendszus M: Iron particle-enhanced visualization of inflammatory central nervous system lesions by high resolution: preliminary data in an animal model. AJNR Am J Neuroradiol 2006, 27: 1225–1229.
Boretius S, Schmelting B, Watanabe T, Merkler D, Tammer R, Czeh B, Michaelis T, Frahm J, Fuchs E: Monitoring of EAE onset and progression in the common marmoset monkey by sequential high-resolution 3D MRI. NMR Biomed 2006, 19: 41–49. 10.1002/nbm.999
Nessler S, Boretius S, Stadelmann C, Bittner A, Merkler D, Hartung HP, Michaelis T, Bruck W, Frahm J, Sommer N, et al.: Early MRI changes in a mouse model of multiple sclerosis are predictive of severe inflammatory tissue damage. Brain 2007, 130: 2186–2198. 10.1093/brain/awm105
Steinbrecher A, Weber T, Neuberger T, Mueller AM, Pedre X, Giegerich G, Bogdahn U, Jakob P, Haase A, Faber C: Experimental autoimmune encephalomyelitis in the rat spinal cord: lesion detection with high-resolution MR microscopy at 17.6 T. AJNR Am J Neuroradiol 2005, 26: 19–25.
Budde MD, Kim JH, Liang HF, Schmidt RE, Russell JH, Cross AH, Song SK: Toward accurate diagnosis of white matter pathology using diffusion tensor imaging. Magn Reson Med 2007, 57: 688–695. 10.1002/mrm.21200
Reinhardt M, Hauff P, Linker RA, Briel A, Gold R, Rieckmann P, Becker G, Toyka KV, Maurer M, Schirner M: Ultrasound derived imaging and quantification of cell adhesion molecules in experimental autoimmune encephalomyelitis (EAE) by Sensitive Particle Acoustic Quantification (SPAQ). Neuroimage 2005, 27: 267–278. 10.1016/j.neuroimage.2005.04.019
McAteer MA, Sibson NR, von Zur MC, Schneider JE, Lowe AS, Warrick N, Channon KM, Anthony DC, Choudhury RP: In vivo magnetic resonance imaging of acute brain inflammation using microparticles of iron oxide. Nat Med 2007, 13: 1253–1258. 10.1038/nm1631
TNF neutralization in MS: results of a randomized, placebo-controlled multicenter study. The Lenercept Multiple Sclerosis Study Group and The University of British Columbia MS/MRI Analysis Group Neurology 1999, 53: 457–465.
Suntharalingam G, Perry MR, Ward S, Brett SJ, Castello-Cortes A, Brunner MD, Panoskaltsis N: Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N Engl J Med 2006, 355: 1018–1028. 10.1056/NEJMoa063842
Liu J, Marino MW, Wong G, Grail D, Dunn A, Bettadapura J, Slavin AJ, Old L, Bernard CC: TNF is a potent anti-inflammatory cytokine in autoimmune-mediated demyelination. Nat Med 1998, 4: 78–83. 10.1038/nm0198-078
Beyersdorf N, Gaupp S, Balbach K, Schmidt J, Toyka KV, Lin CH, Hanke T, Hunig T, Kerkau T, Gold R: Selective targeting of regulatory T cells with CD28 superagonists allows effective therapy of experimental autoimmune encephalomyelitis. J Exp Med 2005, 202: 445–455. 10.1084/jem.20051060
Schmidt J, Elflein K, Stienekemeier M, Rodriguez-Palmero M, Schneider C, Toyka KV, Gold R, Hunig T: Treatment and prevention of experimental autoimmune neuritis with superagonistic CD28-specific monoclonal antibodies. J Neuroimmunol 2003, 140: 143–152. 10.1016/S0165-5728(03)00182-6
Muller N, van den Brandt J, Odoardi F, Tischner D, Herath J, Flugel A, Reichardt HM: A CD28 superagonistic antibody elicits 2 functionally distinct waves of T cell activation in rats. J Clin Invest 2008, 118: 1405–1416. 10.1172/JCI32698
Schilling S, Goelz S, Linker R, Luehder F, Gold R: Fumaric acid esters are effective in chronic experimental autoimmune encephalomyelitis and suppress macrophage infiltration. Clin Exp Immunol 2006, 145: 101–107. 10.1111/j.1365-2249.2006.03094.x
Linker RA, Weller C, Luhder F, Mohr A, Schmidt J, Knauth M, Metselaar JM, Gold R: Liposomal glucocorticosteroids in treatment of chronic autoimmune demyelination: long-term protective effects and enhanced efficacy of methylprednisolone formulations. Exp Neurol 2008, 211: 397–406. 10.1016/j.expneurol.2008.02.005
van Loo G, De Lorenzi R, Schmidt H, Huth M, Mildner A, Schmidt-Supprian M, Lassmann H, Prinz MR, Pasparakis M: Inhibition of transcription factor NF-kappaB in the central nervous system ameliorates autoimmune encephalomyelitis in mice. Nat Immunol 2006, 7: 954–961. 10.1038/ni1372
Steinman L: The gray aspects of white matter disease in multiple sclerosis. Proc Natl Acad Sci USA 2009, 106: 8083–8084. 10.1073/pnas.0903377106
Derfuss T, Parikh K, Velhin S, Braun M, Mathey E, Krumbholz M, Kumpfel T, Moldenhauer A, Rader C, Sonderegger P, et al.: Contactin-2/TAG-1-directed autoimmunity is identified in multiple sclerosis patients and mediates gray matter pathology in animals. Proc Natl Acad Sci USA 2009, 106: 8302–8307. 10.1073/pnas.0901496106
Mathey EK, Derfuss T, Storch MK, Williams KR, Hales K, Woolley DR, Al-Hayani A, Davies SN, Rasband MN, Olsson T, et al.: Neurofascin as a novel target for autoantibody-mediated axonal injury. J Exp Med 2007, 204: 2363–2372. 10.1084/jem.20071053
Ousman SS, Tomooka BH, van Noort JM, Wawrousek EF, O'Connor KC, Hafler DA, Sobel RA, Robinson WH, Steinman L: Protective and therapeutic role for alphaB-crystallin in autoimmune demyelination. Nature 2007, 448: 474–479. 10.1038/nature05935
Babbe H, Roers A, Waisman A, Lassmann H, Goebels N, Hohlfeld R, Friese M, Schroder R, Deckert M, Schmidt S, et al.: Clonal expansions of CD8(+) T cells dominate the T cell infiltrate in active multiple sclerosis lesions as shown by micromanipulation and single cell polymerase chain reaction. J Exp Med 2000, 192: 393–404. 10.1084/jem.192.3.393
Goverman J, Perchellet A, Huseby ES: The role of CD8(+) T cells in multiple sclerosis and its animal models. Curr Drug Targets Inflamm Allergy 2005, 4: 239–245. 10.2174/1568010053586264
Huseby ES, Liggitt D, Brabb T, Schnabel B, Ohlen C, Goverman J: A pathogenic role for myelin-specific CD8(+) T cells in a model for multiple sclerosis. J Exp Med 2001, 194: 669–676. 10.1084/jem.194.5.669
Sun D, Whitaker JN, Huang Z, Liu D, Coleclough C, Wekerle H, Raine CS: Myelin antigen-specific CD8+ T cells are encephalitogenic and produce severe disease in C57BL/6 mice. J Immunol 2001, 166: 7579–7587.
Merkler D, Horvath E, Bruck W, Zinkernagel RM, Del la Torre JC, Pinschewer DD: "Viral deja vu" elicits organ-specific immune disease independent of reactivity to self. J Clin Invest 2006, 116: 1254–1263. 10.1172/JCI27372
Nagaraju K, Raben N, Loeffler L, Parker T, Rochon PJ, Lee E, Danning C, Wada R, Thompson C, Bahtiyar G, et al.: Conditional up-regulation of MHC class I in skeletal muscle leads to self-sustaining autoimmune myositis and myositis-specific autoantibodies. Proc Natl Acad Sci USA 2000, 97: 9209–9214. 10.1073/pnas.97.16.9209
Na SY, Cao Y, Toben C, Nitschke L, Stadelmann C, Gold R, Schimpl A, Hunig T: Naive CD8 T-cells initiate spontaneous autoimmunity to a sequestered model antigen of the central nervous system. Brain 2008, 131: 2353–2365. 10.1093/brain/awn148
Saxena A, Bauer J, Scheikl T, Zappulla J, Audebert M, Desbois S, Waisman A, Lassmann H, Liblau RS, Mars LT: Cutting edge: Multiple sclerosis-like lesions induced by effector CD8 T cells recognizing a sequestered antigen on oligodendrocytes. J Immunol 2008, 181: 1617–1621.
Na SY, Eujen H, Gobel K, Meuth SG, Martens K, Wiendl H, Hunig T: Antigen-specific blockade of lethal CD8 T-cell mediated autoimmunity in a mouse model of multiple sclerosis. J Immunol 2009, 182: 6569–6575. 10.4049/jimmunol.0804200
Lafaille JJ, Nagashima K, Katsuki M, Tonegawa S: High incidence of spontaneous autoimmune encephalomyelitis in immunodeficient anti-myelin basic protein T cell receptor transgenic mice. Cell 1994, 78: 399–408. 10.1016/0092-8674(94)90419-7
Bettelli E, Pagany M, Weiner HL, Linington C, Sobel RA, Kuchroo VK: Myelin oligodendrocyte glycoprotein-specific T cell receptor transgenic mice develop spontaneous autoimmune optic neuritis. J Exp Med 2003, 197: 1073–1081. 10.1084/jem.20021603
Bettelli E, Baeten D, Jager A, Sobel RA, Kuchroo VK: Myelin oligodendrocyte glycoprotein-specific T and B cells cooperate to induce a Devic-like disease in mice. J Clin Invest 2006, 116: 2393–2402. 10.1172/JCI28334
Krishnamoorthy G, Lassmann H, Wekerle H, Holz A: Spontaneous opticospinal encephalomyelitis in a double-transgenic mouse model of autoimmune T cell/B cell cooperation. J Clin Invest 2006, 116: 2385–2392. 10.1172/JCI28330
Krishnamoorthy G, Saxena A, Mars LT, Domingues HS, Mentele R, Ben-Nun A, Lassmann H, Dornmair K, Kurschus FC, Liblau RS, et al.: Myelin-specific T cells also recognize neuronal autoantigen in a transgenic mouse model of multiple sclerosis. Nat Med 2009, 15: 626–632. 10.1038/nm.1975
Pollinger B, Krishnamoorthy G, Berer K, Lassmann H, Bosl MR, Dunn R, Domingues HS, Holz A, Kurschus FC, Wekerle H: Spontaneous relapsing-remitting EAE in the SJL/J mouse: MOG-reactive transgenic T cells recruit endogenous MOG-specific B cells. J Exp Med 2009, 206: 1303–1316. 10.1084/jem.20090299
Madsen LS, Andersson EC, Jansson L, krogsgaard M, Andersen CB, Engberg J, Strominger JL, Svejgaard A, Hjorth JP, Holmdahl R, et al.: A humanized model for multiple sclerosis using HLA-DR2 and a human T-cell receptor. Nat Genet 1999, 23: 343–347. 10.1038/15525
Harkiolaki M, Holmes SL, Svendsen P, Gregersen JW, Jensen LT, McMahon R, Friese MA, van Boxel G, Etzensperger R, Tzartos JS, et al.: T cell-mediated autoimmune disease due to low-affinity crossreactivity to common microbial peptides. Immunity 2009, 30: 348–357. 10.1016/j.immuni.2009.01.009
Friese MA, Jakobsen KB, Friis L, Etzensperger R, Craner MJ, McMahon RM, Jensen LT, Huygelen V, Jones EY, Bell JI, et al.: Opposing effects of HLA class I molecules in tuning autoreactive CD8+ T cells in multiple sclerosis. Nat Med 2008, 14: 1227–1235. 10.1038/nm.1881
Kassmann CM, Lappe-Siefke C, Baes M, Brugger B, Mildner A, Werner HB, Natt O, Michaelis T, Prinz M, Frahm J, et al.: Axonal loss and neuroinflammation caused by peroxisome-deficient oligodendrocytes. Nat Genet 2007, 39: 969–976. 10.1038/ng2070
Maurer M, Kobsar I, Berghoff M, Schmid CD, Carenini S, Martini R: Role of immune cells in animal models for inherited neuropathies: facts and visions. J Anat 2002, 200: 405–414. 10.1046/j.1469-7580.2002.00045.x
Herrmann I, Kellert M, Schmidt H, Mildner A, Hanisch UK, Bruck W, Prinz M, Nau R: Streptococcus pneumoniae Infection aggravates experimental autoimmune encephalomyelitis via Toll-like receptor 2. Infect Immun 2006, 74: 4841–4848. 10.1128/IAI.00026-06
Dirnagl U, Klehmet J, Braun JS, Harms H, Meisel C, Ziemssen T, Prass K, Meisel A: Stroke-induced immunodepression: experimental evidence and clinical relevance. Stroke 2007, 38: 770–773. 10.1161/01.STR.0000251441.89665.bc
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
RAL and DHL performed the literature search and wrote the manuscript.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Linker, R.A., Lee, DH. Models of autoimmune demyelination in the central nervous system: on the way to translational medicine. Exp & Trans Stroke Med 1, 5 (2009). https://doi.org/10.1186/2040-7378-1-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/2040-7378-1-5