
Abstract
Background
The autism spectrum disorders (ASDs) are complex neurodevelopmental disorders that result in severe and pervasive impairment in the development of reciprocal social interaction and verbal and nonverbal communication skills. In addition, individuals with ASD have stereotypical behavior, interests and activities. Rare mutations of some genes, such as neuroligin (NLGN) 3/4, neurexin (NRXN) 1, SHANK3, MeCP2 and NHE9, have been reported to be associated with ASD. In the present study, we investigated whether alterations in mRNA expression levels of these genes could be found in lymphoblastoid cell lines derived from patients with ASD.
Methods
We measured mRNA expression levels of NLGN3/4, NRXN1, SHANK3, MeCP2, NHE9 and AKT1 in lymphoblastoid cells from 35 patients with ASD and 35 healthy controls, as well as from 45 patients with schizophrenia and 45 healthy controls, using real-time quantitative reverse transcriptase polymerase chain reaction assays.
Results
The mRNA expression levels of NLGN3 and SHANK3 normalized by β-actin or TBP were significantly decreased in the individuals with ASD compared to controls, whereas no difference was found in the mRNA expression level of MeCP2, NHE9 or AKT1. However, normalized NLGN3 and SHANK3 gene expression levels were not altered in patients with schizophrenia, and expression levels of NLGN4 and NRXN1 mRNA were not quantitatively measurable in lymphoblastoid cells.
Conclusions
Our results provide evidence that the NLGN3 and SHANK3 genes may be differentially expressed in lymphoblastoid cell lines from individuals with ASD compared to those from controls. These findings suggest the possibility that decreased mRNA expression levels of these genes might be involved in the pathophysiology of ASD in a substantial population of ASD patients.
Similar content being viewed by others
Background
Autism spectrum disorder (ASD), also known as pervasive developmental disorder (PDD), is defined as severe and pervasive impairments in the development of reciprocal social interaction and verbal and nonverbal communication skills. These disorders are also characterized by stereotypical behavior, interests and activities. The lifetime morbidity rate of ASD is 0.2% to 1.0% across studies [1]. In addition, twin and family studies of ASD have demonstrated a high heritability of approximately 90% [2], indicating that ASD is a heterogeneous condition that is likely to result from the combined effects of multiple genetic factors interacting with environmental factors. Recent genetic studies have identified several vulnerability loci and genetic mutations that cause ASD. One of the most striking revelations is the important role of genes that encode proteins at the neuronal synapse [3].
Rare mutations in the neuroligin 3 (NLGN3) and neuroligin 4 (NLGN4) genes, which map to chromosomes Xq13 and Xp22.3, have been reported in some patients with ASD and other neurodevelopmental impairments [4–8]. A particular mutation of NLGN3 (Arg451Cys) is known to cause a defect in protein processing of NLGN3 [9]. In addition, a particular mutation of NLGN4 (1186insT) causes a frameshift mutation that leads to premature termination of NLGN4 (D396X), resulting in a loss of 421 amino acids (51% of the protein) [4]. Neuroligins, which are postsynaptically localized cell adhesion molecules, play a crucial role in organizing excitatory glutamatergic and inhibitory GABAergic synapses in the mammalian brain by interacting with presynaptic β-neurexins (NRXN), thereby triggering the formation of functional presynaptic structures in contacting axons [10]. Mutations of the neurexin 1 (NRXN1) gene, at the chromosome locus 2q32, have been found in individuals with ASD [11–14]. Furthermore, de novo copy number variation analysis revealed deletion of the NRXN1-containing gene region in ASD [15]. The binding of NRXN1 and NLGN genes mediates synaptic development [16]. Interestingly, a mutation of NLGN3 results in a disruption of the ability to bind to NRXN [9]. In addition, neuroligins interact with a postsynaptic scaffolding protein, SHANK3, which is also implicated in ASD [17] and is located on the telomeric terminal of chromosome 22q13.3. Shank proteins couple neurotransmitter receptors, ion channels and other membrane proteins to the actin cytoskeleton and G protein-coupled signaling pathways, and they also play a role in synapse formation and dendritic spine maturation [18]. Deletion or translocation of the genomic locus, which includes the SHANK3 gene, and de novo mutations of the SHANK3 gene result in premature stop codons and have been found in ASD [17, 19, 20].
In a study of consanguineous autism families, Morrow et al. [21] observed a relationship between ASD and alterations in the sodium/hydrogen exchanger 9 (NHE9) gene. Specifically, they found a nonsense mutation in patients with ASD that is a heterozygous CGA-to-TGA transition, changing arginine 423 to a stop codon [21]. The NHE9 gene is located on chromosome 3q24 and is one of the families of Na+/H+ exchangers that regulate ion flux across membranes [22]. Rett syndrome is another PDD, and the methyl-CpG-binding protein 2 (MeCP2) gene is a causal gene for Rett syndrome. MeCP2 is a transcriptional repressor that binds to methylated CpG dinucleotides generally located at gene promoters and recruits histone deacetylase 1 and other proteins involved in chromatin repression [23]. De novo mutations of the MeCP2 gene located on chromosome Xp28 occur in 80% of female patients with Rett syndrome [24]. Some evidence of dysregulation of the phosphatidylinositol 3-kinase (PI3K)/AKT pathway is implicated in ASD, despite the fact that no mutation which causes ASD has been reported in association with the AKT1 gene. The expression and phosphorylation and/or activation of AKT were found to be decreased in the autistic brain [25]. The PTEN gene (phosphatase and tensin homolog deleted on chromosome 10) is a major negative regulator of the PI3K/AKT pathway, and PTEN mutations have been linked to ASD [26].
Recently, several studies have suggested that lymphoblastoid cells can be used to detect biologically plausible correlations between candidate genes and neuropsychiatric diseases, including Rett syndrome [27], nonspecific X-linked mental retardation [28], bipolar disorder [29], fragile X syndrome [30, 31] and dup(15q) [32]. In the present study, we compared mRNA expression levels of various genes in blood-derived lymphoblastoid cells from individuals with ASD and healthy controls.
Methods
Participants
We obtained mRNA samples from patients with ASD, patients with schizophrenia and healthy controls from the research bioresource of the Human Brain Phenotype Consortium in Japan (http://www.sp-web.sakura.ne.jp/consortium.html). The ASD cohort consisted of 35 patients with ASD and healthy controls (Table 1). Patients with ASD and patients with schizophrenia were recruited from both outpatient and inpatient services at Osaka University Hospital. Each ASD patient was diagnosed by at least two trained child psychiatrists and/or child neurologists according to the Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition-Text Revision (DSM-IV-TR) criteria based on unstructured or semistructured behavioral observations of the patients and interviews with the patients and their parents or caregivers. During the interview, the Pervasive Developmental Disorders Autism Society Japan Rating Scale (PARS) [33] and the Japanese version of the Asperger's Questionnaire [34] were used to assist in the evaluation of ASD-specific behaviors and symptoms. PARS is a semistructured interview that is composed of 57 questions in eight domains of the characteristics of children with PDD, which was developed by the Autism Society Japan. The clinicians who diagnosed the individuals were trained in the use of PARS. Twenty individuals met the full criteria for autistic disorder, 11 met the criteria for Asperger syndrome and four for PDD-not otherwise specified (PDD-NOS). Among the patients with ASD, 11 had a low intelligence quotient (IQ) ( < 70). The schizophrenia cohort consisted of 45 patients with schizophrenia and 45 age- and sex-matched healthy controls (Table 2). Each patient with schizophrenia received a consensus diagnosis by at least two trained psychiatrists according to the DSM-IV-TR criteria using the structured clinical interview (SCID) for DSM-IV.
A detailed description of healthy controls was given in previous reports [35, 36]. Briefly, controls were biologically unrelated Japanese participants. Healthy controls were screened using the SCID for the Diagnostic and Statistical Manual, Fourth Edition, Axis I Disorders, Non-Patient version (SCID-I/NP) and were excluded if they (1) had neurological or medical conditions that could potentially affect the central nervous system, (2) had any psychiatric diseases and/or received psychiatric medication, (3) had first- or second-degree relatives with psychiatric disease or (4) presented with an IQ < 70. IQ data were collected using the Japanese version of the full-scale Wechsler Adult Intelligence Scale (WAIS)-III or the full-scale Wechsler Intelligence Scale for Children-Third Edition (WISC-III) [37, 38].
Following description of the study, written informed consent was obtained from each individual (or, when appropriate, his/her guardians). This study was carried out in accordance with the World Medical Association's Declaration of Helsinki and was approved by the ethics committee at Osaka University.
Immortalization of lymphocytes and RNA extraction
Isolation of lymphocytes from blood and lymphocyte immortalization using Epstein-Barr virus (EBV) were entrusted to SRL of Tokyo, Japan. Immortalized, patient-derived lymphocytes were grown in culture media supplemented with 20% fetal bovine serum. Total RNA was extracted from cell pellets using the RNeasy Mini Kit (Qiagen K.K., Tokyo, Japan). The total RNA yield was determined by absorbance at 260 nm, and RNA quality was analyzed using agarose gel electrophoresis.
DNase treatment and reverse transcriptase reaction
Total RNA was treated with DNase to remove contaminating genomic DNA using DNase Treatment & Removal Reagents (Ambion, Austin, TX, USA) according to the manufacturer's protocol. Total RNA (10 μg) treated with DNase was used in a 50-μL reverse transcriptase reaction to synthesize cDNA with the SuperScript First-Strand Synthesis System for RT-PCR (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's protocol. Briefly, total RNA (10 μg) was denatured with 1 mM deoxyribonucleotide triphosphate (dNTP) and 5 ng/μL random hexamers at 65°C for 5 minutes. After the addition of 10xRT buffer (20 mM Tris-HCl (pH 8.4) and 50 mM KCl final concentration; Invitrogen), MgCl2 (5 mM final concentration), dithiothreitol (10 mM final concentration), RNaseOUT Recombinant Ribonuclease Inhibitor (100 U; Invitrogen) and SuperScript III Reverse Transcriptase (125 U; Invitrogen), the reaction mixture was incubated at 25°C for 10 minutes, at 42°C for 40 minutes and at 70°C for 15 minutes. RNase H (5 U) was added to the reaction mixture and incubated at 37°C for 20 minutes to stop the reaction.
Real-time quantitative RT-PCR
The Pre-Developed TaqMan Assay Reagent kit (Applied Biosystems, Foster City, CA, USA) was used to measure mRNA expression levels of NLGN3, NLGN4, NRXN1, SHANK3, MeCP2, NHE9, AKT1 and housekeeping genes (β-actin and TBP). Primers were purchased from Applied Biosystems (gene name: assay ID, transcript ID, target region; NLGN3: Hs01043809_m1, NM_181303.1, Exon4-5; NLGN4: Hs00535592_m1, NM_020742.2, Exon1-2; NRXN1: Hs00985123_m1, NM_001135659.1, Exon22-23; SHANK3: Hs01586468_m1, NM_001080420.1, Exon22-23; MECP2: Hs00172845_m1, NM_004992.3, Exon2-3; NHE9: Hs00543518_m1, NM_173653.3, Exon7-8; AKT1: Hs00920503_m1, NM_001014432.1, Exon13-14; β- actin: 4326315E, NM_001101, no region indicated; TBP: 4326322E, NM_003194, no region indicated). Expression levels of these genes were measured by real-time quantitative reverse transcriptase polymerase chain reaction (qRT-PCR) using an ABI Prism 7900 Sequence Detection System (Applied Biosystems) with a 384-well format as previously described [39, 40]. Each 20-μL PCR reaction contained 6 μL of cDNA, 900 nM concentrations of each primer, a 250 nM concentration of probe and 10 μL of TaqMan Universal PCR Master Mix containing AmpliTaq Gold DNA Polymerase and AmpErase Uracil N-glycosylase (all from Applied Biosystems), as well as dNTP with deoxyuridine triphosphate, passive reference and optimized buffer components. The PCR cycling conditions were 50°C for 2 minutes, 95°C for 10 minutes, 40 cycles of 95°C for 15 seconds and 59°C or 60°C for 1 minute. PCR data were obtained by using Sequence Detector software (SDS version 2.1; Applied Biosystems) and quantified using a standard curve method. This software plotted the real-time fluorescence intensity and selected the threshold within the linear phase of the amplicon profile. The software plotted a standard curve of the cycle at threshold (Ct) (where the fluorescence generated within a reaction crossed the threshold) versus the quantity of RNA. All samples were measured using a single plate per target gene, and their Ct values were in the linear range of the standard curve. Sample quantities were predicted by Ct values. Experiments were typically performed three times in triplicate, and each gene expression level was taken as the average of three independent experiments. The individual expression level of each target gene normalized by a housekeeping gene (raw target gene expression level divided by raw housekeeping gene expression level) was used for statistical analysis.
Statistical analyses
Statistical analyses were carried out using SPSS for Windows version 16.0 software (SPSS Japan Inc., Tokyo, Japan). Group comparisons of demographic data were performed using the c2 test for one categorical variable (sex) or the Mann-Whitney U test for continuous variables as appropriate. Differences in mRNA transcript levels between the groups were analyzed using the Mann-Whitney U test. The Bonferroni correction for multiple tests was applied to assess the mRNA transcript levels on the number of genes (five). All P values reported are based on two-tailed tests. Statistical significance was defined as P < 0.05.
Results
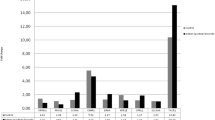
Standard curves for the seven target genes (NLGN3, NLGN4, NRXN1, SHANK3, MeCP2, NHE9 and AKT1) and the two housekeeping genes (β-actin and TBP) were prepared using serial dilutions (1:4) of pooled cDNA from 300 ng of total RNA derived from immortalized lymphoblasts (Figure 1). The R2 values of the standard curves were more than 0.99 (NLGN3, MeCP2, NHE9, AKT1, β-actin and TBP), 0.87 (SHANK3), 0.64 (NRXN1) and 0.63 (NLGN4). Although the SHANK3 gene expression was relatively low, it was measurable in our sample. On the other hand, we did not further analyze NLGN4 and NRXN1 gene expression, as the expression levels of the two genes were too low to quantify using this method.

Standard curves for target genes and housekeeping genes. Standard curves for NLGN3, NLGN4, NRXN1, SHANK3, MeCP2, NHE9, AKT1 and two housekeeping genes (β-actin and TBP). The highest quantity represents an amount of cDNA prepared from 300 ng of total RNA in the polymerase chain reaction.
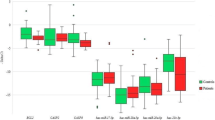
Using immortalized lymphoblastoid cells from 35 individuals with ASD and 35 controls, we quantified the mRNA expression levels of the NLGN3, SHANK3, NHE9, MeCP2 and AKT1 genes normalized by two housekeeping genes, β-actin and TBP (Figure 2). The mRNA expression levels of the NLGN3 gene normalized by β-actin or TBP were decreased by 35% or 26%, respectively, in individuals with ASD (β-actin: P = 0.00024; TBP: P = 0.00089). The mRNA expression levels of the SHANK3 gene normalized by β-actin or TBP were also decreased in individuals with ASD by 39% or 40%, respectively (β-actin: P = 0.000036; TBP: P = 0.0061). The mRNA expression levels of the NHE9 gene were increased by 24% (P = 0.052: normalized by β-actin) and 39% (P = 0.048: normalized by TBP). There was no significant difference in mRNA expression levels of the MeCP2 gene normalized by β-actin or TBP between the two groups (P > 0.1). The mRNA expression levels of the AKT1 gene were decreased by 11% (P = 0.03: normalized by β-actin); however, those levels were not altered when normalized by TBP (P = 0.45). After correction for multiple tests, mRNA expression levels of NLGN3 and SHANK3 remained significantly lower in individuals with ASD than in healthy controls (NLGN3: corrected P = 0.0012, normalized by β-actin, corrected P = 0.0045, normalized by TBP; SHANK3: corrected P = 0.00018, normalized by β-actin, corrected P = 0.03, normalized by TBP). However, the altered expression level of NHE9 or AKT1 was no longer significant after the correction for multiple tests (P > 0.1).

Expression analysis of NLGN3, SHANK3, NHE9, MeCP2 and AKT1 in autism spectrum disorder. Mean relative mRNA expression level scores normalized by housekeeping gene β-actin (a) or TBP (b) in the autism spectrum disorder (ASD) group and the control group are shown. Bars represent the standard error of the mean. Differences between the groups in expression levels of the five genes were analyzed by using the Mann-Whitney U test. Post hoc comparisons were performed by using the Bonferroni correction. **P < 0.01 and ***P < 0.001.
We next measured NLGN3 and SHANK3 mRNA expression levels in immortalized lymphoblastoid cells from 45 patients with schizophrenia and 45 healthy controls to examine the disease specificity of the differential expression levels between patients and healthy controls (Figure 3). We found that the mRNA expression levels for these two genes normalized by β-actin or TBP were not significantly different between patients with schizophrenia and healthy controls (P > 0.2). These results suggest that reduced levels of NLGN3 and SHANK3 mRNA expression might be associated with ASD but not with schizophrenia.

Expression analysis of the NLGN3 and SHANK3 genes in patients with schizophrenia. Mean relative mRNA expression level scores normalized by housekeeping gene β-actin (a) or TBP (b) in the schizophrenia group and the control group are shown. Bars represent standard error of the mean. Differences in expression levels of the two genes between the groups were analyzed by using the Mann-Whitney U test.
Discussion
In this study, we found that the mRNA expression levels of NLGN3 and SHANK3 were significantly lower in individuals with ASD than in healthy controls. Mutations of causal genes are rare, and they have been found to be associated with specific types of ASD. Our findings suggest that not only rare mutations of the causal genes but also functional alterations in the transcriptional activity of these genes might be associated with the pathophysiology of ASD. The NLGN3 and SHANK3 genes are synapse-related genes and were found to be affected in ASD, whereas other genes, including NHE9 and MeCP2, do not play major roles at the synapse and were not found to be affected in ASD. These findings suggest that impairments in synaptic function might be associated with the pathophysiology of ASD.
Reduced expression of the NLGN3 and SHANK3 genes in lymphoblasts of individuals with ASD is consistent with previous reports indicating that mutations of these genes cause reduced expression or loss of function of the protein. Since the NLGN3 gene is located in chromosome X, there may be expressional difference between genders. However, no significant difference of NLGN3 gene expression normalized by β-actin or TBP was observed with regard to gender in healthy controls or individuals with ASD (P > 0.05). This might be due to inactivation of one X chromosome in females [41]. There are several possibilities that might explain the reduced expression of the NLGN3 and SHANK3 genes in ASD. First, our sporadic ASD cases might have mutations, polymorphisms or copy number variations in the NLGN3 or SHANK3 genes, which could result in reduced expression of these genes. Second, mutations or polymorphisms in genes that regulate the expression of NLGN3 or SHANK3 might contribute to the observed reduction in expression of the NLGN3 or SHANK3 genes. To our knowledge, although the regulation of NLGN3 by other genes has not been reported, there are some reports in the literature describing the regulation of SHANK3 gene expression. For example, SHANK3 expression is regulated by DNA methylation [42, 43]. In addition, SHANK3 is one of the predicted targets of dysregulated microRNA (miRNA), and altered miRNA expression levels were found in postmortem brain from autism patients [44]. Further epigenetic analyses might elucidate the mechanisms of reduced SHANK3 expression.
Some findings of gene expression in lymphoblastoid cell lines are in conflict with those of previous studies. For example, Beri et al. [42] reported that SHANK3 is not expressed in EBV-transformed human lymphoblastoid cell lines in an investigation of tissue-specific SHANK3 gene expression and DNA methylation. By using lymphoblastoid cells from autism patients, Talebizadeh et al. [8] detected novel splice isoforms of NLGN4. There are methodological differences between previous studies and our study. SHANK3 gene expression in the previous study [42] was analyzed by using a conventional RT-PCR method; however, we measured the expression levels of SHANK3 gene by using a real-time qRT-PCR method (the TaqMan method). Furthermore, the expression level of SHANK3 was relatively low, which is shown in the standard curve in Figure 1. It is possible that the sensitivity of our real-time qRT-PCR method to detect the SHANK3 gene expression level might be higher than that of a conventional RT-PCR method. On the other hand, we could not quantitatively measure the NLGN4 and NRXN1 genes by using the real-time qRT-PCR method. However, there were slight expressions of these genes in lymphoblastoid cell lines when we used a large quantity of cDNA for the real-time qRT-PCR (Figure 1). Unfortunately, the small expression levels of these genes made it impossible to quantitatively measure the gene expressions in our sample. This may explain possible discrepancies of the gene expression findings of previous studies and our results.
There are several limitations of this study. First, our positive results might have arisen from sample bias due to non-age-matched samples, although the Japanese are a relatively homogeneous population, so the use of non-age-matched samples is unlikely to explain our findings. Second, our sample size might not be small for type I errors but small for type II errors. There is a possibility of type II errors in mRNA expression differences of NHE9, MECP2 and AKT1 between individuals with ASD and healthy controls and expression differences of NLGN3 and SHANK3 between individuals with schizophrenia and healthy controls. In particular, NHE9 might be increased in individuals with ASD, as the expression level of NHE9 was marginally significant before correction for multiple testing. Thus, replication studies using a larger sample size are needed before a firm conclusion can be drawn. Third, we did not perform a mutation search for the examined genes in our sample to replicate the association between the examined genes and ASD and how the causal or risk variants of the genes regulate the gene expression. As the previous evidence for candidate genes of ASD are based on rare mutations and/or copy number variations of the genes, it might be difficult to find a mutation in our 35 individuals with ASD for analysis of the variant effects on the gene expression in this study. A mutation search study of these candidate genes should be done in future studies. Fourth, the IQ scores in the ASD group were lower than those in the healthy control group, so reduced gene expression could be related to lower IQ. However, lower expression of the NLGN3 or SHANK3 genes was not found in individuals with schizophrenia who had lower premorbid IQ scores, and no expression difference was observed in individuals with ASD and mental retardation versus individuals with ASD but without mental retardation (data not shown). Taken together, the reduced gene expression in ASD might be specific to ASD, although other neuropsychiatric diseases, such as attention-deficit/hyperactivity disorder, mental retardation, major depression and bipolar disorder, should be examined in future studies. The ASD cases in this study were consistent with idiopathic autism diagnosed on the basis of clinical features. We did not include individuals with Rett syndrome and the other syndromic autisms, such as multiple sclerosis, which could explain why we did not find altered expression of MeCP2 in this cohort. Our results suggest that the MeCP2 gene may not be associated with the common pathology of ASD, while NLGN3 and SHANK3 may be. Because lymphoblastoid cell lines are not neuronal cells, some of our findings might not reflect the pathophysiology in ASD brains. Further studies investigating these limitations are warranted.
Conclusions
Our study reveals reduced levels of NLGN3 and SHANK3 mRNA expression in lymphoblastoid cell lines derived from individuals with ASD, but not from those of individuals with schizophrenia. These results are consistent with findings that rare mutations of these genes in specific cases cause loss of function, suggesting that reduction of NLGN3 and SHANK3 mRNA expression could be related to the pathophysiology of ASD in a substantial population of patients. Although there are several limitations present in this study, lymphoblastoid cell lines may still allow investigation of the pathophysiology of ASD. Further analyses are required, such as a mutation analysis of the NLGN3 and SHANK3 genes and the genes regulating their expression, in addition to studies designed to elucidate the mechanisms of this reduced expression.
Abbreviations
- ASD:
-
autism spectrum disorder
- DSM-IV-TR:
-
Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition-Text Revision
- F:
-
female
- IQ:
-
intelligence quotient
- JART50:
-
Japanese Adult Reading Test
- M:
-
male
- MeCP2 :
-
methyl-CpG-binding protein 2
- NHE9 :
-
sodium/hydrogen exchanger 9
- NLGN :
-
neuroligin
- NRXN :
-
neurexin
- PARS:
-
Pervasive Developmental Disorders Autism Society Japan Rating Scale
- PDD:
-
pervasive developmental disorder
- PDD-NOS:
-
pervasive developmental disorder not otherwise specified
- SCID:
-
structured clinical interview
- SCID-I/NP:
-
Diagnostic and Statistical Manual, Fourth Edition, Axis I Disorders, Non-Patient version
- SD:
-
standard deviation
- WAIS-III:
-
Wechsler Adult Intelligence Scale-III
- WISC-III:
-
Wechsler Intelligence Scale for Children-Third Edition.
References
Levy SE, Mandell DS, Schultz RT: Autism. Lancet. 2009, 374: 1627-1638. 10.1016/S0140-6736(09)61376-3.
Lichtenstein P, Carlström E, Råstam M, Gillberg C, Anckarsäter H: The genetics of autism spectrum disorders and related neuropsychiatric disorders in childhood. Am J Psychiatry. 2010, 167: 1357-1363. 10.1176/appi.ajp.2010.10020223.
Walsh CA, Morrow EM, Rubenstein JL: Autism and brain development. Cell. 2008, 135: 396-400. 10.1016/j.cell.2008.10.015.
Jamain S, Quach H, Betancur C, Råstam M, Colineaux C, Gillberg IC, Soderstrom H, Giros B, Leboyer M, Gillberg C, Bourgeron T: Paris Autism Research International Sibpair Study: Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat Genet. 2003, 34: 27-29. 10.1038/ng1136.
Laumonnier F, Bonnet-Brilhault F, Gomot M, Blanc R, David A, Moizard MP, Raynaud M, Ronce N, Lemonnier E, Calvas P, Laudier B, Chelly J, Fryns JP, Ropers HH, Hamel BC, Andres C, Barthélémy C, Moraine C, Briault S: X-linked mental retardation and autism are associated with a mutation in the NLGN4 gene, a member of the neuroligin family. Am J Hum Genet. 2004, 74: 552-557. 10.1086/382137.
Lawson-Yuen A, Saldivar JS, Sommer S, Picker J: Familial deletion within NLGN4 associated with autism and Tourette syndrome. Eur J Hum Genet. 2008, 16: 614-618. 10.1038/sj.ejhg.5202006.
Yan J, Oliveira G, Coutinho A, Yang C, Feng J, Katz C, Sram J, Bockholt A, Jones IR, Craddock N, Cook EH, Vicente A, Sommer SS: Analysis of the neuroligin 3 and 4 genes in autism and other neuropsychiatric patients. Mol Psychiatry. 2005, 10: 329-332. 10.1038/sj.mp.4001629.
Talebizadeh Z, Lam DY, Theodoro MF, Bittel DC, Lushington GH, Butler MG: Novel splice isoforms for NLGN3 and NLGN4 with possible implications in autism. J Med Genet. 2006, 43: e21-
Comoletti D, De Jaco A, Jennings LL, Flynn RE, Gaietta G, Tsigelny I, Ellisman MH, Taylor P: The Arg451Cys-neuroligin-3 mutation associated with autism reveals a defect in protein processing. J Neurosci. 2004, 24: 4889-4893. 10.1523/JNEUROSCI.0468-04.2004.
Craig AM, Kang Y: Neurexin-neuroligin signaling in synapse development. Curr Opin Neurobiol. 2007, 17: 43-52. 10.1016/j.conb.2007.01.011.
Feng J, Schroer R, Yan J, Song W, Yang C, Bockholt A, Cook EH, Skinner C, Schwartz CE, Sommer SS: High frequency of neurexin 1β signal peptide structural variants in patients with autism. Neurosci Lett. 2006, 409: 10-13. 10.1016/j.neulet.2006.08.017.
Kim HG, Kishikawa S, Higgins AW, Seong IS, Donovan DJ, Shen Y, Lally E, Weiss LA, Najm J, Kutsche K, Descartes M, Holt L, Braddock S, Troxell R, Kaplan L, Volkmar F, Klin A, Tsatsanis K, Harris DJ, Noens I, Pauls DL, Daly MJ, MacDonald ME, Morton CC, Quade BJ, Gusella JF: Disruption of neurexin 1 associated with autism spectrum disorder. Am J Hum Genet. 2008, 82: 199-207. 10.1016/j.ajhg.2007.09.011.
Autism Genome Project Consortium, Szatmari P, Paterson AD, Zwaigenbaum L, Roberts W, Brian J, Liu XQ, Vincent JB, Skaug JL, Thompson AP, Senman L, Feuk L, Qian C, Bryson SE, Jones MB, Marshall CR, Scherer SW, Vieland VJ, Bartlett C, Mangin LV, Goedken R, Segre A, Pericak-Vance MA, Cuccaro ML, Gilbert JR, Wright HH, Abramson RK, Betancur C, Bourgeron T, Gillberg C: Mapping autism risk loci using genetic linkage and chromosomal rearrangements. Nat Genet. 2007, 39: 319-328. 10.1038/ng1985.
Yan J, Noltner K, Feng J, Li W, Schroer R, Skinner C, Zeng W, Schwartz CE, Sommer SS: Neurexin 1α structural variants associated with autism. Neurosci Lett. 2008, 438: 368-370. 10.1016/j.neulet.2008.04.074.
Glessner JT, Wang K, Cai G, Korvatska O, Kim CE, Wood S, Zhang H, Estes A, Brune CW, Bradfield JP, Imielinski M, Frackelton EC, Reichert J, Crawford EL, Munson J, Sleiman PM, Chiavacci R, Annaiah K, Thomas K, Hou C, Glaberson W, Flory J, Otieno F, Garris M, Soorya L, Klei L, Piven J, Meyer KJ, Anagnostou E, Sakurai T: Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature. 2009, 459: 569-573. 10.1038/nature07953.
Dean C, Dresbach T: Neuroligins and neurexins: linking cell adhesion, synapse formation and cognitive function. Trends Neurosci. 2006, 29: 21-29. 10.1016/j.tins.2005.11.003.
Durand CM, Betancur C, Boeckers TM, Bockmann J, Chaste P, Fauchereau F, Nygren G, Rastam M, Gillberg IC, Anckarsäter H, Sponheim E, Goubran-Botros H, Delorme R, Chabane N, Mouren-Simeoni MC, de Mas P, Bieth E, Rogé B, Héron D, Burglen L, Gillberg C, Leboyer M, Bourgeron T: Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat Genet. 2007, 39: 25-27. 10.1038/ng1933.
Vessey JP, Karra D: More than just synaptic building blocks: scaffolding proteins of the post-synaptic density regulate dendritic patterning. J Neurochem. 2007, 102: 324-332. 10.1111/j.1471-4159.2007.04662.x.
Bonaglia MC, Giorda R, Borgatti R, Felisari G, Gagliardi C, Selicorni A, Zuffardi O: Disruption of the ProSAP2 gene in a t(12;22)(q24.1;q13.3) is associated with the 22q13.3 deletion syndrome. Am J Hum Genet. 2001, 69: 261-268. 10.1086/321293.
Moessner R, Marshall CR, Sutcliffe JS, Skaug J, Pinto D, Vincent J, Zwaigenbaum L, Fernandez B, Roberts W, Szatmari P, Scherer SW: Contribution of SHANK3 mutations to autism spectrum disorder. Am J Hum Genet. 2007, 81: 1289-1297. 10.1086/522590.
Morrow EM, Yoo SY, Flavell SW, Kim TK, Lin Y, Hill RS, Mukaddes NM, Balkhy S, Gascon G, Hashmi A, Al-Saad S, Ware J, Joseph RM, Greenblatt R, Gleason D, Ertelt JA, Apse KA, Bodell A, Partlow JN, Barry B, Yao H, Markianos K, Ferland RJ, Greenberg ME, Walsh CA: Identifying autism loci and genes by tracing recent shared ancestry. Science. 2008, 321: 218-223. 10.1126/science.1157657.
Nakamura N, Tanaka S, Teko Y, Mitsui K, Kanazawa H: Four Na+/H+ exchanger isoforms are distributed to Golgi and post-Golgi compartments and are involved in organelle pH regulation. J Biol Chem. 2005, 280: 1561-1572.
Chahrour M, Zoghbi HY: The story of Rett syndrome: from clinic to neurobiology. Neuron. 2007, 56: 422-437. 10.1016/j.neuron.2007.10.001.
Renieri A, Meloni I, Longo I, Ariani F, Mari F, Pescucci C, Cambi F: Rett syndrome: the complex nature of a monogenic disease. J Mol Med. 2003, 81: 346-354.
Sheikh AM, Malik M, Wen G, Chauhan A, Chauhan V, Gong CX, Liu F, Brown WT, Li X: BDNF-Akt-Bcl2 antiapoptotic signaling pathway is compromised in the brain of autistic subjects. J Neurosci Res. 88: 2641-2647.
Zhou J, Blundell J, Ogawa S, Kwon CH, Zhang W, Sinton C, Powell CM, Parada LF: Pharmacological inhibition of mTORC1 suppresses anatomical, cellular, and behavioral abnormalities in neural-specific Pten knock-out mice. J Neurosci. 2009, 29: 1773-1783. 10.1523/JNEUROSCI.5685-08.2009.
Horike S, Cai S, Miyano M, Cheng JF, Kohwi-Shigematsu T: Loss of silent-chromatin looping and impaired imprinting of DLX5 in Rett syndrome. Nat Genet. 2005, 37: 31-40. 10.1038/ng1570.
Meloni I, Muscettola M, Raynaud M, Longo I, Bruttini M, Moizard MP, Gomot M, Chelly J, des Portes V, Fryns JP, Ropers HH, Magi B, Bellan C, Volpi N, Yntema HG, Lewis SE, Schaffer JE, Renieri A: FACL4, encoding fatty acid-CoA ligase 4, is mutated in nonspecific X-linked mental retardation. Nat Genet. 2002, 30: 436-440. 10.1038/ng857.
Iwamoto K, Kakiuchi C, Bundo M, Ikeda K, Kato T: Molecular characterization of bipolar disorder by comparing gene expression profiles of postmortem brains of major mental disorders. Mol Psychiatry. 2004, 9: 406-416. 10.1038/sj.mp.4001437.
Brown V, Jin P, Ceman S, Darnell JC, O'Donnell WT, Tenenbaum SA, Jin X, Feng Y, Wilkinson KD, Keene JD, Darnell RB, Warren ST: Microarray identification of FMRP-associated brain mRNAs and altered mRNA translational profiles in fragile X syndrome. Cell. 2001, 107: 477-487. 10.1016/S0092-8674(01)00568-2.
Nishimura Y, Martin CL, Vazquez-Lopez A, Spence SJ, Alvarez-Retuerto AI, Sigman M, Steindler C, Pellegrini S, Schanen NC, Warren ST, Geschwind DH: Genome-wide expression profiling of lymphoblastoid cell lines distinguishes different forms of autism and reveals shared pathways. Hum Mol Genet. 2007, 16: 1682-1698. 10.1093/hmg/ddm116.
Baron CA, Tepper CG, Liu SY, Davis RR, Wang NJ, Schanen NC, Gregg JP: Genomic and functional profiling of duplicated chromosome 15 cell lines reveal regulatory alterations in UBE3A-associated ubiquitin-proteasome pathway processes. Hum Mol Genet. 2006, 15: 853-869. 10.1093/hmg/ddl004.
Yamada A, Suzuki M, Kato M, Tanaka S, Shindo T, Taketani K, Akechi T, Furukawa TA: Emotional distress and its correlates among parents of children with pervasive developmental disorders. Psychiatry Clin Neurosci. 2007, 61: 651-657. 10.1111/j.1440-1819.2007.01736.x.
Wakabayashi A, Tojo Y, Baron-Cohen S, Wheelwright S: [The Autism-Spectrum Quotient (AQ) Japanese version: evidence from high-functioning clinical group and normal adults] [in Japanese]. Shinrigaku Kenkyu. 2004, 75: 78-84.
Yasuda Y, Hashimoto R, Ohi K, Fukumoto M, Takamura H, Iike N, Yoshida T, Hayashi N, Takahashi H, Yamamori H, Morihara T, Tagami S, Okochi M, Tanaka T, Kudo T, Kamino K, Ishii R, Iwase M, Kazui H, Takeda M: Association study of KIBRA gene with memory performance in a Japanese population. World J Biol Psychiatry. 2010, 11: 852-857. 10.3109/15622971003797258.
Hashimoto R, Ohi K, Yasuda Y, Fukumoto M, Iwase M, Iike N, Azechi M, Ikezawa K, Takaya M, Takahashi H, Yamamori H, Okochi T, Tanimukai H, Tagami S, Morihara T, Okochi M, Tanaka T, Kudo T, Kazui H, Iwata N, Takeda M: The impact of a genome-wide supported psychosis variant in the ZNF804A gene on memory function in schizophrenia. Am J Med Genet B Neuropsychiatr Genet. 2010, 153B: 1459-1464. 10.1002/ajmg.b.31123.
Committee JW-IP: Japanese Wechsler Intelligence Scale for Children. 1998, Tokyo: Nihon Bunka Kagakusha
Wechsler D: Wechsler Intelligence Scale for Children-Third Edition Manual. 1991, New York: Psychological Corp
Hashimoto R, Straub RE, Weickert CS, Hyde TM, Kleinman JE, Weinberger DR: Expression analysis of neuregulin-1 in the dorsolateral prefrontal cortex in schizophrenia. Mol Psychiatry. 2004, 9: 299-307. 10.1038/sj.mp.4001434.
Chiba S, Hashimoto R, Hattori S, Yohda M, Lipska B, Weinberger DR, Kunugi H: Effect of antipsychotic drugs on DISC1 and dysbindin expression in mouse frontal cortex and hippocampus. J Neural Transm. 2006, 113: 1337-1346. 10.1007/s00702-005-0414-1.
Willard HF: X chromosome inactivation and X-linked mental retardation. Am J Med Genet. 1996, 64: 21-26. 10.1002/(SICI)1096-8628(19960712)64:1<21::AID-AJMG2>3.0.CO;2-U.
Beri S, Tonna N, Menozzi G, Bonaglia MC, Sala C, Giorda R: DNA methylation regulates tissue-specific expression of Shank3. J Neurochem. 2007, 101: 1380-1391. 10.1111/j.1471-4159.2007.04539.x.
Maunakea AK, Nagarajan RP, Bilenky M, Ballinger TJ, D'Souza C, Fouse SD, Johnson BE, Hong C, Nielsen C, Zhao Y, Turecki G, Delaney A, Varhol R, Thiessen N, Shchors K, Heine VM, Rowitch DH, Xing X, Fiore C, Schillebeeckx M, Jones SJ, Haussler D, Marra MA, Hirst M, Wang T, Costello JF: Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature. 2010, 466: 253-257. 10.1038/nature09165.
Abu-Elneel K, Liu T, Gazzaniga FS, Nishimura Y, Wall DP, Geschwind DH, Lao K, Kosik KS: Heterogeneous dysregulation of microRNAs across the autism spectrum. Neurogenetics. 2008, 9: 153-161. 10.1007/s10048-008-0133-5.
Acknowledgements
This work was funded in part by Grants-in-Aid from the Japanese Ministry of Health, Labor and Welfare (H19-kokoro-002, H22-seishin-ippan-001, H22-rinken-ippan-002, H22-sinkei-ippan-001); the Japanese Ministry of Education, Culture, Sports, Science and Technology (18689030, 22390225); CREST (Core Research for Evolutionary Science and Technology) of JST (Japan Science and Technology Agency), Grant-in-Aid for Scientific Research on Priority Areas, Research on the Pathomechanisms of Brain Disorders, from the Ministry of Education, Culture, Sports, Science, and Technology (18023045); the Japan Foundation for Neuroscience and Mental Health and the Meiji Yasuda Mental Health Foundation; and the Osaka University Program for the Support of Networking among Present and Future Researchers. The study sponsors had no further role in the study design; the collection, analysis and interpretation of data; the writing of the report; or the decision to submit the manuscript for publication.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
RH supervised the entire project; collected the data; wrote the manuscript; was critically involved in the design, analysis and interpretation of the data; and was responsible for performing the literature review. YY was critically involved in the collection and analysis of the data, contributed to the editing of the final manuscript and contributed intellectually to the interpretation of the data. HY, SU and AI were involved in the mRNA measurements and collection of the majority of the data. KO, MF, IM, MTan and MTak were heavily involved in the collection of the majority of the data and contributed intellectually to the interpretation of the data. All authors reviewed the manuscript before submission and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Yasuda, Y., Hashimoto, R., Yamamori, H. et al. Gene expression analysis in lymphoblasts derived from patients with autism spectrum disorder. Molecular Autism 2, 9 (2011). https://doi.org/10.1186/2040-2392-2-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/2040-2392-2-9