
Abstract
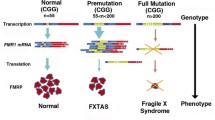
Fragile X syndrome (FXS) is caused by an expanded CGG repeat (> 200 repeats) in the 5' untranslated portion of the fragile mental retardation 1 gene (FMR1), leading to deficiency or absence of the FMR1 protein (FMRP). FMRP is an RNA carrier protein that controls the translation of several other genes that regulate synaptic development and plasticity. Autism occurs in approximately 30% of FXS cases, and pervasive developmental disorder, not otherwise specified (PDD-NOS) occurs in an additional 30% of cases. Premutation repeat expansions (55 to 200 CGG repeats) may also give rise to autism spectrum disorders (ASD), including both autism and PDD-NOS, through a different molecular mechanism that involves a direct toxic effect of the expanded CGG repeat FMR1 mRNA. RNA toxicity can also lead to aging effects including tremor, ataxia and cognitive decline, termed fragile X-associated tremor ataxia syndrome (FXTAS), in premutation carriers in late life. In studies of mice bearing premutation expansions, there is evidence of early postnatal neuronal cell toxicity, presenting as reduced cell longevity, decreased dendritic arborization and altered synaptic morphology. There is also evidence of mitochondrial dysfunction in premutation carriers. Many of the problems with cellular dysregulation in both premutation and full mutation neurons also parallel the cellular abnormalities that have been documented in autism without fragile X mutations. Research regarding dysregulation of neurotransmitter systems in FXS, including the metabotropic glutamate receptor (mGluR)1/5 pathway and γ aminobutyric acid (GABA)A pathways, have led to new targeted treatments for FXS. Preliminary evidence suggests that these new targeted treatments will also be beneficial in non-fragile X forms of autism.
Similar content being viewed by others


Introduction
Fragile X syndrome (FXS) is an important subtype of autism, both because of its frequency and because knowledge of the molecular mechanisms involved in its pathogenesis has facilitated the development of targeted treatments with the potential to reverse or dramatically improve both behavioral and cognitive deficits. Because FXS is the most common single gene cause of autism, responsible for 2% to 6% of all cases of autism, it is clinically recommended that all individuals diagnosed with autism or ASD should have the FX DNA test (both PCR and Southern blot) when the etiology of their autism is not known [1–4]. FXS is nearly always caused by a trinucleotide (CGG) repeat expansion, located in the 5' untranslated region of the FMR1 gene, to a length of greater than 200 repeats (full mutation range). Full mutation expansions typically lead to methylation of the gene, reduced or absent transcription, and consequent decreased reduction in translation of the FMR1 protein (FMRP), the proximal basis of FXS. FMRP levels are correlated with the degree of clinical involvement including physical, cognitive and structural/functional brain involvement [5–10].
Approximately 30% of males with FXS have full autism, as determined by the standardized criteria of the Autism Diagnostic Observation Scale (ADOS) and the Autism Diagnostic Interview (ADI-R) [11–15]. An additional 30% of boys have pervasive developmental disorder, not otherwise specified (PDD-NOS) [11]. Among the remaining patients with FXS, of those who do not meet the criteria for an autism spectrum disorder (ASD) diagnosis, the majority have one or more autistic features, such as hand flapping, poor eye contact and tactile defensiveness [11].
A premutation CGG-repeat range (55 to 200 repeats) was initially defined in terms of an increased frequency of expansion of the CGG repeat to the full mutation range when transmitted by a premutation (carrier) woman. All children with the full mutation have a carrier mother, although a female patient with a premutation could have received this mutation from either her mother or her father. Moreover, the propensity for transmission of a full mutation allele increases with increasing CGG repeat number in the mother [16]. A father who is a carrier of either a premutation or full mutation allele will pass only a premutation to all of his daughters, presumably due to selective production of premutation allele-bearing sperm [17].
Carriers of premutation alleles were generally considered to be clinically uninvolved until premature ovarian failure, recently renamed FX-associated primary ovarian insufficiency (FXPOI), was reported [18]. Subsequently, the late onset neurodegenerative disorder, FX-associated tremor ataxia syndrome (FXTAS), was described [19, 20], further establishing clinical involvement among premutation carriers. It is now evident that a spectrum of neurodevelopmental and aging/neurological problems are associated with premutations, including autism and ASD [21–26]. Most individuals with a premutation are neither developmentally disabled nor do they have autism; however, a subgroup does experience cognitive, emotional and/or behavioral involvement. There is a negative correlation between CGG repeat number and the level of FMRP in a premutation range [27], which predisposes individuals in the high end of a premutation range to cognitive and behavioral impairment. In addition, all individuals with a premutation have elevated FMR1 mRNA, whereas the opposite occurs in the full mutation [27]. Thus, the cognitive and behavioral impairments in a premutation and full mutation ranges are likely to have both distinct and overlapping mechanisms.
Clinical and molecular involvement in FXS, and association with autism
The basis for incomplete penetrance of autism (30%) or PDD-NOS (30%) among individuals with FXS is not known. However, there is evidence that patients with additional medical disorders that affect the CNS, such as seizures or additional genetic problems, have an increased risk for autism compared with patients with FXS alone [28–30]. For those with both FXS and autism, there is a spectrum of involvement with significant heterogeneity, both cognitively and behaviorally, with IQ values ranging from severely intellectually impaired to normal, particularly in females. However, there is a strong association between low IQ and the autism diagnosis in both males and females with FXS [11–14, 31–35]. The cause of this heterogeneity is related to background genetic effects and environmental effects that influence IQ, social abilities, anxiety, attention deficit hyperactive disorder and additional features that are components of the FXS phenotype (Figure 1). Background genetic effects include additional pathological mutations (FXS has been reported with sex chromosome disorders, Down syndrome, Tourette syndrome and other conditions [28, 29, 36], allelic variants [37], and gene expression changes [38]). An example of the later condition is the Prader-Willi phenotype (PWP) of FXS, in which there is no structural or methylation change at 15q 11-13; rather, there is significant downregulation of expression of CYFIP 1, which is located at the 15q locus in Prader-Willi syndrome (PWS) [38]. Males with the PWP of FXS have severe obesity, hyperphagia and hypogenitalia, and a higher rate of ASD than those with FXS without the PWP [38].

Overview of the behavioral/cognitive phenotype of fragile X syndrome (FXS). The interrelationships among cognitive, behavioral and attentional deficits in FXS are modified by additional environmental influences and genetic background effects. Environmental influences include seizures, trauma, abuse and socioeconomic status. Genetic influences include allelic variations, additional genetic disorders and variation in the expression levels of genes important for the phenotype of FXS.
Environmental influences on the phenotype of FXS include exposures to toxins (for example, alcohol, leading to fetal alcohol syndrome and FXS), abuse (physical or sexual), neglect, perinatal asphyxia, head trauma, seizures and socioeconomic status. Additional environmental exposures leading to further toxicity are just beginning to be explored in both premutation and full mutation involvement, as they are in idiopathic autism [39–42]. Such studies are occurring at a cellular level in premutation neurons; these neurons die earlier than do control neurons, with increased cell death documented by 21 days in culture [43]. In addition, mitochondrial dysfunction has been documented in fibroblasts and brain tissue in premutation carriers both with and without FXTAS [44]. We hypothesize that premutation neurons are more vulnerable to environmental toxins, and clinical case reports appear to support this notion [42, 45].
The absence of FMRP in individuals with FXS has significant consequences in the translation of dozens and probably hundreds of proteins. Because FMRP usually suppresses translation, its absence leads to broad translational upregulation in the hippocampus [46]. Recent studies by Darnell et al.[47] and others have demonstrated linkage between FMRP and many proteins that are related to autism, including neuroligin 3 and 4, neurorexin, PDP (postsynaptic density protein) 95, CYFIP (cytoplasmic FMR1 interacting protein) 1 and 2, SHANK (Src homology 3 and multiple ankyrin repeat domains)3, Arc, PTEN (phosphatase and tensin homolog), MAPK (mitogen activated protein kinase), JAKMIP (janus kinase and microtubule interacting protein)1 and HERC (homologous to the E6-AP carboxyl terminus) and regulator of chromosome condensation (RCC)1-like domain-containing protein) 2, among others [2, 4–50]. Most of these proteins are associated with synapse formation and plasticity; however, the PTEN gene encodes a dual specificity phosphatase effecting G1 cell cycle arrest and/or apoptosis, and 17% (3/18) of individuals with autism and macrocephaly were found to have a PTEN mutation [51]. Macrocephaly also occurs in FXS, often with a broad forehead remarkably similar to the broad foreheads described by Butler and colleagues; this characteristic is hypothesized to be related to the downregulation of PTEN that occurs in FXS [47, 52]. Expression of JAKMIP1 and the G protein coupled receptor (GRP)155 were both altered by reduction of FMRP (seen in FXS) or induction of CYFIP1 (seen in the 15q duplication form of autism) in vitro[53]. These proteins were also dysregulated in boys with idiopathic ASD relative to their unaffected siblings [53]. Both CYFIP1 (a partner protein to FMRP and regulated by FMRP) and JAKMIP1 are involved with the RacGTPase system, which modulates the neurite development that is crucial for proper brain connectivity [54]. There is also evidence for upregulation of the mammalian target of rapamycin (mTOR) pathway in the hippocampus of the knockout (KO)mouse [55] and in studies of humans with FXS [56]. The mTOR pathway is dysregulated in several other genetic disorders that are associated with autism, such as tuberous sclerosis (TS) [57]. These findings have stimulated targeted treatments using rapamycin to downregulate the mTOR system in patients with TS, with initially positive results. The overlap of molecular mechanisms in those with a premutation or the full mutation and idiopathic autism is shown in Figure 2.

Molecular overlap between autism, fragile x syndrome (FXS) and premutation disorders. Absence of the FMR1 protein (FMRP) leads to the dysregulation of several proteins including those involved with synapse formation and plasticity, glutamate and γ aminobuyric acid (GABA) neurotransmission and mammalian target of rapamycin (mTOR) and phosphatase and tensin homolog (PTEN) pathways. A premutation is associated with elevation of FMR1 mRNA, leading to sequestration of proteins and mitochondrial dysfunction. Many of these same molecular changes can also occur in some types of autism. Some patients with FXS have mosaicism of premutation and full cells, so there is overlap of the molecular mechanisms among all three disorders.
Recently, a number of studies have directly compared patients with FXS and those with autism without FXS. There are unique structural differences in the central nervous system (CNS) between the two disorders even when both disorders have comparable degrees of autism as assessed by standardized behavioral measures [58]. Those with FXS have an enlarged caudate compared with typically developing individuals and those with autism, whereas those with autism have a larger amygdala compared with FXS or controls [58]. These differences continue to evolve with age, as does the severity of the autistic features in FXS [59]. Therefore, from early in life, and probably in utero, there are structural CNS changes that are related to the lack of FMRP. The dysregulation of proteins that are important for synaptic plasticity and connectivity in the brain leads to the gradual deficits in socialization, behavior and cognition that characterize the FXS phenotype [60, 61]. Although eye contact problems are usually not present during the first year of life, they evolve over time, as do the sensory hyperarousal, anxiety, motor and social deficits. Hoeft et al.[62] have reported that the early trajectory of brain growth abnormalities in FXS becomes more exaggerated over time and includes enhanced growth of the caudate, nucleus basalis and thalamus, compared with controls. Those authors also documented enhanced white matter volume, particularly of the striatal-frontal regions, becoming more dramatic in the early years (1 to 3 years of age), which suggests axonal pathology as opposed to secondary connectional dysregulation [62]. Their work further suggests that the earlier the intervention is begun, the better the outcome for an individual with FXS. These findings provide neurobiological support for initiating interventions as early in the lifespan as possible, although further clinical studies are needed. A summary of treatment for FXS was reviewed by Hagerman et al.[63].
RNA toxicity and a premutation carrier
The discovery of the neurodegenerative disorder, FXTAS, in older adult carriers of premutation alleles, coupled with increased FMR1 transcriptional activity in the premutation range, led to the recognition of an entirely distinct pathogenic mechanism associated with the FMR1 gene: RNA toxicity [64–68]. A range of studies on the adverse consequences of expressing the expanded CGG repeat in human, animal and cell models has helped to establish an RNA toxicity model involving a toxic gain of function of premutation FMR1 RNA [69–79]. However, although carriers of premutation alleles have elevated FMR1 mRNA [27, 80, 81], the strongest argument for an RNA-based toxicity mechanism in both FXTAS and FXPOI [82–84], is that these clinical syndromes are limited to a premutation repeat range, where the gene is active; that is, low levels (or absence) of FMRP do not cause either FXTAS or FXPOI. However, moderately lowered FMRP levels in the upper premutation range may compound the effects of elevated RNA levels (a mechanistic issue that still needs to be resolved) but the primary effect appears to be expression of the expanded CGG-repeat RNA. A supporting argument for an RNA-based mechanism is that the FMR1 mRNA is found within the characteristic intranuclear neuronal and astrocytic inclusions of FXTAS [85, 86].
FXTAS was originally described as a late adult-onset neurodegenerative disorder; however, there is an emerging view that FXTAS, and probably also FXPOI, is the end stage of a process that actually begins in early development, and that may be responsible for the emotional and behavioral problems, cognitive impairment, ASD and seizure activity experienced by children who are carriers of premutation alleles [21, 25, 87]. This view is based on a combination of animal and cell-based studies for early abnormalities resulting from expression of a premutation allele. In particular, Chen et al.[43] demonstrated that in cultured hippocampal neurons from day 1 postnatal premutation (knock-in; KI) mice, there were CGG repeat-dependent decreases in both the number of branches and the interbranch lengths, and decreased longevity in culture. Moreover, Garcia-Arocena et al.[88] observed abnormal lamin A/C architecture, with loss of ring-like nuclear staining, in embryonic fibroblasts from the KI mouse. In behavioral studies with the KI mice, there were progressive deficits in spatial processing (but no motor involvement) in mice as young as 12 weeks [70, 89]. These observations, plus elevated levels of FMR1 mRNA in children with premutation alleles [90], support the presence of an early developmental component of FMR1 mRNA-associated toxicity.
Based on the toxic RNA gain of function model for myotonic dystrophy, in which disease pathogenesis involves the sequestration of one or more proteins by an expanded rCUG repeat in the 3' untranslated region of the myotonic dystrophy protein kinase (DMPK) gene [91, 92], the first view of FXTAS envisioned a similar, direct-RNA mechanism in which proteins would be sequestered by the expanded CGG repeat [19, 65, 67, 69]. A growing number of animal and cell- l-based studies support this 'direct RNA' model [71, 72, 93, 94]. Recently, Sellier et al.[94]presented evidence for both sequestration of an RNA processing protein, Sam68 and the consequent altered splice-site regulation of several RNAs whose splicing is known to be regulated by Sam68. In addition to their demonstration of the functional consequences of Sam68 sequestration, Sellier et al. demonstrated that the incorporation of the protein into nuclear aggregates displayed a CGG-repeat cutoff that meant aggregation only occurred for expansions exceeding ~40 CGG repeats. More recently, Sellier et al.[95] also reported that a consequence of this sequestration is dysregulation of microRNAs, which may be related to the clinical problems of premutation carriers.
It should be noted that although the sequestration model remains the most viable mechanism for RNA toxicity, the clinical data only support the requirement for transcription. Thus, a role for other mechanisms such as RNA-triggered signaling or co-transcriptional mechanisms cannot be discounted [68] (Figure 3). Evidence for a direct RNA-based (for example, sequestration) model cannot exclude the possibility that co-transcriptional RNA, or even DNA, has a role in the pathogenesis. Entezam and Usdin [74] observed that the DNA-repair protein ATR is recruited to CGG expansions, and the fact that another DNA-repair protein, γ-H2AX[96], is found in the intranuclear FXTAS inclusions [97], suggests that transcription-induced DNA damage could also trigger the pathogenesis of premutation-associated disorders.

Potential mechanisms of FMR1 mRNA toxicity. Although numerous studies point to RNA toxicity as the underlying pathogenic trigger in fragile X-associated tremor ataxia syndrome (FXTAS), the specific mechanism for such toxicity is not known. Possibilities include (1) sequestration of one or more proteins that bind to the RNA, thus attenuating their other cell functions; (2) protein activation upon binding to the CGG-repeat RNA, leading to dysregulation of one or more signaling cascades; and (3) various co-transcriptional process, such as R-loop formation, that lead to DNA damage/repair signaling and consequent cellular dysregulation.
Recent work from the laboratory of Guilivi has demonstrated mitochondrial dysfunction in fibroblasts and brain samples in premutation carriers both with and without FXTAS [44]. Mitochondrial dysfunction in carriers included uncoupling between electron transport and synthesis of ATP in addition to decreased levels of mitochondrial proteins including the ATPase β-subunit (ATPB) from complex V, the cytochrome c oxidase subunit from complex IV (CCOIV) and manganese superoxide dismutase as part of the mitochondrial antioxidant defense. The levels of the mitochondrial proteins correlated inversely with the CGG repeat numbers in the premutation range. These protein changes increased oxidative stress and oxidatively modified mitochondrial proteins, and activated the unfolded protein response and phosphorylation of the alpha subunit of the heterotrimeric eukaryotic translational initiation factor 2 (eIF2α), resulting in a decrease in protein translation. Similar types of mitochondrial abnormalities have been seen in those with autism without a FX mutation (Giulivi et al. unpublished data) [98, 99]. Specifically, Olivera et al.[98] reported that 14 of 69 patients with autism had hyperlactacidemia, and in 5 of 11 of these patients who underwent a deltoid muscle biopsy, there was a mitochondrial respiratory chain disorder with enzyme function that was <20% of normal mean activity, including complex I, complex IV and complex V abnormalities. Weissman et al. studied 25 patients with autism and evidence of oxidative phosphorylation abnormalities, and found 19 with elevated lactate levels, 64% with complex I deficiency and 20% with complex III deficiency. Two of the patients had pathological mitochondrial DNA mutations [99]. Other reports of mitochondrial gene mutations in children with autism have also been reported [100–102].
Clinical involvement of some premutation carriers
Although autism and other clinical involvement in a subgroup of young premutation carriers was initially thought to be only an occasional occurrence [26, 103–106], research cohorts demonstrated that approximately 14% of boys and 5% of girls with a premutation had ASD [107]. More recent studies demonstrated a high rate of ASD (73%) in boys with a premutation who were referred clinically to the UC Davis MIND Institute, although this was much lower in premutation males who were identified by cascade testing (7%) compared with their brothers who did not have a premutation (0%) [108]. Although there is clearly a bias towards an ASD phenotype in those who present clinically, a recent online family questionnaire completed by over 1200 families affected by FXS found that 19% of 57 males with a premutation had a diagnosis of autism, which was significantly higher than control boys (5%). In this survey 1% of 199 females with a premutation also had a diagnosis of autism [87]. This same survey found that 33% of boys with a premutation had developmental delays, which was significantly higher than in a group of age-matched boys without a premutation (1.8%). A completely unbiased population of premutation carriers that should be followed carefully are those diagnosed by screening as newborns; three studies are currently in progress in the USA.
Studies of neuropsychological deficits in premutation carriers during adulthood have been complicated because of the subclinical CNS changes that can occur related to the development of FXTAS [109–111]. Studies have detected deficits in executive function in a subgroup of males with a premutation, but not in the corresponding group of females [112–117]. In contrast to these four reports, Hunter et al.[118] found no neuropsychological deficits in 54 men with a premutation who were aged under 50 years, although the Behavioral Dyscontrol Scale (BDS) [113, 119], which was found to be most sensitive to executive dysfunction in older male carriers [112], was not used. Clearly, recruitment bias is likely to affect adult premutation studies in neuropsychological testing and in emotional assessments. In contrast to the neuropsychological testing, standardized emotional assessments have demonstrated problems with anxiety and/or depression in both males and females with a premutation, both with and without FXTAS, compared with controls at multiple centers [22, 120–124]
An emerging phenotype includes the finding of autoimmune problems in a subgroup of women with a premutation. These problems include fibromyalgia, hypothyroidism and multiple sclerosis, and they can occur in women with a premutation both with and without FXTAS [24, 125–127]. Hunter et al.[82] found that women with irregular cycles reported higher rates of thyroid disease in addition to depression/anxiety. The molecular process leading to the autoimmune problems are unknown, although they are most likely related to the RNA toxicity. Predisposing factors leading to autoimmune disease in some females are likely to be genetic, because in our clinical experience, they cluster in families. Because of concern about the genetic factors that underlie both autoimmune disease and autism, we studied whether there is an increase in ASD with FXS in the children of female carriers who have autoimmune disease, compared with carriers who do not have autoimmune disease [128]. The odds ratio (OR) for ASD was 1.27 (P = 0.51) which was not significant; however, the ORs for seizures and tics in the offspring were 3.81 (P = 0.031) and 2.94 (P = 0.019) respectively. These results raise the possibility that there are intergenerational autoimmune factors or perhaps auto-antibodies that affect the prevalence of seizures and tics in the offspring of mothers with a premutation and autoimmune disease [128].
FMRP function throughout life leading to targeted treatments for FXS
FMRP is an mRNA-binding protein that is important for mRNA transport, mRNA stabilization and translation of mRNA into protein at the synapse [129–131]. FMRP is also a factor in the regulation of adult neurogenesis, so in the absence of FMRP there is dysregulation of glycogen synthase kinase (GSK)3β, reduced β-catenin and defective Wnt signaling. These alterations lead to downregulation of neurogenin 1, which is an early initiator of neuronal differentiation and an inhibitor of astrocyte differentiation [132]. Therefore, FMRP is important throughout life and there is a high incidence of motor problems, including Parkinson disease (PD), with aging in those with FXS [133]. In addition, in neuropathologic studies, there is evidence of migration problems in the hippocampus and in the cerebellum in those with FXS (Greco et al,. unpublished data), which are similar to those reported in individuals with autism [134]. These problems may be related to dysregulation of Wnt signaling in both FXS and autism.
Perhaps the most important change in protein expression in the absence of FMRP is the excess basal translation of proteins involved in the metabotropic glutamate receptor (mGluR) 5 receptor pathway [135]. Bear and colleagues have proposed the mGluR theory of FX, suggesting that the deficits associated with FXS are related to upregulation of the downstream effectors of the mGluR5 pathway, leading to enhanced long-term depression (LTD), and that treatment with an mGluR5 antagonist could be a targeted treatment for FXS [135, 136]. Both FMRP and mGluRs play important roles in synaptogenesis and synaptic plasticity, and in the absence of FMRP there are long, thin and immature dendritic spines in both human and animal models of FXS [137–142]. There are also enhanced, abnormal epileptiform discharges consistent with an enhanced rate of clinical seizures in FXS [143, 144].
Support for the 'mGluR theory' has been shown by generating FMR1 mutant mice with a 50% reduction in mGluR5 expression [145]. The mGluR5 deficiency rescued most of the KO mouse abnormalities including altered ocular dominance plasticity, increased density of dendritic spines on cortical pyramidal neurons, increased basal protein synthesis in the hippocampus, exaggerated inhibitory avoidance extinction, audiogenic seizures and accelerated body growth. However, macroorchidism was not rescued. This work is supportive of the proposal by Bear et al.[146]that excessive mGluR5 signaling is responsible for the psychiatric and neurological symptoms of FXS, including cognitive deficits, seizures, anxiety, perseverative movements and social deficits.
Use of mGluR5 antagonists in animal models of FXS further supports the mGluR theory. MPEP (2-methyl-6-phenylethynyl pyridine hydrochloride) is a potent, highly selective antagonist of mGluR5 receptors [147]. In vitro, both MPEP and fenobam, another mGluR5 antagonist, were able to rescue hippocampal dendritic abnormalities in the KO mice [148, 149]. MPEP has reversed audiogenic seizures, epileptiform discharges, open field hyperactivity and the defect in prepulse inhibition (PPI) of the startle response in KO mice [148–150]. When MPEP and lithium, a partial mGluR5 antagonist that also blocks GSK3β, were given to dfmr1 loss of function Drosophila mutants, the flies had restored normal courtship behavior, memory and brain structural abnormalities through the reduction of mGluR activity [151]. MPEP is toxic to humans, so other mGluR5 antagonists including fenobam have been studied in FXS [152, 153]. Fenobam was found to be safe in a single dose trial in 12 adults with FXS. There were improvements in hyperactivity and anxiety, and 50% showed at least a 20% improvement in PPI [152]. Currently there are two additional mGluR5 antagonists undergoing trials in adults with FXS at multiple centers [153].
Other mechanisms to downregulate glutamate release and modulate mGluR overactivity have been investigated. γ Aminobutyric acid (GABA)B receptor agonists, such as baclofen, inhibit both presynaptic release of glutamate and postsynaptic transmission and/or intracellular signaling downstream from mGluR5 [154–156]. Baclofen has been shown to be efficacious in treating hyperactivity [157], marble burying (Seaside Therapeutics, unpublished data) and audiogenic seizure phenotypes in FX KO mice [158]. A double-blind, placebo-controlled, crossover trial of arbaclofen, the right sided isomer of baclofen that is significantly more potent than regular baclofen as a GABA agent, has just been completed at multiple centers,and involved over 60 individuals with FXS (aged 6 years and older). The preliminary safety and efficacy results are positive, with improvement in the Clinical Global Impression Improvement scale in those with the most severe baseline ratings [159]. There are also preliminary studies that are taking place involving individuals with autism without FXS, and these studies have also produced preliminary positive results. Therefore, further studies on both FXS and autism are set to take place.
The GABAergic system is also dysregulated in FXS, and GABA agents are important to consider for targeted treatment studies in FXS. GABA is a major inhibitory neurotransmitter receptor in the brain, which is important in anxiety, depression, epilepsy, insomnia, and learning and memory [160]. GABA-mediated inhibition is important for terminating ictal discharges and the spread of hyperexcitability, which can lead to seizures [161].
There are two main subtypes of GABA receptors: GABAA and GABAB. The main difference between them is that the first is a ligand gated Cl- channel that gives fast inhibition, whereas the latter is a G-protein coupled receptor which gives slower and more prolonged inhibitory signals [162, 163]. The metabotropic GABAB receptor can either be presynaptic and inhibit the release of neurotransmitters through downregulation of high-voltage activated Ca2+-channels; or, when postsynaptic, decrease neuronal excitability through its influence on K+ channels. Thus, GABAB agonists such as arbaclofen mediate their downregulating effects on either side of the synapse. The ionotropic GABAA receptor is usually localized postsynaptically, and their activation leads to opening of Cl- channels and hyperpolarization of the membrane potential, thus making it difficult for excitatory neurotransmitters such as glutamate to generate an action potential. GABAA receptors are more abundant than GABAB receptors in mammalian brain, and disorders such as epilepsy, sleep disorders and anxiety are now being treated using drugs that act on the GABAA receptor[164].
Direct binding between FMRP and the mRNA of the delta subunit of the GABAA receptor has been shown [165]. Reduced expression and dysfunction of several subunits of the GABAA receptor (α1, α3, α4; β1, β2; γ1, γ2 and δ) have been shown in FX animal models [166–168]. FMR1 Drosophila mutants destined to die from glutamate toxicity were rescued after administering molecules involved in the GABAergic pathway [166]. In addition, abnormal male courtship behavior and mushroom body abnormalities were rescued by GABA agents [166].
There is a profound reorganization of neocortical inhibitory circuits of GABAergic intraneurons in the KO mouse [164, 167–173]. Recent evidence indicates that deficits in GABA-mediated inhibition may underlie many of the key symptoms in FXS, including the seizures, anxiety and autistic-like behaviors [167, 169, 173]. The neocortex in KO mice exhibits a marked reduction in the density of GABAergic interneurons that stain with parvalbumin. Moreover, electrophysiological studies in brain slices from these animals exhibit impaired GABAA receptor-mediated inhibitory function [174]. In addition to a gross reduction in GABA-mediated inhibition caused by the maldevelopment of inhibitory circuits and the loss of GABAergic interneurons, there is also evidence of altered GABAA receptor subunit expression in the FX KO mouse [167]. In particular, there appears to be a selective reduction in the expression of δ subunits [167, 168]. Global expression analysis by means of the differential display in the FX mouse model revealed consistent underexpression of only three genes, one of which was the GABAA receptor subunit δ. As GABAA receptors are the major inhibitory receptors in the brain, and are specifically involved in processes that are disturbed in FX, including neuronal excitability (leading to enhanced seizure susceptibility), anxiety, sleep and learning, enhancement of the function of GABAA receptors may have major therapeutic benefits for FXS. Kooy and colleagues [175] have demonstrated that use of the GABAA agonist ganaxolone (3α-hydroxy-3β-methyl-5α-pregnan-20-one) improved seizures in the KO mouse model of FXS. Ganaxolone is a 3β-methylated synthetic analog of the progesterone metabolite allopregnanolone, which is itself a neuroactive steroid. Unlike progesterone, neither allopregnanolone nor ganaxolone have direct hormonal activity via progesterone receptor activation, and cannot cause hormonal side-effects. However, allopregnanolone and ganaxolone are powerful positive allosteric modulators of GABAA receptors [161]. Human trials indicate that ganaxolone is well tolerated and that it may be efficacious in the treatment of diverse forms of epilepsy in children and adults [176–180]. Plans for studies on ganaxolone are currently underway in children and adults with FXS.
Minocycline, a widely used antibiotic used to treat acne and skin infections, is another promising drug that may target core symptoms of FXS and autism. Minocycline inhibits matrix metalloproteinase (MMP)-9 and reduces inflammation in the central nervous system. MMPs are enzymes involved in synaptic plasticity, and are associated with immature dendritic spine morphology [140, 181]; MMP-9 is elevated in FXS. When minocycline was administered to FMR1 KO mice, their hippocampal neurons exhibited mature dendritic spines, and behaviorally, they showed decreased anxiety and improved exploration skills [140]. Off-label use of minocycline to treat 50 individuals with FXS resulted in two-thirds of families noticing positive improvements in their child's language, attention and/or behavioral improvements while on the medication [182]. An open-label trial is ongoing to investigate the effects of minocycline on children with regressive autism at the National Institute of Mental Health (NIMH). Paribello reported beneficial effects on the CGI and the Aberrant Behavior checklist in an open trial of minocycline involving patients with FXS who were aged 13 and older [183]. Currently, a double-blind, placebo-controlled clinical trial is in progress at the Medical Investigation of Neurodevelopmental Disorders (MIND) Institute for individuals with FXS who are aged 3.5 to 16 years
FXS has led the way for targeted treatments in neurodevelopmental disorders, and many of the treatments guided by molecular abnormalities in FXS may also be helpful for non-FX autism. The treatment trials will now combine targeted treatments, which strengthen synaptic connections, with enhanced educational and behavioral interventions to further develop appropriate synaptic connections in FXS. These targeted treatments combined with educational interventions look promising for reversing the intellectual and behavioral problems of FXS. Because of the shared neurobiological and molecular pathways, these interventions will hopefully also prove helpful in a subset of patients with idiopathic autism
Conclusions
FX syndrome and autism are intertwined, because FMRP regulates the translation of many messages that affect synaptic plasticity and connectivity in the central nervous system. The absence of FMRP also leads to upregulation of mGluR5 pathways and downregulation of GABAA pathways. Targeted treatments to reverse these problems are currently being studied in patients with FXS. Many of these targeted treatments may also be helpful for ASD without FXS.
A premutation can also cause ASD, particularly in a subset of young males, and the mechanism of involvement relates to elevated mRNA levels causing dysregulation of numerous proteins, early neuronal cell death in culture, mitochondrial dysfunction and vulnerability to environmental toxicity. Targeted treatments are currently being developed for premutation involvement in early childhood, and also for neurodegenerative problems including FXTAS in aging individuals.
References
Miller DT, Adam MP, Aradhya S, Biesecker LG, Brothman AR, Carter NP, Church DM, Crolla JA, Eichler EE, Epstein CJ, Faucett WA, Feuk L, Friedman JM, Hamosh A, Jackson L, Kaminsky EB, Kok K, Krantz ID, Kuhn RM, Lee C, Ostell JM, Rosenberg C, Scherer SW, Spinner NB, Stavropoulos DJ, Tepperberg JH, Thorland EC, Vermeesch JR, Waggoner DJ, Watson MS, Martin CL, Ledbetter DH: Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet. 2010, 86: 749-764. 10.1016/j.ajhg.2010.04.006.
Nishimura Y, Martin CL, Vazquez-Lopez A, Spence SJ, Alvarez-Retuerto AI, Sigman M, Steindler C, Pellegrini S, Schanen NC, Warren ST, Geschwind DH: Genome-wide expression profiling of lymphoblastoid cell lines distinguishes different forms of autism and reveals shared pathways. Hum Mol Genet. 2007, 16: 1682-1698. 10.1093/hmg/ddm116.
Reddy KS: Cytogenetic abnormalities and fragile-X syndrome in Autism Spectrum Disorder. BMC Med Genet. 2005, 6: 3-10.1186/1471-2350-6-3.
van Karnebeek CD, Jansweijer MC, Leenders AG, Offringa M, Hennekam RC: Diagnostic investigations in individuals with mental retardation: a systematic literature review of their usefulness. Eur J Hum Genet. 2005, 13: 6-25. 10.1038/sj.ejhg.5201279.
Lightbody AA, Reiss AL: Gene, brain, and behavior relationships in fragile X syndrome: evidence from neuroimaging studies. Dev Disabil Res Rev. 2009, 15: 343-352. 10.1002/ddrr.77.
Tassone F, Hagerman RJ, Ikle DN, Dyer PN, Lampe M, Willemsen R, Oostra BA, Taylor AK: FMRP expression as a potential prognostic indicator in fragile X syndrome. Am J Med Genet. 1999, 84: 250-261. 10.1002/(SICI)1096-8628(19990528)84:3<250::AID-AJMG17>3.0.CO;2-4.
Miller LJ, McIntosh DN, McGrath J, Shyu V, Lampe M, Taylor AK, Tassone F, Neitzel K, Stackhouse T, Hagerman RJ: Electrodermal responses to sensory stimuli in individuals with fragile X syndrome: a preliminary report. Am J Med Genet. 1999, 83: 268-279. 10.1002/(SICI)1096-8628(19990402)83:4<268::AID-AJMG7>3.0.CO;2-K.
Loesch DZ, Huggins RM, Hagerman RJ: Phenotypic variation and FMRP levels in fragile X. Ment Retard Dev Disabil Res Rev. 2004, 10: 31-41. 10.1002/mrdd.20006.
Gothelf D, Furfaro JA, Hoeft F, Eckert MA, Hall SS, O'Hara R, Erba HW, Ringel J, Hayashi KM, Patnaik S, Golianu B, Kraemer HC, Thompson PM, Piven J, Reiss AL: Neuroanatomy of fragile X syndrome is associated with aberrant behavior and the fragile X mental retardation protein (FMRP). Ann Neurol. 2008, 63: 40-51. 10.1002/ana.21243.
Hoeft F, Hernandez A, Parthasarathy S, Watson CL, Hall SS, Reiss AL: Fronto-striatal dysfunction and potential compensatory mechanisms in male adolescents with fragile X syndrome. Hum Brain Mapp. 2007, 28: 543-554. 10.1002/hbm.20406.
Harris SW, Hessl D, Goodlin-Jones B, Ferranti J, Bacalman S, Barbato I, Tassone F, Hagerman PJ, Herman H, Hagerman RJ: Autism profiles of males with fragile X syndrome. Am J Ment Retard. 2008, 113: 427-438. 10.1352/2008.113:427-438.
Rogers SJ, Wehner EA, Hagerman RJ: The behavioral phenotype in fragile X: Symptoms of autism in very young children with fragile X syndrome, idiopathic autism, and other developmental disorders. J Dev Behav Pediatr. 2001, 22: 409-417.
Kaufmann WE, Cortell R, Kau AS, Bukelis I, Tierney E, Gray RM, Cox C, Capone GT, Stanard P: Autism spectrum disorder in fragile X syndrome: communication, social interaction, and specific behaviors. Am J Med Genet. 2004, 129A: 225-234. 10.1002/ajmg.a.30229.
Hatton DD, Sideris J, Skinner M, Mankowski J, Bailey DB, Roberts JE, Mirrett P: Autistic behavior in children with fragile X syndrome: prevalence, stability, and the impact of FMRP. Am J Med Genet A. 2006, 140: 1804-1813.
Hall SS, Lightbody AA, Reiss AL: Compulsive, self-injurious, and autistic behavior in children and adolescents with fragile X syndrome. Am J Ment Retard. 2008, 113: 44-53. 10.1352/0895-8017(2008)113[44:CSAABI]2.0.CO;2.
McConkie-Rosell A, Abrams L, Finucane B, Cronister A, Gane LW, Coffey SM, Sherman S, Nelson LM, Berry-Kravis E, Hessl D, Chiu S, Street N, Vatave A, Hagerman RJ: Recommendations from multi-disciplinary focus groups on cascade testing and genetic counseling for fragile X-associated disorders. J Genet Couns. 2007, 16: 593-606. 10.1007/s10897-007-9099-y.
Reyniers E, Vits L, De Boulle K, Van Roy B, Van Velzen D, de Graaff E, Verkerk AJ, Jorens HZ, Darby JK, Oostra B: The full mutation in the FMR-1 gene of male fragile X patients is absent in their sperm. Nat Genet. 1993, 4: 143-146. 10.1038/ng0693-143.
Cronister A, Schreiner R, Wittenberger M, Amiri K, Harris K, Hagerman RJ: Heterozygous fragile X female: historical, physical, cognitive, and cytogenetic features. Am J Med Genet. 1991, 38: 269-274. 10.1002/ajmg.1320380221.
Hagerman RJ, Leehey M, Heinrichs W, Tassone F, Wilson R, Hills J, Grigsby J, Gage B, Hagerman PJ: Intention tremor, parkinsonism, and generalized brain atrophy in male carriers of fragile X. Neurology. 2001, 57: 127-130.
Jacquemont S, Hagerman RJ, Leehey M, Grigsby J, Zhang L, Brunberg JA, Greco C, Des Portes V, Jardini T, Levine R, Berry-Kravis E, Brown WT, Schaeffer S, Kissel J, Tassone F, Hagerman PJ: Fragile X premutation tremor/ataxia syndrome: molecular, clinical, and neuroimaging correlates. Am J Hum Genet. 2003, 72: 869-878. 10.1086/374321.
Aziz M, Stathopulu E, Callias M, Taylor C, Turk J, Oostra B, Willemsen R, Patton M: Clinical features of boys with fragile X premutations and intermediate alleles. Am J Med Genet B Neuropsychiatr Genet. 2003, 121: 119-127. 10.1002/ajmg.b.20030.
Bourgeois JA, Coffey SM, Rivera SM, Hessl D, Gane LW, Tassone F, Greco C, Finucane B, Nelson L, Berry-Kravis E, Grigsby J, Hagerman PJ, Hagerman RJ: A review of fragile X premutation disorders: expanding the psychiatric perspective. J Clin Psychiatry. 2009, 70: 852-862. 10.4088/JCP.08r04476.
Chonchaiya W, Schneider A, Hagerman RJ: Fragile X: a family of disorders. Adv Pediatr. 2009, 56: 165-186. 10.1016/j.yapd.2009.08.008.
Coffey SM, Cook K, Tartaglia N, Tassone F, Nguyen DV, Pan R, Bronsky HE, Yuhas J, Borodyanskaya M, Grigsby J, Doerflinger M, Hagerman PJ, Hagerman RJ: Expanded clinical phenotype of women with the FMR1 premutation. Am J Med Genet A. 2008, 146A: 1009-1016. 10.1002/ajmg.a.32060.
Farzin F, Perry H, Hessl D, Loesch D, Cohen J, Bacalman S, Gane L, Tassone F, Hagerman P, Hagerman R: Autism spectrum disorders and attention-deficit/hyperactivity disorder in boys with the fragile X premutation. J Dev Behav Pediatr. 2006, 27: S137-S144. 10.1097/00004703-200604002-00012.
Goodlin-Jones B, Tassone F, Gane LW, Hagerman RJ: Autistic spectrum disorder and the fragile X premutation. J Dev Behav Pediatr. 2004, 25: 392-398. 10.1097/00004703-200412000-00002.
Tassone F, Hagerman RJ, Taylor AK, Gane LW, Godfrey TE, Hagerman PJ: Elevated levels of FMR1 mRNA in carrier males: a new mechanism of involvement in the fragile-X syndrome. Am J Hum Genet. 2000, 66: 6-15. 10.1086/302720.
Garcia-Nonell C, Ratera ER, Harris S, Hessl D, Ono MY, Tartaglia N, Marvin E, Tassone F, Hagerman RJ: Secondary medical diagnosis in fragile X syndrome with and without autism spectrum disorder. Am J Med Genet A. 2008, 146A: 1911-1916. 10.1002/ajmg.a.32290.
Stevens L, Tartaglia N, Hagerman R, Riley K: Clinical report: a male with Down syndrome, fragile X syndrome, and autism. J Dev Behav Pediatr. 31: 333-337. 10.1097/DBP.0b013e3181d5aa56.
Berry-Kravis E, Raspa M, Loggin-Hester L, Bishop E, Holiday D, D B: Epilepsy in fragile X syndrome: characteristics and co-morbid diagnoses. Am J Intellect Dev Disabil.
Leigh MJ, Tassone F, Mendoza-Morales G, Nguyen D, Boyd A, Brodovsky J, Ruiz C, Hessl D, Hagerman RJ: Evaluation of autism spectrum disorders in females with Fragile X syndrome [abstract]. International Meeting for Autism Research; 2010, May 20-22; Philadelphia, PA. 2010, 375-376. Abstract # 118.169
Bailey DB, Hatton DD, Mesibov GB, Ament N, Skinner M: Early development, temperament and functional impairment in autism and fragile X syndrome. J Autism Dev Disord. 2000, 30: 49-59. 10.1023/A:1005412111706.
Bailey DB, Hatton DD, Skinner M, Mesibov GB: Autistic behavior, FMR1 protein, and developmental trajectories in young males with fragile X syndrome. J Autism Dev Disord. 2001, 31: 165-174. 10.1023/A:1010747131386.
Loesch DZ, Bui QM, Dissanayake C, Clifford S, Gould E, Bulhak-Paterson D, Tassone F, Taylor AK, Hessl D, Hagerman R, Huggins RM: Molecular and cognitive predictors of the continuum of autistic behaviours in fragile X. Neurosci Biobehav Rev. 2007, 31: 315-326. 10.1016/j.neubiorev.2006.09.007.
Lewis P, Abbeduto L, Murphy M, Richmond E, Giles N, Bruno L, Schroeder S, Anderson J, Orsmond G: Psychological well-being of mothers of youth with fragile X syndrome: syndrome specificity and within-syndrome variability. J Intellect Disabil Res. 2006, 50: 894-904. 10.1111/j.1365-2788.2006.00907.x.
Hagerman RJ: Physical and behavioral phenotype. Fragile X syndrome: Diagnosis, treatment and research. Edited by: Hagerman RJ, Hagerman PJ. 2002, Baltimore: The Johns Hopkins University Press, 3-109. 3
Hessl D, Tassone F, Cordeiro L, Koldewyn K, McCormick C, Green C, Wegelin J, Yuhas J, Hagerman RJ: Brief report: aggression and stereotypic behavior in males with fragile X syndrome--moderating secondary genes in a "single gene" disorder. J Autism Dev Disord. 2008, 38: 184-189. 10.1007/s10803-007-0365-5.
Nowicki ST, Tassone F, Ono MY, Ferranti J, Croquette MF, Goodlin-Jones B, Hagerman RJ: The Prader-Willi phenotype of fragile X syndrome. J Dev Behav Pediatr. 2007, 28: 133-138. 10.1097/01.DBP.0000267563.18952.c9.
Kenet T, Froemke RC, Schreiner CE, Pessah IN, Merzenich MM: Perinatal exposure to a noncoplanar polychlorinated biphenyl alters tonotopy, receptive fields, and plasticity in rat primary auditory cortex. Proc Natl Acad Sci USA. 2007, 104: 7646-7651. 10.1073/pnas.0701944104.
Waly M, Olteanu H, Banerjee R, Choi SW, Mason JB, Parker BS, Sukumar S, Shim S, Sharma A, Benzecry JM, Power-Charnitsky VA, Deth RC: Activation of methionine synthase by insulin-like growth factor-1 and dopamine: a target for neurodevelopmental toxins and thimerosal. Mol Psychiatry. 2004, 9: 358-370. 10.1038/sj.mp.4001476.
Roberts EM, English PB, Grether JK, Windham GC, Somberg L, Wolff C: Maternal residence near agricultural pesticide applications and autism spectrum disorders among children in the California Central Valley. Environ Health Perspect. 2007, 115: 1482-1489.
Paul R, Pessah IN, Gane L, Ono M, Hagerman PJ, Brunberg JA, Tassone F, Bourgeois JA, Adams PE, Nguyen DV, Hagerman R: Early onset of neurological symptoms in fragile X premutation carriers exposed to neurotoxins. Neurotoxicology. 2010, 31: 399-402. 10.1016/j.neuro.2010.04.002.
Chen Y, Tassone F, Berman RF, Hagerman PJ, Hagerman RJ, Willemsen R, Pessah IN: Murine hippocampal neurons expressing Fmr1 gene premutations show early developmental deficits and late degeneration. Hum Mol Genet. 2010, 19: 196-208. 10.1093/hmg/ddp479.
Ross-Inta C, Omanska-Klusek A, Wong S, Barrow C, Garcia-Arocena D, Iwahashi C, Berry-Kravis E, Hagerman RJ, Hagerman PJ, Giulivi C: Mitochondrial dysfunction in fragile X-associated tremor/ataxia syndrome. J Biol Chem. 2010, 429: 545-52.
O'Dwyer JP, Clabby C, Crown J, Barton DE, Hutchinson M: Fragile X-associated tremor/ataxia syndrome presenting in a woman after chemotherapy. Neurology. 2005, 65: 331-332. 10.1212/01.wnl.0000168865.36352.53.
Qin M, Kang J, Burlin TV, Jiang C, Smith CB: Postadolescent changes in regional cerebral protein synthesis: an in vivo study in the FMR1 null mouse. J Neurosci. 2005, 25: 5087-5095. 10.1523/JNEUROSCI.0093-05.2005.
Darnell JC, van Dreische S, Zhang C, Mele A, Zang JB, Fak JJ, S-W C, Richter J, Darnell RB: HITS-CLIP identifies specific neuronal mRNA targets of translational repression by the fragile X mental retardation protein, FMRP [abstract]. Keystone Symposia; Snowbird, UT. 2010, 56-Abstract # 016
Dahlhaus R, El-Husseini A: Altered neuroligin expression is involved in social deficits in a mouse model of the fragile X syndrome. Behav Brain Res. 2010, 208: 96-105. 10.1016/j.bbr.2009.11.019.
Darnell JC, Mostovetsky O, Darnell RB: FMRP RNA targets: identification and validation. Genes Brain Behav. 2005, 4: 341-349. 10.1111/j.1601-183X.2005.00144.x.
Miyashiro KY, Beckel-Mitchener A, Purk TP, Becker KG, Barret T, Liu L, Carbonetto S, Weiler IJ, Greenough WT, Eberwine J: RNA cargoes associating with FMRP reveal deficits in cellular functioning in Fmr1 null mice. Neuron. 2003, 37: 417-431. 10.1016/S0896-6273(03)00034-5.
Butler MG, Dasouki MJ, Zhou XP, Talebizadeh Z, Brown M, Takahashi TN, Miles JH, Wang CH, Stratton R, Pilarski R, Eng C: Subset of individuals with autism spectrum disorders and extreme macrocephaly associated with germline PTEN tumour suppressor gene mutations. J Med Genet. 2005, 42: 318-321. 10.1136/jmg.2004.024646.
Chiu S, Wegelin JA, Blank J, Jenkins M, Day J, Hessl D, Tassone F, Hagerman R: Early acceleration of head circumference in children with fragile x syndrome and autism. J Dev Behav Pediatr. 2007, 28: 31-35. 10.1097/01.DBP.0000257518.60083.2d.
Nishimura Y, Martin CL, Vazquez-Lopez A, Spence SJ, Alvarez-Retuerto AI, Sigman M, Steindler C, Pellegrini S, Schanen NC, Warren ST, Geschwind DH: Genome-wide expression profiling of lymphoblastoid cell lines distinguishes different forms of autism and reveals shared pathways. Hum Mol Genet. 2007, 16: 1682-1698. 10.1093/hmg/ddm116.
Schenck A, Bardoni B, Langmann C, Harden N, Mandel JL, Giangrande A: CYFIP/Sra-1 controls neuronal connectivity in Drosophila and links the Rac1 GTPase pathway to the fragile X protein. Neuron. 2003, 38: 887-898. 10.1016/S0896-6273(03)00354-4.
Sharma A, Hoeffer CA, Takayasu Y, Miyawaki T, McBride SM, Klann E, Zukin RS: Dysregulation of mTOR signaling in fragile X syndrome. J Neurosci. 2010, 30: 694-702. 10.1523/JNEUROSCI.3696-09.2010.
Tassone F: mTOR up-regulation in patients with FXS [abstract]. FRAXA Investigators meeting May 2nd - 5th; Durham, NH. 2010
de Vries PJ: Targeted treatments for cognitive and neurodevelopmental disorders in tuberous sclerosis complex. Neurotherapeutics. 2010, 7: 275-282. 10.1016/j.nurt.2010.05.001.
Hazlett HC, Poe MD, Lightbody AA, Gerig G, MacFall JR, Ross AK, Provenzale J, Martin A, Reiss AL, Piven J: Teasing apart the heterogeneity of autism: Same behavior, different brains in toddlers with fragile X syndrome and autism. J Neurodevelop Disord. 2009, 1: 81-90. 10.1007/s11689-009-9009-8.
Hernandez RN, Feinberg RL, Vaurio R, Passanante NM, Thompson RE, Kaufmann WE: Autism spectrum disorder in fragile X syndrome: a longitudinal evaluation. Am J Med Genet A. 2009, 149A: 1125-1137. 10.1002/ajmg.a.32848.
Belmonte MK, Bourgeron T: Fragile X syndrome and autism at the intersection of genetic and neural networks. Nat Neurosci. 2006, 9: 1221-1225. 10.1038/nn1765.
Harlow EG, Till SM, Russell TA, Wijetunge LS, Kind P, Contractor A: Critical period plasticity is disrupted in the barrel cortex of FMR1 knockout mice. Neuron. 65: 385-398. 10.1016/j.neuron.2010.01.024.
Hoeft F, Carter JC, Lightbody AA, Cody Hazlett H, Piven J, Reiss AL: Region-specific alterations in brain development in one- to three-year-old boys with fragile X syndrome. Proc Natl Acad Sci USA. 2010, 107: 9335-9339. 10.1073/pnas.1002762107.
Hagerman RJ, Berry-Kravis E, Kaufmann WE, Ono MY, Tartaglia N, Lachiewicz A, Kronk R, Delahunty C, Hessl D, Visootsak J, Picker J, Gane L, Tranfaglia M: Advances in the treatment of fragile X syndrome. Pediatrics. 2009, 123: 378-390. 10.1542/peds.2008-0317.
Amiri K, Hagerman RJ, Hagerman PJ: Fragile X-associated tremor/ataxia syndrome: an aging face of the fragile X gene. Arch Neurol. 2008, 65: 19-25. 10.1001/archneurol.2007.30.
Brouwer JR, Willemsen R, Oostra BA: The FMR1 gene and fragile X-associated tremor/ataxia syndrome. Am J Med Genet B Neuropsychiatr Genet. 2009, 150B: 782-798. 10.1002/ajmg.b.30910.
Dick KA, Margolis JM, Day JW, Ranum LP: Dominant non-coding repeat expansions in human disease. Genome Dyn. 2006, 1: 67-83. full_text.
Tan H, Li H, Jin P: RNA-mediated pathogenesis in fragile X-associated disorders. Neurosci Lett. 2009, 466: 103-108. 10.1016/j.neulet.2009.07.053.
Garcia-Arocena D, Hagerman PJ: Advances in understanding the molecular basis of FXTAS. Hum Mol Genet. 2010, 19: R83-89. 10.1093/hmg/ddq166.
Hagerman PJ, Hagerman RJ: Fragile X-associated tremor/ataxia syndrome (FXTAS). Ment Retard Dev Disabil Res Rev. 2004, 10: 25-30. 10.1002/mrdd.20005.
Berman RF, Willemsen R: Mouse models of fragile X-associated tremor ataxia. J Investig Med. 2009, 57: 837-841.
Jin P, Duan R, Qurashi A, Qin Y, Tian D, Rosser TC, Liu H, Feng Y, Warren ST: Pur alpha binds to rCGG repeats and modulates repeat-mediated neurodegeneration in a Drosophila model of fragile X tremor/ataxia syndrome. Neuron. 2007, 55: 556-564. 10.1016/j.neuron.2007.07.020.
Sofola OA, Jin P, Qin Y, Duan R, Liu H, de Haro M, Nelson DL, Botas J: RNA-binding proteins hnRNP A2/B1 and CUGBP1 suppress fragile X CGG premutation repeat-induced neurodegeneration in a Drosophila model of FXTAS. Neuron. 2007, 55: 565-571. 10.1016/j.neuron.2007.07.021.
Willemsen R, Hoogeveen-Westerveld M, Reis S, Holstege J, Severijnen LA, Nieuwenhuizen IM, Schrier M, van Unen L, Tassone F, Hoogeveen AT, Hagerman PJ, Mientjes EJ, Oostra BA: The FMR1 CGG repeat mouse displays ubiquitin-positive intranuclear neuronal inclusions; implications for the cerebellar tremor/ataxia syndrome. Hum Mol Genet. 2003, 12: 949-959. 10.1093/hmg/ddg114.
Entezam A, Biacsi R, Orrison B, Saha T, Hoffman GE, Grabczyk E, Nussbaum RL, Usdin K: Regional FMRP deficits and large repeat expansions into the full mutation range in a new Fragile X premutation mouse model. Gene. 2007, 395: 125-134. 10.1016/j.gene.2007.02.026.
Handa V, Saha T, Usdin K: The fragile X syndrome repeats form RNA hairpins that do not activate the interferon-inducible protein kinase, PKR, but are cut by Dicer. Nucleic Acids Res. 2003, 31: 6243-6248. 10.1093/nar/gkg818.
Hashem V, Galloway JN, Mori M, Willemsen R, Oostra BA, Paylor R, Nelson DL: Ectopic expression of CGG containing mRNA is neurotoxic in mammals. Hum Mol Genet. 2009, 18: 2443-2451. 10.1093/hmg/ddp182.
Jin P, Zarnescu DC, Zhang F, Pearson CE, Lucchesi JC, Moses K, Warren ST: RNA-mediated neurodegeneration caused by the fragile X premutation rCGG repeats in Drosophila. Neuron. 2003, 39: 739-747. 10.1016/S0896-6273(03)00533-6.
Sofola OA, Jin P, Botas J, Nelson DL: Argonaute-2-dependent rescue of a Drosophila model of FXTAS by FRAXE premutation repeat. Hum Mol Genet. 2007, 16: 2326-2332. 10.1093/hmg/ddm186.
Van Dam D, Errijgers V, Kooy RF, Willemsen R, Mientjes E, Oostra BA, De Deyn PP: Cognitive decline, neuromotor and behavioural disturbances in a mouse model for fragile-X-associated tremor/ataxia syndrome (FXTAS). Behav Brain Res. 2005, 162: 233-239. 10.1016/j.bbr.2005.03.007.
Allen E, He W, Yadav-Shah M, Sherman S: A study of the distributional characteristics of FMR1 transcript levels in 238 individuals. Human Genetics. 2004, 114: 439-447. 10.1007/s00439-004-1086-x.
Kenneson A, Zhang F, Hagedorn CH, Warren ST: Reduced FMRP and increased FMR1 transcription is proportionally associated with CGG repeat number in intermediate-length and premutation carriers. Hum Mol Genet. 2001, 10: 1449-1454. 10.1093/hmg/10.14.1449.
Hunter JE, Rohr JK, Sherman SL: Co-occurring diagnoses among FMR1 premutation allele carriers. Clin Genet. 2010, 77: 374-381. 10.1111/j.1399-0004.2009.01317.x.
Sullivan AK, Marcus M, Epstein MP, Allen EG, Anido AE, Paquin JJ, Yadav-Shah M, Sherman SL: Association of FMR1 repeat size with ovarian dysfunction. Hum Reprod. 2005, 20: 402-412. 10.1093/humrep/deh635.
Wittenberger MD, Hagerman RJ, Sherman SL, McConkie-Rosell A, Welt CK, Rebar RW, Corrigan EC, Simpson JL, Nelson LM: The FMR1 premutation and reproduction. Fertil Steril. 2007, 87: 456-465. 10.1016/j.fertnstert.2006.09.004.
Greco CM, Hagerman RJ, Tassone F, Chudley AE, Del Bigio MR, Jacquemont S, Leehey M, Hagerman PJ: Neuronal intranuclear inclusions in a new cerebellar tremor/ataxia syndrome among fragile X carriers. Brain. 2002, 125: 1760-1771. 10.1093/brain/awf184.
Tassone F, Iwahashi C, Hagerman PJ: FMR1 RNA within the intranuclear inclusions of fragile X-associated tremor/ataxia syndrome (FXTAS). RNA Biol. 2004, 1: 103-105.
Bailey DB, Raspa M, Olmsted M, Holiday DB: Co-occurring conditions associated with FMR1 gene variations: findings from a national parent survey. Am J Med Genet A. 2008, 146A: 2060-2069. 10.1002/ajmg.a.32439.
Garcia-Arocena D, Yang JE, Brouwer JR, Tassone F, Iwahashi C, Berry-Kravis EM, Goetz CG, Sumis AM, Zhou L, Nguyen DV, Campos L, Howell E, Ludwig A, Greco C, Willemsen R, Hagerman RJ, Hagerman PJ: Fibroblast phenotype in male carriers of FMR1 premutation alleles. Hum Mol Genet. 2010, 19: 299-312. 10.1093/hmg/ddp497.
Hunsaker MR, Wenzel HJ, Willemsen R, Berman RF: Progressive spatial processing deficits in a mouse model of the fragile X premutation. Behav Neurosci. 2009, 123: 1315-1324. 10.1037/a0017616.
Tassone F, Adams J, Berry-Kravis EM, Cohen SS, Brusco A, Leehey MA, Li L, Hagerman RJ, Hagerman PJ: CGG repeat length correlates with age of onset of motor signs of the fragile X-associated tremor/ataxia syndrome (FXTAS). Am J Med Genet B Neuropsychiatr Genet. 2007, 144: 566-569.
Lee JE, Cooper TA: Pathogenic mechanisms of myotonic dystrophy. Biochem Soc Trans. 2009, 37: 1281-1286. 10.1042/BST0371281.
Wheeler TM, Thornton CA: Myotonic dystrophy: RNA-mediated muscle disease. Curr Opin Neurol. 2007, 20: 572-576. 10.1097/WCO.0b013e3282ef6064.
Arocena DG, Iwahashi CK, Won N, Beilina A, Ludwig AL, Tassone F, Schwartz PH, Hagerman PJ: Induction of inclusion formation and disruption of lamin A/C structure by premutation CGG-repeat RNA in human cultured neural cells. Hum Mol Genet. 2005, 14: 3661-3671. 10.1093/hmg/ddi394.
Sellier C, Rau F, Liu Y, Tassone F, Hukema RK, Gattoni R, Schneider A, Richard S, Willemsen R, Elliott DJ, Hagerman PJ, Charlet-Berguerand N: Sam68 sequestration and partial loss of function are associated with splicing alterations in FXTAS patients. EMBO J. 2010, 29: 1248-1261. 10.1038/emboj.2010.21.
Sellier C, Hagerman P, Willemsen R, Charlet-Berguerand N: DROSHA/DGCR8 sequestration by expanded CGG repeats leads to global micro-RNA processing alteration in FXTAS patients [abstract]. 12th International Fragile X Conference; July 21-25; Detroit, MI. 2010
Mah LJ, El-Osta A, Karagiannis TC: gammaH2AX: a sensitive molecular marker of DNA damage and repair. Leukemia. 2010, 24: 679-686. 10.1038/leu.2010.6.
Iwahashi CK, Yasui DH, An HJ, Greco CM, Tassone F, Nannen K, Babineau B, Lebrilla CB, Hagerman RJ, Hagerman PJ: Protein composition of the intranuclear inclusions of FXTAS. Brain. 2006, 129: 256-271. 10.1093/brain/awh650.
Oliveira G, Diogo L, Grazina M, Garcia P, Ataide A, Marques C, Miguel T, Borges L, Vicente AM, Oliveira CR: Mitochondrial dysfunction in autism spectrum disorders: a population-based study. Dev Med Child Neurol. 2005, 47: 185-189. 10.1017/S0012162205000332.
Weissman JR, Kelley RI, Bauman ML, Cohen BH, Murray KF, Mitchell RL, Kern RL, Natowicz MR: Mitochondrial disease in autism spectrum disorder patients: a cohort analysis. PLoS One. 2008, 3: e3815-10.1371/journal.pone.0003815.
Graf WD, Marin-Garcia J, Gao HG, Pizzo S, Naviaux RK, Markusic D, Barshop BA, Courchesne E, Haas RH: Autism associated with the mitochondrial DNA G8363A transfer RNA(Lys) mutation. J Child Neurol. 2000, 15: 357-361. 10.1177/088307380001500601.
Pons R, Andreu AL, Checcarelli N, Vila MR, Engelstad K, Sue CM, Shungu D, Haggerty R, de Vivo DC, DiMauro S: Mitochondrial DNA abnormalities and autistic spectrum disorders. J Pediatr. 2004, 144: 81-85. 10.1016/j.jpeds.2003.10.023.
Filipek PA, Juranek J, Smith M, Mays LZ, Ramos ER, Bocian M, Masser-Frye D, Laulhere TM, Modahl C, Spence MA, Gargus JJ: Mitochondrial dysfunction in autistic patients with 15q inverted duplication. Ann Neurol. 2003, 53: 801-804. 10.1002/ana.10596.
Hagerman RJ, Staley LW, O'Connor R, Lugenbeel K, Nelson D, McLean SD, Taylor A: Learning-disabled males with a fragile X CGG expansion in the upper premutation size range. Pediatrics. 1996, 97: 122-126.
Loesch DZ, Hay DA, Mulley J: Transmitting males and carrier females in fragile X--revisited. Am J Med Genet. 1994, 51: 392-399. 10.1002/ajmg.1320510418.
Tassone F, Hagerman RJ, Taylor AK, Mills JB, Harris SW, Gane LW, Hagerman PJ: Clinical involvement and protein expression in individuals with the FMR1 premutation. Am J Med Genet. 2000, 91: 144-152. 10.1002/(SICI)1096-8628(20000313)91:2<144::AID-AJMG14>3.0.CO;2-V.
Aziz M, Stathopulu E, Callias M, Taylor C, Turk J, Oostra B, Willemsen R, Patton M: Clinical features of boys with fragile X premutations and intermediate alleles. Am J Med Genet. 2003, 121B: 119-127. 10.1002/ajmg.b.20030.
Clifford S, Dissanayake C, Bui QM, Huggins R, Taylor AK, Loesch DZ: Autism spectrum phenotype in males and females with fragile X full mutation and premutation. J Autism Dev Disord. 2007, 37: 738-747. 10.1007/s10803-006-0205-z.
Farzin F, Perry H, Hessl D, Loesch D, Cohen J, Bacalman S, Gane L, Tassone F, Hagerman P, Hagerman R: Autism spectrum disorders and attention-deficit/hyperactivity disorder in boys with the fragile X premutation. J Dev Behav Pediatr. 2006, 27: S137-144. 10.1097/00004703-200604002-00012.
Loesch DZ, Cook M, Litewka L, Gould E, Churchyard A, Tassone F, Slater HR, Storey E: A low symptomatic form of neurodegeneration in younger carriers of the FMR1 premutation, manifesting typical radiological changes. J Med Genet. 2008, 45: 179-181. 10.1136/jmg.2007.054171.
Hashimoto RI, Backer KC, Tassone F, Hagerman RJ, Rivera SM: An fMRI study of the prefrontal activity during the performance of a working memory task in premutation carriers of the fragile X mental retardation 1 gene with and without fragile X-associated tremor/ataxia syndrome (FXTAS). J Psychiatr Res. 2010
Hashimoto R, Srivastava S, Tassone F, Hagerman RJ, Rivera SM: Diffusion tensor imaging in male premutation carriers of the fragile X mental retardation gene. Movement Disord.
Loesch DZ, Bui QM, Grigsby J, Butler E, Epstein J, Huggins RM, Taylor AK, Hagerman RJ: Effect of the fragile X status categories and the fragile X mental retardation protein levels on executive functioning in males and females with fragile X. Neuropsychology. 2003, 17: 646-657. 10.1037/0894-4105.17.4.646.
Grigsby J, Brega AG, Engle K, Leehey MA, Hagerman RJ, Tassone F, Hessl D, Hagerman PJ, Cogswell JB, Bennett RE, Cook K, Hall DA, Bounds LS, Paulich MJ, Reynolds A: Cognitive profile of fragile X premutation carriers with and without fragile X-associated tremor/ataxia syndrome. Neuropsychology. 2008, 22: 48-60. 10.1037/0894-4105.22.1.48.
Moore CJ, Daly EM, Schmitz N, Tassone F, Tysoe C, Hagerman RJ, Hagerman PJ, Morris RG, Murphy KC, Murphy DG: A neuropsychological investigation of male premutation carriers of fragile X syndrome. Neuropsychologia. 2004, 42: 1934-1947. 10.1016/j.neuropsychologia.2004.05.002.
Cornish KM, Li L, Kogan CS, Jacquemont S, Turk J, Dalton A, Hagerman RJ, Hagerman PJ: Age-dependent cognitive changes in carriers of the fragile X syndrome. Cortex. 2008, 44: 628-636. 10.1016/j.cortex.2006.11.002.
Cornish KM, Kogan CS, Li L, Turk J, Jacquemont S, Hagerman RJ: Lifespan changes in working memory in fragile X premutation males. Brain Cogn. 2009, 69: 551-558. 10.1016/j.bandc.2008.11.006.
Kogan CS, Cornish KM: Mapping self-reports of working memory deficits to executive dysfunction in fragile X mental retardation 1 (FMR1) gene premutation carriers asymptomatic for FXTAS. Brain Cogn. 2010, 73: 236-243. 10.1016/j.bandc.2010.05.008.
Hunter JE, Allen EG, Abramowitz A, Rusin M, Leslie M, Novak G, Hamilton D, Shubeck L, Charen K, Sherman SL: No evidence for a difference in neuropsychological profile among carriers and noncarriers of the FMR1 premutation in adults under the age of 50. Am J Hum Genet. 2008, 83: 692-702. 10.1016/j.ajhg.2008.10.021.
Grigsby J, Hills J, Wilson R, Leehey M, Hagerman RJ, Tassone F, Hagerman PJ: Dysexecutive syndrome in older men with action tremor and the fragile X premutation. J Int Neuropsychol Soc. 2002, 8: 282-
Roberts J, Mazzocco MM, Murphy MM, Hoehn-Saric R: Arousal modulation in females with fragile X or Turner syndrome. J Autism Dev Disord. 2008, 38: 20-27. 10.1007/s10803-007-0356-6.
Rodriguez-Revenga L, Madrigal I, Alegret M, Santos M, Mila M: Evidence of depressive symptoms in fragile-X syndrome premutated females. Psychiatr Genet. 2008, 18: 153-155. 10.1097/YPG.0b013e3282f97e0b.
Franke P, Leboyer M, Gansicke M, Weiffenbach O, Biancalana V, Cornillet-Lefebre P, Croquette MF, Froster U, Schwab SG, Poustka F, Hautzinger M, Maier W: Genotype-phenotype relationship in female carriers of the premutation and full mutation of FMR-1. Psychiatry Res. 1998, 80: 113-127. 10.1016/S0165-1781(98)00055-9.
Hessl D, Tassone F, Loesch DZ, Berry-Kravis E, Leehey MA, Gane LW, Barbato I, Rice C, Gould E, Hall DA, Grigsby J, Wegelin JA, Harris S, Lewin F, Weinberg D, Hagerman PJ, Hagerman RJ: Abnormal elevation of FMR1 mRNA is associated with psychological symptoms in individuals with the fragile X premutation. Am J Med Genet B Neuropsychiatr Genet. 2005, 139B: 115-121. 10.1002/ajmg.b.30241.
Bourgeois J, Seritan A, Casillas E, Hessl D, Schneider A, Yang Y, Kaur I, Cogswell J, Nguyen D, Hagerman R: Lifetime prevalence of mood and anxiety disorders in fragile X premutation carriers. J Clin Psychiatry. 2010, 10.4088/JCP.4009m05407blu
Rodriguez-Revenga L, Madrigal I, Pagonabarraga J, Xuncla M, Badenas C, Kulisevsky J, Gomez B, Mila M: Penetrance of FMR1 premutation associated pathologies in fragile X syndrome families. Eur J Hum Genet. 2009, 17: 1359-1362. 10.1038/ejhg.2009.51.
Zhang L, Coffey S, Lua LL, Greco CM, Schafer JA, Brunberg J, Borodyanskaya M, Agius MA, Apperson M, Leehey M, Tartaglia N, Tassone F, Hagerman PJ, Hagerman RJ: FMR1 premutation in females diagnosed with multiple sclerosis. J Neurol Neurosurg Psychiatry. 2009, 80: 812-814. 10.1136/jnnp.2008.160960.
Greco CM, Tassone F, Garcia-Arocena D, Tartaglia N, Coffey SM, Vartanian TK, Brunberg JA, Hagerman PJ, Hagerman RJ: Clinical and neuropathologic findings in a woman with the FMR1 premutation and multiple sclerosis. Arch Neurol. 2008, 65: 1114-1116. 10.1001/archneur.65.8.1114.
Chonchaiya W, Tassone F, Ashwood P, Hessl D, Schneider A, Campos L, Nguyen D, Au J, Hagerman R: Autoimmune disease in mothers with the FMR1 premutation is associated with seizures in their children with fragile X syndrome. J Hum Genet.
Zalfa F, Eleuteri B, Dickson KS, Mercaldo V, De Rubeis S, di Penta A, Tabolacci E, Chiurazzi P, Neri G, Grant SG, Bagni C: A new function for the fragile X mental retardation protein in regulation of PSD-95 mRNA stability. Nat Neurosci. 2007, 10: 578-587. 10.1038/nn1893.
Penagarikano O, Mulle JG, Warren ST: The pathophysiology of fragile X syndrome. Annu Rev Genomics Hum Genet. 2007, 8: 109-129. 10.1146/annurev.genom.8.080706.092249.
Bassell GJ, Warren ST: Fragile X syndrome: loss of local mRNA regulation alters synaptic development and function. Neuron. 2008, 60: 201-214. 10.1016/j.neuron.2008.10.004.
Luo Y, Shan G, Guo W, Smrt RD, Johnson EB, Li X, Pfeiffer RL, Szulwach KE, Duan R, Barkho BZ, Li W, Liu C, Jin P, Zhao X: Fragile x mental retardation protein regulates proliferation and differentiation of adult neural stem/progenitor cells. PLoS Genet. 2010, 6: e1000898-10.1371/journal.pgen.1000898.
Utari A, Adams E, Berry-Kravis E, Chavez A, Scaggs F, Ngotran L, Boyd A, Hessl D, Gane LW, Tassone F, Tartaglia N, Leehey MA, Hagerman RJ: Aging in fragile X syndrome. J Neurodev Disord. 2010, 2: 70-76. 10.1007/s11689-010-9047-2.
Wegiel J, Kuchna I, Nowicki K, Imaki H, Wegiel J, Marchi E, Ma SY, Chauhan A, Chauhan V, Bobrowicz TW, de Leon M, Louis LA, Cohen IL, London E, Brown WT, Wisniewski T: The neuropathology of autism: defects of neurogenesis and neuronal migration, and dysplastic changes. Acta Neuropathol. 2010, 119: 755-770. 10.1007/s00401-010-0655-4.
Bear MF, Huber KM, Warren ST: The mGluR theory of fragile X mental retardation. Trends Neurosci. 2004, 27: 370-377. 10.1016/j.tins.2004.04.009.
Dolen G, Bear MF: Role for metabotropic glutamate receptor 5 (mGluR5) in the pathogenesis of fragile X syndrome. J Physiol. 2008, 586: 1503-1508. 10.1113/jphysiol.2008.150722.
Comery TA, Harris JB, Willems PJ, Oostra BA, Irwin SA, Weiler IJ, Greenough WT: Abnormal dendritic spines in fragile X knockout mice: maturation and pruning deficits. Proc Natl Acad Sci USA. 1997, 94: 5401-5404. 10.1073/pnas.94.10.5401.
Nimchinsky EA, Oberlander AM, Svoboda K: Abnormal development of dendritic spines in FMR1 knock-out mice. J Neurosci. 2001, 21: 5139-5146.
Irwin SA, Patel B, Idupulapati M, Harris JB, Crisostomo RA, Larsen BP, Kooy F, Willems PJ, Cras P, Kozlowski PB, Swain RA, Weiler IJ, Greenough WT: Abnormal dendritic spine characteristics in the temporal and visual cortices of patients with fragile-X syndrome: a quantitative examination. Am J Med Genet. 2001, 98: 161-167. 10.1002/1096-8628(20010115)98:2<161::AID-AJMG1025>3.0.CO;2-B.
Bilousova TV, Dansie L, Ngo M, Aye J, Charles JR, Ethell DW, Ethell IM: Minocycline promotes dendritic spine maturation and improves behavioural performance in the fragile X mouse model. J Med Genet. 2009, 46: 94-102. 10.1136/jmg.2008.061796.
Hinton VJ, Brown WT, Wisniewski K, Rudelli RD: Analysis of neocortex in three males with the fragile X syndrome. Am J Med Genet. 1991, 41: 289-294. 10.1002/ajmg.1320410306.
Irwin SA, Galvez R, Greenough WT: Dendritic spine structural anomalies in fragile-X mental retardation syndrome. Cerebral Cortex. 2000, 10: 1038-1044. 10.1093/cercor/10.10.1038.
Chuang SC, Zhao W, Bauchwitz R, Yan Q, Bianchi R, Wong RK: Prolonged epileptiform discharges induced by altered group I metabotropic glutamate receptor-mediated synaptic responses in hippocampal slices of a fragile X mouse model. J Neurosci. 2005, 25: 8048-8055. 10.1523/JNEUROSCI.1777-05.2005.
Berry-Kravis E: Epilepsy in fragile X syndrome. Developmental Medicine and Child Neurology. 2002, 44: 724-728. 10.1111/j.1469-8749.2002.tb00277.x.
Dolen G, Osterweil E, Rao BS, Smith GB, Auerbach BD, Chattarji S, Bear MF: Correction of fragile X syndrome in mice. Neuron. 2007, 56: 955-962. 10.1016/j.neuron.2007.12.001.
Bear MF: Therapeutic implications of the mGluR theory of fragile X mental retardation. Genes Brain Behav. 2005, 4: 393-398. 10.1111/j.1601-183X.2005.00135.x.
Gasparini F, Lingenhohl K, Stoehr N, Flor PJ, Heinrich M, Vranesic I, Biollaz M, Allgeier H, Heckendorn R, Urwyler S, Varney MA, Johnson EC, Hess SD, Rao SP, Sacaan AI, Santori EM, Veliçelebi G, Kuhn R: 2-Methyl-6-(phenylethynyl)-pyridine (MPEP), a potent, selective and systemically active mGlu5 receptor antagonist. Neuropharmacology. 1999, 38: 1493-1503. 10.1016/S0028-3908(99)00082-9.
Yan QJ, Rammal M, Tranfaglia M, Bauchwitz RP: Suppression of two major fragile X syndrome mouse model phenotypes by the mGluR5 antagonist MPEP. Neuropharmacology. 2005, 49: 1053-1066. 10.1016/j.neuropharm.2005.06.004.
de Vrij FM, Levenga J, van der Linde HC, Koekkoek SK, De Zeeuw CI, Nelson DL, Oostra BA, Willemsen R: Rescue of behavioral phenotype and neuronal protrusion morphology in Fmr1 KO mice. Neurobiol Dis. 2008, 31: 127-132. 10.1016/j.nbd.2008.04.002.
Bauchwitz RP, Yan Q, Rammal M: Modulation of mGluR5 activity in vivo can ameliorate phenotypic markers of fragile X syndrome in mice. Society for Neuroscience; October 23-27; San Diego, CA. 2004, Society for Neuroscience, 583.520
McBride SM, Choi CH, Wang Y, Liebelt D, Braunstein E, Ferreiro D, Sehgal A, Siwicki KK, Dockendorff TC, Nguyen HT, McDonald TV, Jongens TA: Pharmacological rescue of synaptic plasticity, courtship behavior, and mushroom body defects in a Drosophila model of fragile X syndrome. Neuron. 2005, 45: 753-764. 10.1016/j.neuron.2005.01.038.
Berry-Kravis E, Hessl D, Coffey S, Hervey C, Schneider A, Yuhas J, Hutchison J, Snape M, Tranfaglia M, Nguyen DV, Hagerman R: A pilot open label, single dose trial of fenobam in adults with fragile X syndrome. J Med Genet. 2009, 46: 266-271. 10.1136/jmg.2008.063701.
Wang LW, Berry-Kravis E, Hagerman RJ: Fragile X: leading the way for targeted treatments in autism. Neurotherapeutics. 2010, 7: 264-274. 10.1016/j.nurt.2010.05.005.
Isaacson JS, Hille B: GABA(B)-mediated presynaptic inhibition of excitatory transmission and synaptic vesicle dynamics in cultured hippocampal neurons. Neuron. 1997, 18: 143-152. 10.1016/S0896-6273(01)80053-2.
Scanziani M, Capogna M, Gahwiler BH, Thompson SM: Presynaptic inhibition of miniature excitatory synaptic currents by baclofen and adenosine in the hippocampus. Neuron. 1992, 9: 919-927. 10.1016/0896-6273(92)90244-8.
Sohn JW, Lee D, Cho H, Lim W, Shin HS, Lee SH, Ho WK: Receptor-specific inhibition of GABAB-activated K+ currents by muscarinic and metabotropic glutamate receptors in immature rat hippocampus. J Physiol. 2007, 580: 411-422. 10.1113/jphysiol.2006.125914.
Zupan B, Toth M: Inactivation of the maternal fragile X gene results in sensitization of GABAB receptor function in the offspring. J Pharmacol Exp Ther. 2008, jpet.108.143990
Pacey LK, Heximer SP, Hampson DR: Increased GABA(B) receptor-mediated signaling reduces the susceptibility of fragile X knockout mice to audiogenic seizures. Mol Pharmacol. 2009, 76: 18-24. 10.1124/mol.109.056127.
Berry-Kravis E, Cherubini M, Zarevics P, Rathmell B, Wang PP, Carpenter R, Bear M, Hagerman R: Arbaclofen for the treatment of children and adults with fragile X syndrome: results of a phase 2, randomized, double-blind, placebo-controlled, crossover study [Abstract]. International Meeting for Autism Research; Philadelphia, PA. 2010, 741-Abstract # 140.004
Mihalek RM, Banerjee PK, Korpi ER, Quinlan JJ, Firestone LL, Mi ZP, Lagenaur C, Tretter V, Sieghart W, Anagnostaras SG, Sage JR, Fanselow MS, Guidotti A, Spigelman I, Li Z, DeLorey TM, Olsen RW, Homanics GE: Attenuated sensitivity to neuroactive steroids in gamma-aminobutyrate type A receptor delta subunit knockout mice. Proc Natl Acad Sci USA. 1999, 96: 12905-12910. 10.1073/pnas.96.22.12905.
Carter RB, Wood PL, Wieland S, Hawkinson JE, Belelli D, Lambert JJ, White HS, Wolf HH, Mirsadeghi S, Tahir SH, Bolger MB, Lan NC, Gee KW: Characterization of the anticonvulsant properties of ganaxolone (CCD 1042; 3alpha-hydroxy-3beta-methyl-5alpha-pregnan-20-one), a selective, high-affinity, steroid modulator of the gamma-aminobutyric acid(A) receptor. J Pharmacol Exp Ther. 1997, 280: 1284-1295.
Bormann J: The 'ABC' of GABA receptors. Trends Pharmacol Sci. 2000, 21: 16-19. 10.1016/S0165-6147(99)01413-3.
Chebib M, Johnston GA: The 'ABC' of GABA receptors: a brief review. Clin Exp Pharmacol Physiol. 1999, 26: 937-940. 10.1046/j.1440-1681.1999.03151.x.
D'Hulst C, Kooy RF: The GABAA receptor: a novel target for treatment of fragile X?. Trends Neurosci. 2007, 30: 425-431. 10.1016/j.tins.2007.06.003.
Miyashiro KY, Beckel-Mitchener A, Purk TP, Becker KG, Barret T, Liu L, Carbonetto S, Weiler IJ, Greenough WT, Eberwine J: RNA cargoes associating with FMRP reveal deficits in cellular functioning in Fmr1 null mice. Neuron. 2003, 37: 417-431. 10.1016/S0896-6273(03)00034-5.
Chang S, Bray SM, Li Z, Zarnescu DC, He C, Jin P, Warren ST: Identification of small molecules rescuing fragile X syndrome phenotypes in Drosophila. Nat Chem Biol. 2008, 4: 256-263. 10.1038/nchembio.78.
D'Hulst C, De Geest N, Reeve SP, Van Dam D, De Deyn PP, Hassan BA, Kooy RF: Decreased expression of the GABAA receptor in fragile X syndrome. Brain Res. 2006, 1121: 238-245. 10.1016/j.brainres.2006.08.115.
Gantois I, Vandesompele J, Speleman F, Reyniers E, D'Hooge R, Severijnen LA, Willemsen R, Tassone F, Kooy RF: Expression profiling suggests underexpression of the GABA(A) receptor subunit delta in the fragile X knockout mouse model. Neurobiol Dis. 2006, 21: 346-357. 10.1016/j.nbd.2005.07.017.
Selby L, Zhang C, Sun QQ: Major defects in neocortical GABAergic inhibitory circuits in mice lacking the fragile X mental retardation protein. Neurosci Lett. 2007, 412: 227-232. 10.1016/j.neulet.2006.11.062.
D'Hulst C, Heulens I, Brouwer JR, Willemsen R, De Geest N, Reeve SP, De Deyn PP, Hassan BA, Kooy RF: Expression of the GABAergic system in animal models for fragile X syndrome and fragile X associated tremor/ataxia syndrome (FXTAS). Brain Research. 2009, 1253: 176-183. 10.1016/j.brainres.2008.11.075.
Curia G, Papouin T, Seguela P, Avoli M: Downregulation of tonic GABAergic inhibition in a mouse model of fragile X syndrome. Cereb Cortex. 2009, 19: 1515-1520. 10.1093/cercor/bhn159.
El Idrissi A, Ding XH, Scalia J, Trenkner E, Brown WT, Dobkin C: Decreased GABA(A) receptor expression in the seizure-prone fragile X mouse. Neurosci Lett. 2005, 377: 141-146. 10.1016/j.neulet.2004.11.087.
Kooy RF: Of mice and the fragile X syndrome. Trends Genet. 2003, 19: 148-154. 10.1016/S0168-9525(03)00017-9.
D'Antuono M, Merlo D, Avoli M: Involvement of cholinergic and gabaergic systems in the fragile X knockout mice. Neuroscience. 2003, 119: 9-13. 10.1016/S0306-4522(03)00103-9.
Kooy F, Heulens I, D'Hulst C, Van der Aa N, Bagni C, Hassan B, De Deyn P: The GABAA receptor as a potential target for therapy of the fragile X syndrome [abstract]. NFXF 12th International FX Conference; July 21 - 25; Detroit, MI. 2010
Reddy DS: Pharmacology of endogenous neuroactive steroids. Crit Rev Neurobiol. 2003, 15: 197-234. 10.1615/CritRevNeurobiol.v15.i34.20.
Bialer M, Johannessen SI, Kupferberg HJ, Levy RH, Perucca E, Tomson T: Progress report on new antiepileptic drugs: a summary of the Eighth Eilat Conference (EILAT VIII). Epilepsy Res. 2007, 73: 1-52. 10.1016/j.eplepsyres.2006.10.008.
Rogawski MA, Reddy DS: Neurosteroids: endogenous modulators of seizure susceptibility. Epilepsy: Scientific Foundations of Clinical Practice. Edited by: Rho JM, Sankar R, Cavazos J. 2004, New York: Marcel Dekker, 319-355.
Laxer K, Blum D, Abou-Khalil BW, Morrell MJ, Lee DA, Data JL, Monaghan EP: Assessment of ganaxolone's anticonvulsant activity using a randomized, double-blind, presurgical trial design. Ganaxolone Presurgical Study Group. Epilepsia. 2000, 41: 1187-1194. 10.1111/j.1528-1157.2000.tb00324.x.
Kerrigan JF, Shields WD, Nelson TY, Bluestone DL, Dodson WE, Bourgeois BF, Pellock JM, Morton LD, Monaghan EP: Ganaxolone for treating intractable infantile spasms: a multicenter, open-label, add-on trial. Epilepsy Res. 2000, 42: 133-139. 10.1016/S0920-1211(00)00170-4.
Bilousova TV, Rusakov DA, Ethell DW, Ethell IM: Matrix metalloproteinase-7 disrupts dendritic spines in hippocampal neurons through NMDA receptor activation. J Neurochem. 2006, 97: 44-56. 10.1111/j.1471-4159.2006.03701.x.
Utari A, Chonchaiya W, Rivera SM, Schneider A, Hagerman RJ, Faradz SM, Ethell IM, Nguyen DV: Side effects of minocycline treatment in patients with fragile X syndrome and exploration of outcome measures. Am J Intellect Dev Disabil. 2010, 115: 433-443. 10.1352/1944-7558-115.5.433.
Paribello : Open label add on treatment trial of minocycline in patients with fragile X syndrome [abstract]. FRAXA Investigators Meeting May 2nd - 5th; Durham, NH. 2010
Acknowledgements
This work was supported by National Institute of Health grants HD036071, HD02274, DE019583, DA024854, AG032119, AG032115, UL1DE019583; National Center for Research Resources UL1 RR024146; support from the Health and Human Services Administration of Developmental Disabilities grant 90DD05969 and the Norwegian Research Council, through The Medical Student Research Program.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
RH has received funding from Seaside Therapeutics, Novartis, Roche, Forest, Johnson & Johnson and Curemark for clinical trials, and also consults with Novartis and Roche regarding clinical trials in fragile X syndrome. PH is an unpaid consultant with Asuragen, and has a filed patent application for an FMR1 genotyping method. GH has no conflicts of interest.
Authors' contributions
All authors helped draft the manuscript, and all authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Hagerman, R., Hoem, G. & Hagerman, P. Fragile X and autism: Intertwined at the molecular level leading to targeted treatments. Molecular Autism 1, 12 (2010). https://doi.org/10.1186/2040-2392-1-12
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/2040-2392-1-12