
Abstract
Methanobacterium formicicum BRM9 was isolated from the rumen of a New Zealand Friesan cow grazing a ryegrass/clover pasture, and its genome has been sequenced to provide information on the phylogenetic diversity of rumen methanogens with a view to developing technologies for methane mitigation. The 2.45 Mb BRM9 chromosome has an average G + C content of 41%, and encodes 2,352 protein-coding genes. The genes involved in methanogenesis are comparable to those found in other members of the Methanobacteriaceae with the exception that there is no [Fe]-hydrogenase dehydrogenase (Hmd) which links the methenyl-H4MPT reduction directly with the oxidation of H2. Compared to the rumen Methanobrevibacter strains, BRM9 has a much larger complement of genes involved in determining oxidative stress response, signal transduction and nitrogen fixation. BRM9 also has genes for the biosynthesis of the compatible solute ectoine that has not been reported to be produced by methanogens. The BRM9 genome has a prophage and two CRISPR repeat regions. Comparison to the genomes of other Methanobacterium strains shows a core genome of ~1,350 coding sequences and 190 strain-specific genes in BRM9, most of which are hypothetical proteins or prophage related.
Similar content being viewed by others

Introduction
Ruminants have evolved an efficient digestive system in which microbes ferment the plant material that constitutes the animal’s diet to produce short chain fatty acids, principally acetic, propionic and butyric acids, and other products [1]. This fermentation is carried out by a complex microbial community which includes bacteria, ciliate protozoa, anaerobic fungi, and methanogenic archaea, and has been the focus of numerous studies. The role of the methanogenic archaea in the rumen environment is important as they use hydrogen (H2) derived from microbial fermentation as their energy source and combine it with carbon dioxide (CO2) to form methane (CH4), which is belched from the animal and released to the atmosphere. Other fermentation end-products including formate and methyl-containing compounds can also be substrates for methanogenesis [2].
Methane is a potent greenhouse gas contributing to global climate change, and ruminant derived CH4 accounts for about one quarter of all anthropogenic CH4 emissions [3]. Development of strategies to reduce CH4 emissions from farmed animals are currently being investigated, and methanogen genome sequence information has already been used to inform CH4 mitigation strategies based on vaccines and small-molecule inhibitors [4, 5]. CH4 mitigation technologies should target features that are conserved across all rumen methanogens, and be methanogen-specific so that other rumen microbes can continue their normal digestive functions. To address this we are sequencing the genomes of cultures that represent the phylogenetic diversity of rumen methanogens to define their conserved features as targets for developing CH4 mitigation technologies [4, 6, 7], and to understand their role in the rumen environment, and interactions with other members of the rumen microbiome.
Organism information
Methanobacterium sp. BRM9 was isolated from the rumen of a New Zealand Friesan cow grazing a ryegrass/clover pasture [8]. It was described as a Gram positive non-motile, short rod which becomes a long, irregular rod at later growth stages. It is able to grow and produce methane from formate and H2/CO2, but not from acetate, alcohols or methylamines. Growth occurred over a wide temperature range (25–45°C) and at pH 6–8. Rumen fluid was required for growth. The 16S rRNA from BRM9 is 99.8% similar to the M. formicicum type strain DSM 1535 [Figure 1] which was isolated from a sewage sludge digester [9, 10] and as such BRM9 can be considered as a strain of M. formicicum. M. formicicum is found at high densities in anaerobic digesters and freshwater sediments, and has previously been isolated from the rumen [11], although Methanobacterium species only occur at low density in this environment [2]. Isolates have also been obtained as endosymbionts of anaerobic amoebae and ciliate protozoa species. Electron microscopic studies of M. formicicum show a long rod shaped morphology, and cells characterized by numerous cytoplasmic membrane bodies believed to be formed by invagination of the cell membrane [12, 13]. Characteristics of M. formicicum BRM9 are shown in Table 1 and Additional file 1: Table S1

Phylogenetic tree showing the position of Methanobacterium sp. BRM9 relative to type strains of other Methanobacterium species. The strains and their corresponding accession numbers are shown. The evolutionary history was inferred using the Neighbor-Joining method [14] with Methanothermus fervidus used as an outgroup. The optimal tree with the sum of branch length = 0.34833139 is shown. The percentage of replicate trees (>90%) in which the associated taxa clustered together in the bootstrap test (1000 replicates) are shown next to the branches [15]. The tree is drawn to scale, with branch lengths in the same units as those of the evolutionary distances used to infer the phylogenetic tree. The evolutionary distances were computed using the Kimura 2-parameter method [16] and are in the units of the number of base substitutions per site. The analysis involved 19 nucleotide sequences. All positions containing gaps and missing data were eliminated. There were a total of 1168 positions in the final dataset. Evolutionary analyses were conducted in MEGA5 [17].
Genome sequencing information
Genome project history
Methanobacterium formicicum BRM9 was selected for genome sequencing on the basis of its phylogenetic position relative to other methanogens belonging to the family Methanobacteriaceae. Table 2 presents the project information and its association with MIGS version 2.0 compliance [27].
Growth conditions and DNA isolation
BRM9 was grown in BY medium [28] with added SL10 Trace Elements solution (1 ml added l−1) [29], Selenite/Tungstate solution (final concentration of selenite and tungstate are 3 and 4 μg l−1 respectively), [30] and Vitamin 10 solution (0.1 ml added to 10 ml culture before inoculation) [6]. H2 was supplied as the energy source by pumping the culture vessels to 180 kPa over pressure with an 80:20 mixture of H2:CO2. Genomic DNA was extracted from freshly grown cells using a modified version of a liquid N2 and grinding method [31]. Briefly, BRM9 cultures were harvested by centrifugation at 20,000 × g for 20 min at 4°C and cell pellets combined into 40 ml Oakridge centrifuge tubes and frozen at −80°C. The frozen cell pellets were placed in a sterile, pre-cooled (−85°C) mortar and ground to a powder with periodic addition of liquid N2. Buffer B1 (5 ml Qiagen Genomic-Tip 500 Maxi kit, Qiagen, Hilden, Germany) containing RNase (2 μg ml−1 final concentration) was added to the powdered cell pellet to create a slurry which was then removed to a 15 ml Falcon tube. An additional 6 ml of B1 buffer was used to rinse the remaining material from the mortar and pestle and combined with the cell slurry, which was then treated following the Qiagen Genomic-Tip 500/G Maxi kit instructions. Finally, the genomic DNA was precipitated by the addition of 0.7 vol isopropanol, and collected by centrifugation at 12,000 × g for 10 min at room temperature. The supernatant was removed, and the DNA pellet was washed in 70% ethanol, re-dissolved in TE buffer (10 mM Tris–HCl, 1 mM EDTA pH 7.5) and stored at −20°C until required.
Genome sequencing and assembly
The complete genome sequence of BRM9 was determined using pyrosequencing of 3Kb mate paired-end sequence libraries using a 454 GS FLX platform with Titanium chemistry (Macrogen, Korea). Pyrosequencing reads provided 97× coverage of the genome and were assembled using the Newbler assembler version 2.0 (Roche 454 Life Sciences, USA). The Newbler assembly resulted in 85 contigs across 9 scaffolds. Gap closure was managed using the Staden package [32] and gaps were closed using additional Sanger sequencing by standard and inverse PCR based techniques. A total of 219 additional reactions were used to close gaps and to improve the quality of the genome sequence to ensure correct assembly and to resolve any remaining base-conflicts. Assembly validation was confirmed by pulsed-field gel electrophoresis as described previously [6], using the enzyme AscI which cuts the BRM9 chromosome at 6 sites.
Genome annotation
A GAMOLA/ARTEMIS [33, 34] software suite was used to manage genome annotation. Protein-encoding open reading frames (ORFs) were identified using the ORF-prediction program Glimmer [35] and BLASTX [36, 37]. A manual inspection was performed to verify or, if necessary, redefine the start and stop codons of each ORF. Assignment of protein function to ORFs was performed manually using results from the following sources; BLASTP [36] to both a non-redundant protein database provided by the National Centre for Biotechnology Information (NCBI) [38] and Clusters of Orthologous Groups (COG) database [39]. HMMER [40] was used to identify protein motifs to both the PFAM [41] and TIGRFAM [42] libraries. TMHMM [43, 44] was used to predict transmembrane sequences, and SignalP, version 4.1 [45] was used for the prediction of signal peptides. Ribosomal RNA genes were detected on the basis of BLASTN searches to a custom GAMOLA ribosomal database. Transfer RNA genes were identified using tRNAscan-SE [46]. Miscellaneous-coding RNAs were identified using the Rfam database [47] utilizing the INFERNAL software package [48]. The genome sequence was prepared for NCBI submission using Sequin [49]. The adenine residue of the start codon of the Cdc6-1 replication initiation protein (BRM9_0001) gene was chosen as the first base for the BRM9 genome. The nucleotide sequence of the Methanobacterium formicicum BRM9 chromosome has been deposited in Genbank under accession number CP006933.
Genome properties
The genome of Methanobacterium formicicum BRM9 consists of a single 2,449,988 basepair (bp) circular chromosome with an average G + C content of 41%. A total of 2,418 genes were predicted, 2,352 of which were protein-coding genes, representing 83% of the total genome sequence. A putative function was assigned to 1,715 of the protein-coding genes, with the remainder annotated as hypothetical proteins. The properties and statistics of the genome are summarized in Tables 3, 4 and 5. The BRM9 genome has a 37 Kb prophage (BRM9_1642-1689) with several genes that have best matches to those from other prophage. The phage ORFs are flanked by 22 bp sequences indicative of attL and attR sites. In addition there are several CRISPR genes associated with two CRISPR repeat regions of 7178 and 11914 bp, as well as the components of a type I restriction-modification system.
Insights from the genome
The genes involved in methanogenesis are comparable to those found in other members of the Methanobacteriaceae with the exception that there is no [Fe]-hydrogenase dehydrogenase (Hmd) which links the methenyl-H4MPT reduction directly with the oxidation of H2. BRM9 has the methyl coenzyme M reductase II genes (mrt AGDB, BRM9_2153-2156), unlike Methanobrevibacter strains M1 and AbM4 [6, 7]. BRM9 has a cysteinyl-tRNA synthetase (cys S), but also encodes the alternative tRNA-dependent cysteine biosynthesis pathway (sep S/psc S) found in Methanocaldococcus jannaschii and other methanogens [50] but not in Methanobrevibacter sp. BRM9 also has a carbon monoxide dehydrogenase/acetyl-coenzyme A synthase (CODH/ACS, or Cdh) to fix CO2 and form acetyl-CoA, and several acetyl-CoA synthetases one of which is located next to a possible acetate permease (BRM9_1255). Like many other methanogens, the CODH/ACS genes in BRM9 are found in a single cluster (BRM9_0795-0801). There is also a NAD-dependent malic enzyme (BRM9_2358) able to catalyse the oxidative decarboxylation of malate to form pyruvate and CO2. This is found in three other Methanobacterium strains (MBC34, PP1, SWAN-1) but not in other members of the Methanobacteriaceae.
The cell walls of members of the Methanobacteriaceae consist of pseudomurein and while the pathway for pseudomurein biosynthesis and its primary structure have been elucidated the enzymes involved have not been characterized. The predicted pseudomurein biosynthesis genes are similar to those found in Methanobrevibacter species [6], but there are differences in the other cell wall glycopolymers. BRM9 has several proteins with multiple copies of the PMBR domain (Pfam accession PF09373) predicted to be involved in binding to pseudomurein. There are four clusters of genes involved in polysaccharide biosynthesis and two oligosaccharyl transferases, but BRM9 does not have homologues of neuA/neuB found in other methanogen strains including M. formicicum DSM 3637 [51]. BRM9 has fewer cell surface proteins than do Methanobrevibacter species, and these contain a range of different repeat domains.
Compared to the rumen Methanobrevibacter species BRM9 has a much larger complement of activities involved in oxidative stress response with a superoxide dismutase, a catalase/peroxidase and a peroxiredoxin (alkyl hydroperoxide reductase). BRM9 also has the three ectoine biosynthetic genes (ect ABC, BRM9_2205-2207) that encode production of the compatible solute ectoine that is normally found in halophilic or halotolerant organisms but has not been reported to be produced by methanogens [52]. The ectoine biosynthetic genes in BRM9 show no BLAST matches to other methanogens but have significant matches to Dehalogenimonas lykanthroporepellens, a dehalogenating bacterium from the phylum Chloroflexi isolated from contaminated groundwater [53]. The ect B and ect C genes also show homology to those from the rumen bacterium Wolinella succinogenes. Unlike the Methanobrevibacter species BRM9 has a large number of genes encoding components of histidine kinase/response regulator signal transduction systems. Many of these proteins include 1–5 PAS domains. These are believed to monitor changes in redox potential, oxygen, and the overall energy level of the cell [54].
The metabolism of nitrogen by BRM9 is somewhat different from Methanobrevibacter M1 and AbM4. BRM9 has two ammonium transporters and encodes the glutamine synthase (GS)/glutamate synthase (glutamine:2-oxoglutarate aminotransferase, GOGAT) pathway of ammonium assimilation. Methanobacterium formicicum has been reported to fix nitrogen [55] and BRM9 contains a nif operon similar to that found in Methanococcus maripaludis and composed of nitrogenase and nitrogenase cofactor biosynthesis genes. Nitrogen assimilation genes are regulated by NrpR which represses transcription of nitrogen fixation genes, glutamine synthase, ammonium transporters and some other genes in M. maripaludis [56]. NrpR binds to inverted repeat operators in the promoter regions of these genes. The inverted repeat sequence recognized is GGAAN6TTCC and occurs in BRM9 upstream from the starts of gln A, nif H, pdx T, amt 1 and amt 2.
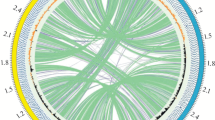
The genome of M. formicicum BRM9 is compared with those of other sequenced methanogens from the genus Methanobacterium in Table 6. The genome atlas of M. formicicum BRM9 is shown in Figure 2 and indicates that the gene content of these Methanobacterium strains is highly similar. Comparison of the ORFeome of BRM9 with those of other sequenced Methanobacterium species [Figure 3] shows a core genome of ~1,350 genes. There are 190 strain-specific genes in BRM9, which include the ectoine biosynthesis genes, CRISPR and prophage-related genes as well as numerous hypothetical proteins.

Genome atlas of Methanobacterium formicum BRM9. The circles from the outside represent: (1) forward and reverse coding domain sequences (CDS), the color coding of the CDS represent different Clusters of Orthologous Groups (COG) categories; (2) Reciprocal BLAST results with Methanobacterium strains MBC34; (3) PP1; (4) AL-21; (5) SWAN-1; (6) MB1; (7) rRNA, tRNA and CRISPR regions; (8) % GC plot; (9) GC skew [(GC)/(G + C)].

Flower plot illustrating the number of shared and specific genes based on OrthoMCL [ [61] ] analysis of Methanobacterium genomes.
Conclusions
This is the first report of a genome sequence for a Methanobacterium formicicum strain of rumen origin. The genus Methanobacterium consists of mesophilic methanogens from diverse anaerobic environments, but they only constitute a small proportion of the methanogen diversity in the rumen. However, the similarity in gene content between BRM9 and strains from other environments implies that BRM9 is not particularly adapted to the rumen and may struggle in competition with the better adapted Methanobrevibacter species. The conserved nature of the M. formicicum BRM9 genes for methanogenesis, central metabolism and pseudomurein cell wall formation suggest that this species will be amenable to inhibition by the small molecule inhibitor and vaccine-based methane mitigation technologies that are being developed for the other genera of methanogens found in the rumen.
References
Clauss M, Hume ID, Hummel J: Evolutionary adaptations of ruminants and their potential relevance for modern production systems. Animal 2010, 4: 979–992. 10.1017/S1751731110000388
Janssen PH, Kirs M: Structure of the archaeal community of the rumen. Appl Environ Microbiol 2008, 74: 3619–3625. 10.1128/AEM.02812-07
Thorpe A: Enteric fermentation and ruminant eructation: the role (and control?) of methane in the climate change debate. Climate Change 2009, 93: 407–431. 10.1007/s10584-008-9506-x
Leahy SC, Kelly WJ, Ronimus RS, Wedlock N, Altermann E, Attwood GT: Genome sequencing of rumen bacteria and archaea and its application to methane mitigation strategies. Animal 2013,7(Suppl 2):235–243.
Wedlock N, Janssen PH, Leahy S, Shu D, Buddle BM: Progress in the development of vaccines against rumen methanogens. Animal 2013,7(Suppl 2):244–252.
Leahy SC, Kelly WJ, Altermann E, Ronimus RS, Yeoman CJ, Pacheco DM, Li D, Kong Z, McTavish S, Sang C, Lambie SC, Janssen PH, Dey D, Attwood GT: The genome sequence of the rumen methanogen Methanobrevibacter ruminantium reveals new possibilities for controlling ruminant methane emissions. PLoS One 2010, 5: e8926. 10.1371/journal.pone.0008926
Leahy SC, Kelly WJ, Li D, Li Y, Altermann E, Lambie SC, Cox F, Attwood GT: The complete genome sequence of Methanobrevibacter sp. AbM4. Stand Genomic Sci 2013, 8: 215–227. 10.4056/sigs.3977691
Jarvis GN, Strömpl C, Burgess DM, Skillman LC, Moore ERB, Joblin KN: Isolation and identification of ruminal methanogens from grazing cattle. Curr Microbiol 2000, 40: 327–332. 10.1007/s002849910065
Bryant MP, Boone DR: Isolation and characterization of Methanobacterium formicicum MF. Int J Syst Bacteriol 1987, 37: 171. 10.1099/00207713-37-2-171
Boone DR: Replacement of the type strain of Methanobacterium formicicum and reinstatement of Methanobacterium bryantii sp. nov. nom. rev. (ex Balch and Wolfe, 1981) with M.o.H. (DSM 863) as the type strain. Int J Syst Bacteriol 1987, 37: 172–173. 10.1099/00207713-37-2-172
Oppermann RA, Nelson WO, Brown RE: In vitro studies of methanogenic rumen bacteria. J Dairy Sci 1957, 40: 779–788. 10.3168/jds.S0022-0302(57)94554-X
Langenberg KF, Bryant MP, Wolfe RS: Hydrogen-oxidizing methane bacteria II. Electron microscopy. J Bacteriol 1968, 95: 1124–1129.
Zeikus JG, Bowen VG: Comparative ultrastructure of methanogenic bacteria. Can J Microbiol 1975, 21: 121–129. 10.1139/m75-019
Saitou N, Nei M: The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol Biol Evol 1987, 4: 406–425.
Felsenstein J: Confidence limits on phylogenies: An approach using the bootstrap. Evolution 1985, 39: 783–791. 10.2307/2408678
Kimura M: A simple method for estimating evolutionary rate of base substitutions through comparative studies of nucleotide sequences. J Mol Evol 1980, 16: 111–120. 10.1007/BF01731581
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S: MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 2011, 28: 2731–2739. 10.1093/molbev/msr121
Woese CR, Kandler O, Wheelis ML: Towards a natural system of organisms: proposal for the domains Archaea, Bacteria, and Eucarya. Proc Natl Acad Sci U S A 1990, 87: 4576–4579. 10.1073/pnas.87.12.4576
Garrity GM, Phylum HJG, AII: Euryarchaeota phy. nov. In Bergey’s Manual of Systematic Bacteriology. Volume 1. 2nd edition. Edited by: Garrity GM, Boone DR, Castenholz RW. New York: Springer; 2001:211–355.
Validation List no. 85. Validation of publication of new names and new combinations previously effectively published outside the IJSEM Int J Syst Evol Microbiol 2002, 52: 685–690.
Boone DR, Class I: Methanobacteria class. nov. In Bergey’s Manual of Systematic Bacteriology. Volume 1. 2nd edition. Edited by: Garrity GM, Boone DR, Castenholz RW. New York: Springer; 2001:213–234.
List Editor. Validation List no. 6. Validation of the publication of new names and new combinations previously effectively published outside the IJSB Int J Syst Bacteriol 1981, 31: 215–218.
Balch WE, Fox GE, Magrum LJ, Woese CR, Wolfe RS: Methanogens: reevaluation of a unique biological group. Microbiol Rev 1979, 43: 260–296.
Judicial Commission of the International Committee on Systematics of Prokaryotes: The nomenclatural types of the orders Acholeplasmatales , Halanaerobiales , Halobacteriales , Methanobacteriales , Methanococcales , Methanomicrobiales , Planctomycetales , Prochlorales , Sulfolobales , Thermococcales , Thermoproteales and Verrucomicrobiales are the genera Acholeplasma , Halanaerobium , Halobacterium , Methanobacterium , Methanococcus , Methanomicrobium , Planctomyces , Prochloron , Sulfolobus , Thermococcus , Thermoproteus and Verrucomicrobium , respectively. Opinion 79. Int J Syst Evol Microbiol 2005, 55: 517–518.
Skerman VBD, McGowan V, Sneath PHA: Approved Lists of Bacterial Names. Int J Syst Bacteriol 1980, 30: 225–420. 10.1099/00207713-30-1-225
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G: Gene Ontology: tool for the unification of biology. Nat Genet 2000, 25: 25–29. 10.1038/75556
Field D, Garrity G, Gray T, Morrison N, Selengut J, Sterk P, Tatusova T, Thomson N, Allen MJ, Angiuoli SV, Ashburner M, Axelrod N, Baldauf S, Ballard S, Boore J, Cochrane G, Cole J, Dawyndt P, De Vos P, DePamphilis C, Edwards R, Faruque N, Feldman R, Gilbert J, Gilna P, Glöckner FO, Goldstein P, Guralnick R, Haft D, Hancock D, Hermjakob H, Hertz-Fowler C, et al.: The minimum information about a genome sequence (MIGS) specification. Nat Biotechnol 2008, 26: 541–547. 10.1038/nbt1360
Joblin KN, Naylor GE, Williams AG: Effect of Methanobrevibacter smithii on xylanolytic activity of anaerobic ruminal fungi. Appl Environ Microbiol 1990, 56: 2287–2295.
Widdel F, Kohring G, Mayer F: Studies on dissimilatory sulfate-reducing bacteria that decompose fatty acids III. Characterization of the filamentous gliding Desulfonema limicola gen. nov. sp. nov., and Desulfonema magnum sp. nov. Arch Microbiol 1983, 134: 286–294. 10.1007/BF00407804
Tschech A, Pfennig N: Growth yield increase linked to caffeate reduction in Acetobacterium woodii . Arch Microbiol 1984, 137: 163–167. 10.1007/BF00414460
Jarrell KF, Faguy D, Herbert AM, Kalmakoff ML: A general method of isolating high molecular weight DNA from methanogenic archaea (archaebacteria). Can J Microbiol 1992, 38: 65–68. 10.1139/m92-010
Staden R, Beal KF, Bonfield JK: The Staden package, 1998. Methods Mol Biol 2000, 132: 115–130.
Altermann E, Klaenhammer T: GAMOLA: a new local solution for sequence annotation and analyzing draft and finished prokaryotic genomes. OMICS 2003, 7: 161–169. 10.1089/153623103322246557
Rutherford K, Parkhill J, Crook J, Horsnell T, Rice P, Ranjandream MA, Barrell B: Artemis: sequence visualization and annotation. Bioinformatics 2000, 16: 944–945. 10.1093/bioinformatics/16.10.944
Delcher AL, Harmon D, Kasif S, White O, Salzberg S: Improved microbial gene identification with GLIMMER. Nucleic Acids Res 1999, 27: 4636–4641. 10.1093/nar/27.23.4636
Altschul SF, Gish W, Miller W, Myers E, Lipman D: Basic local alignment search tool. J Mol Biol 1990, 215: 403–410. 10.1016/S0022-2836(05)80360-2
Gish W, States D: Identification of protein coding regions by database similarity search. Nat Genet 1993, 3: 266–272. 10.1038/ng0393-266
NCBI Resource Coordinators: Database resources of the National Center for Biotechnology Information. Nucleic Acids Res 2013, 41: D8-D20.
Tatusov RL, Galperin M, Natale D, Koonin E: The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res 2000, 28: 33–36. 10.1093/nar/28.1.33
Eddy SR: Profile hidden Markov models. Bioinformatics 1998, 14: 755–763. 10.1093/bioinformatics/14.9.755
Punta M, Coggill PC, Eberhardt RY, Mistry J, Tate J, Boursnell C, Pang N, Forslund K, Ceric G, Clements J, Heger A, Holm L, Sonnhammer EL, Eddy SR, Bateman A, Finn RD: The Pfam protein families data-base. Nucleic Acids Res 2012, 40: D290-D301. 10.1093/nar/gkr1065
Haft DH, Selengut JD, Richter RA, Harkins D, Basu MK, Beck E: TIGRFAMs and genome properties in 2013. Nucleic Acids Res 2013, 41: D387-D395. 10.1093/nar/gks1234
Krogh A, Larsson B, von Heijne G, Sonnhammer E: Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol 2001, 305: 567–580. 10.1006/jmbi.2000.4315
Petersen TN, Brunak S, von Heijne G, Nielsen H: SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods 2011, 8: 785–786. 10.1038/nmeth.1701
Lowe TM, Eddy S: tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 1997, 25: 955–964. 10.1093/nar/25.5.0955
Burge SW, Daub J, Eberhardt R, Tate J, Barquist L, Nawrocki EP, Eddy SR, Gardner PP, Bateman A: Rfam 11.0: 10 years of RNA families. Nucleic Acids Res 2013, 41: D226-D232. 10.1093/nar/gks1005
Eddy SR: A memory-efficient dynamic programing algorithm for optimal alignment of a sequence to an RNA secondary structure. BMC Bioinformatics 2002, 3: 18. 10.1186/1471-2105-3-18
Benson DA, Cavanaugh M, Clark K, Karsch-Mizrachi I, Lipman DJ, Ostell J, Sayers EW: GenBank. Nucleic Acids Res 2013, 41: D36-D42. 10.1093/nar/gks1195
Helgadóttir S, Rosas-Sandoval G, Söll D, Graham DE: Biosynthesis of phosphoserine in the Methanococcales . J Bacteriol 2007, 189: 575–582. 10.1128/JB.01269-06
Kandiba L, Eichler J: Analysis of putative nonulosonic acid biosynthesis pathways in Archaea reveals a complex evolutionary history. FEMS Microbiol Lett 2013, 345: 110–120. 10.1111/1574-6968.12193
Lo C, Bonner CA, Xie G, D’Souza M, Jensen RA: Cohesion group approach for evolutionary analysis of aspartokinase, an enzyme that feeds a branched network of many biochemical pathways. Microbiol Mol Biol Rev 2009, 73: 594–651. 10.1128/MMBR.00024-09
Moe WM, Yan J, Nobre MF, da Costa MS, Rainey FA: Dehalogenimonas lykanthroporepellens gen. nov., sp. nov., a reductively dehalogenating bacterium isolated from chlorinated solvent-contaminated groundwater. Int J Syst Evol Microbiol 2009, 59: 2692–2697. 10.1099/ijs.0.011502-0
Taylor BL, Zhulin IB: PAS domains: internal sensors of oxygen, redox potential, and light. Microbiol Mol Biol Rev 1999, 63: 479–506.
Magingo FSS, Stumm CK: Nitrogen fixation by Methanobacterium formicicum . FEMS Microbiol Lett 1991, 81: 273–278. 10.1111/j.1574-6968.1991.tb04771.x
Lie TJ, Hendrickson EL, Niess UM, Moore BC, Haydock AK, Leigh JA: Overlapping repressor binding sites regulate expression of the Methanococcus maripaludis glnK 1 operon. Mol Microbiol 2010, 75: 755–762.
Gutiérrez G: Draft genome sequence of Methanobacterium formicicum DSM 3637, an archaebacterium isolated from the methane producer amoeba Pelomyxa palustris . J Bacteriol 2012, 194: 6967–6968. 10.1128/JB.01829-12
Rosewarne CP, Greenfield P, Li D, Tran-Dinh N, Midgley DJ, Hendry P: Draft genome sequence of Methanobacterium sp. Maddingley, reconstructed from metagenomic sequencing of a methanogenic microbial consortium enriched from coal-seam gas formation water. Genome Announc 2013, 1: e00082–12.
Cadillo-Quiroz H, Bräuer SL, Goodson N, Yavitt JB, Zinder SH: Methanobacterium paludis sp. nov. and a novel strain of Methanobacterium lacus isolated from northern peatlands. Int J Syst Evol Microbiol 2014, 64: 1473–1480. 10.1099/ijs.0.059964-0
Maus I, Wibberg D, Stantscheff R, Cibis K, Eikmeyer F-G, König H, Pühler A, Schlüter A: Complete genome sequence of the hydrogenotrophic archaeon Methanobacterium sp. Mb1 isolated from a production-scale biogas plant. J Biotechnol 2013, 168: 734–736. 10.1016/j.jbiotec.2013.10.013
Li L, Stoeckert CJ Jr, Roos DS: OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res 2003, 13: 2178–2189. 10.1101/gr.1224503
Acknowledgements
The BRM9 genome sequencing project was funded by the New Zealand Pastoral Greenhouse Gas Research Consortium.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
WJK, SCL, GTA, EA conceived and designed the experiments. SCL, DL, RP, SCLa performed the sequencing and assembly experiments. WJK, SCL, EA performed the annotation and comparative studies. WJK, SCL wrote the manuscript. All authors commented on the manuscript before submission. All authors read and approved the final manuscript.
Electronic supplementary material
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Kelly, W.J., Leahy, S.C., Li, D. et al. The complete genome sequence of the rumen methanogen Methanobacterium formicicum BRM9. Stand in Genomic Sci 9, 15 (2014). https://doi.org/10.1186/1944-3277-9-15
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1944-3277-9-15