
Abstract
Roseovarius mucosus Biebl et al. 2005 is a bacteriochlorophyll a-producing representative of the marine Roseobacter group within the alphaproteobacterial family Rhodobacteraceae, which was isolated from the dinoflagellate Alexandrium ostenfeldii. The marine Roseobacter group was found to be abundant in the ocean and plays an important role for global and biogeochemical processes. Here we describe the features of the R. mucosus strain DFL-24T together with its genome sequence and annotation generated from a culture of DSM 17069T. The 4,247,724 bp containing genome sequence encodes 4,194 protein-coding genes and 57 RNA genes. In addition to the presence of four plasmids, genome analysis revealed the presence of genes associated with host colonization, DMSP utilization, cytotoxins, and quorum sensing that could play a role in the interrelationship of R. mucosus with the dinoflagellate A. ostenfeldii and other marine organisms. Furthermore, the genome encodes genes associated with mixotrophic growth, where both reduced inorganic compounds for lithotrophic growth and a photoheterotrophic lifestyle using light as additional energy source could be used.
Similar content being viewed by others


Introduction
The Roseobacter group was shown to constitute a major component within the marine environments, encompassing around 20% of the bacterial community in coastal areas and 15% in mixed ocean-layers [1, 2]. Strain DFL-24T (= DSM 17069T = NCIMB 14077T = KACC 12996T) is the type strain of R. mucosus, originally isolated from a culture of the dinoflagellate Alexandrium ostenfeldii KO287, where it was found to be attached to the dinoflagellate surface [3]. R. mucosus DFL-24T is one of currently seventeen species within the genus Roseovarius with a validly published name [4, 5]. Whereas many Roseovarius species were named after their origin of isolation or their tolerances, the species epithet of strain DFL-24T was chosen to reflect its slimy colony appearance [3].
Current PubMed records do not indicate any follow-up research with strain DFL-24T after an overview about isolated aerobic anoxygenic phototrophs of the Roseobacter group from different marine habitats [6] and the initial description of R. mucosus[3]. Here, we analyzed the genome sequence of R. mucosus DSM 17069T. We present a description of the genome sequencing; its annotation and a summary classification together with a set of features for strain DFL-24T, including novel aspects of its phenotype.
Organism information
Classification and features
Figure 1 shows the phylogenetic neighborhood of R. mucosus in a 16S rRNA gene sequence based tree. The sequence of the single 16S rRNA gene copy in the genome is identical with the previously published 16S rRNA gene sequence (AJ534215).

Phylogenetic tree highlighting the position of R. mucosus relative to the type strains of the other species within the genus Roseovarius . The tree was inferred from 1,386 aligned characters [7, 8] of the 16S rRNA gene sequence under the maximum likelihood criterion [9]. Rooting was done initially using the midpoint method [10] and then checked for its agreement with the current classification (Table 1). The branches are scaled in terms of the expected number of substitutions per site. Numbers adjacent to the branches are support values from 1,000 ML bootstrap replicates [11] (left) and from 1,000 maximum-parsimony bootstrap replicates [12] (right) if larger than 60%. Lineages with type strain genome sequencing projects registered in GOLD [13] are labeled with one asterisk.
The 16S rRNA gene sequence was compared using NCBI BLAST [14] under default settings (e.g., considering only the high-scoring segment pairs (HSPs) from the best 250 hits) with the most recent release of the Greengenes database [15] and the relative frequencies of taxa and keywords (reduced to their stem [16]) were determined, weighted by BLAST scores. The highest-scoring environmental sequence was HM591393 (Greengenes short name ‘Change microbial structure microfiltration and sand filter pretreatment systems seawater reverse osmosis process clone MF-July-41’), which showed an identity of 99.8% and a HSP coverage of 50.2%. The most frequently occurring keywords within the labels of all environmental samples which yielded hits were ‘microbi’ (3.4%), ‘lake’ (3.2%), ‘tin’ (2.9%), ‘xiaochaidan’ (2.7%) and ‘sediment’ (2.4%) (378 hits in total). The most frequently occurring keyword within the labels of those environmental samples which yielded hits of a higher score than the highest scoring species was ‘chang, filter, microbi, microfiltr, osmosi, pretreat, process, revers, sand, seawat, structur, system’ (8.3%) (1 hit in total). These keywords partially fit to the marine origin of strain DFL-24T.
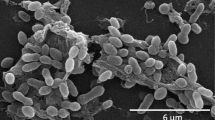
Cells of strain DFL-24T [3] are rod-shaped with characteristic pointed cell poles, 1.3-3.0 μm in length and 0.5-0.7 μm in width (Figure 2). Colonies on marine agar 2216 (BD Difco) are circular, convex and glistening, sometimes slimy, and white to slightly pinkish in color [3]. Growth occurs at a temperature range between 15 and 43°C, a pH range between 6.0 and 8.8 and a salinity between 1 and 7% sea salts [3]. Substrates for growth are acetate, butyrate, fumarate, glutamate, glycerol, lactate, pyruvate, malate and succinate; growth does not occur with citrate, glucose, fructose, ethanol and methanol [3]. Cells are positive for catalase and oxidase [3]. A summary of features can be found in Table 1.

Micrograph of strain R. mucosus DSM 17069 T .
Strain R. mucosus DFL-24T was described as showing no real motility [3]. However, in the course of this study cells were observed that show a characteristic movement conferred by flagella.
The utilization of carbon compounds by R. mucosus DSM 17069T grown at 28°C was also determined for this study using Generation-III microplates in an OmniLog phenotyping device (BIOLOG Inc., Hayward, CA, USA). The microplates were inoculated with a cell suspension at a cell density of 95-96% turbidity and dye IF-A. Further additives were vitamin, micronutrient and sea-salt solutions. The plates were sealed with parafilm to avoid a loss of fluid. The exported measurement data were further analyzed with the opm package for R [28], using its functionality for statistically estimating parameters from the respiration curves such as the maximum height, and automatically translating these values into negative, ambiguous, and positive reactions. The reactions were recorded in three individual biological replicates.
According to the Generation-III plates, the strain was positive for pH 6, 1% NaCl, 4% NaCl, 8% NaCl, D-galactose, D-fucose, L-fucose, L-rhamnose, 1% sodium lactate, myo-inositol, glycerol, D-aspartic acid, troleandomycin, rifamycin SV, glycyl-L-proline, L-alanine, L-arginine, L-aspartic acid, L-glutamic acid, L-histidine, L-pyroglutamic acid, L-serine, lincomycin, niaproof, quinic acid, vancomycin, methyl-pyruvate, L-lactic acid, citric acid, α-keto-glutaric acid, D-malic acid, L-malic acid, nalidixic acid, lithium chloride, potassium tellurite, γ-amino-n-butyric acid, β-hydroxy-butyric acid, acetoacetic acid, propionic acid, acetic acid, sodium formate, butyric acid and the positive control.
The strain was negative for the negative control (i.e. a well without substrate), dextrin, D-maltose, D-trehalose, D-cellobiose, β-gentiobiose, sucrose, D-turanose, stachyose, pH 5, D-raffinose, α-D-lactose, D-melibiose, β-methyl-D-galactoside, D-salicin, N-acetyl-D-glucosamine, N-acetyl-β-D-mannosamine, N-acetyl-D-galactosamine, N-acetyl-neuraminic acid, D-glucose, D-mannose, D-fructose, 3-O-methyl-D-glucose, inosine, fusidic acid, D-serine, D-sorbitol, D-mannitol, D-arabitol, D-glucose-6-phosphate, D-fructose-6-phosphate, D-serine, minocycline, gelatin, guanidine hydrochloride, pectin, D-galacturonic acid, L-galactonic acid-γ-lactone, D-gluconic acid, D-glucuronic acid, glucuronamide, mucic acid, D-saccharic acid, tetrazolium violet, tetrazolium blue, p-hydroxy-phenylacetic acid, D-lactic acid methyl ester, bromo-succinic acid, tween 40, α-hydroxy-butyric acid, α-keto-butyric acid, aztreonam and sodium bromate.
As far as present on Generation-III microplates, all growth tests conducted by Biebl et al. [3] were confirmed, and a number of additional sugars and amino acids were shown to be utilized by R. mucosus DSM 17069T. In accordance with the growth results, R. mucosus DSM 17069T prefers carboxylic acids as the main carbon sources in the respiratory measurements. In contrast to the results recorded in [3], the Phenotype Microarray reaction to citrate was positive. This may be due to respiratory measurements being more sensitive than growth measurements [29].
Chemotaxonomy
The principal cellular fatty-acid composition of strain DFL-24T was determined by Biebl et al. [3]. The major fatty acids (>10% of total) identified in strain DFL-24T are C18:1 ω7c and C16:0 and were found to be similar within the genus Roseovarius [25]. As predominant isoprenoid quinone ubiquinone-10 (Q-10) was found [3], which is typical for the genus Roseovarius [25] and the majority of the class Alphaproteobacteria.
Genome sequencing and annotation
Genome project history
The genome of R. mucosus DSM 17069T was sequenced within the project “Ecology, Physiology and Molecular Biology of the Roseobacter clade: Towards a Systems Biology Understanding of a Globally Important Clade of Marine Bacteria”. The strain was chosen for genome sequencing according the Genomic Encyclopedia of Bacteria and Archaea criteria [30, 31].
Project information is stored at the Genomes OnLine Database [13]. The Whole Genome Shotgun (WGS) sequence was produced using state of the art sequencing technology [32] and can be found at GenBank and the Integrated Microbial Genomes database [33]. A summary of the project information is shown in Table 2.
Growth conditions and DNA isolation
A culture of strain DSM 17069T was grown aerobically in DSMZ medium 514 [34] at 28°C. Genomic DNA was isolated using Jetflex Genomic DNA Purification Kit (GENOMED 600100) following the standard protocol provided by the manufacturer but modified by an incubation time of 60 min, incubation on ice overnight on a shaker, the use of additional 50 μl proteinase K, and the addition of 100 μl protein precipitation buffer. DNA is available from the DSMZ through the DNA Network [35].
Genome sequencing and assembly
The genome was sequenced using a combination of two libraries (Table 2). Illumina sequencing was performed on a GA IIx platform with 150 cycles. The paired-end library contained inserts of an average insert size of 350 bp. The first run on an Illumina GAII platform delivered 3.0 million reads. A second Illumina run was performed on a Miseq platform to gain a higher sequencing depth. To achieve longer reads, the library was sequenced in one direction for 300 cycles, providing another 2.0 million reads. Error correction and clipping were performed by fastq-mcf [36] and quake [37]. The data was assembled using velvet [38]. The first draft assembly from 2,822,784 filtered reads with an average read length of 165 bp resulted in more than 120 unordered contigs.
To improve the assembly, an additional 454 run was performed. The paired-end jumping library of 3 kb insert size was sequenced on a 1/8 lane. Pyrosequencing resulted in 92,601 reads with an average read length of 371 bp assembled in Newbler (Roche Diagnostics).
Both draft assemblies (Illumina and 454 sequences) were fractionated into artificial Sanger reads of 1000 bp in length plus 75 bp overlap on each site. These artificial reads served as an input for the phred/phrap/consed package [39]. By manual editing the number of contigs could be reduced to 26, localized in 17 scaffolds. The combined sequences provided a 191 × coverage of the genome.
Genome annotation
Genes were identified using Prodigal [40] as part of the JGI genome annotation pipeline. The predicted CDSs were translated and used to search the National Center for Biotechnology Information\nonredundant database, UniProt, TIGR-Fam, Pfam, PRIAM, KEGG, COG, and InterPro databases. Identification of RNA genes were carried out by using HMMER 3.0rc1 [41] (rRNAs) and tRNAscan-SE 1.23 [42] (tRNAs). Other non-coding genes were predicted using INFERNAL 1.0.2 [43]. Additional gene prediction analysis and functional annotation was performed within the Integrated Microbial Genomes - Expert Review platform [44]. CRISPR elements were detected using CRT [45] and PILER-CR [46]. The annotation on the IMG-ER platform was used for the genome analysis and its genome description; the annotation of the NCBI deposit of the genome as well as later version on IMG might slightly differ from the figures given below.
Genome properties
The genome statistics are provided in Table 3 and Figure 3. The genome of strain DSM 17069T has a total length of 4,247,724 bp and a G + C content of 61.9%. Of the 4,251 genes predicted, 4,194 were identified as protein-coding genes, and 57 as RNAs. The majority of the protein-coding genes were assigned a putative function (81.6%) while the remaining ones were annotated as hypothetical proteins. The distribution of genes into COGs functional categories is presented in Table 4.

Graphical map of the largest scaffold. From bottom to the top: Genes on forward strand (colored by COG categories), Genes on reverse strand (colored by COG categories), RNA genes (tRNAs green, rRNAs red, other RNAs black), GC content (black), GC skew (purple/olive).
Insights from the genome sequence
Genome comparisons and extrachromosomal elements
The two other Roseovarius genomes, Roseovarius nubinhibens ISM (IMG taxon ID 638341183 = AALY00000000; total number of genes: 3605) and Roseovarius sp. TM1035 (IMG taxon ID 640963035 = ABCL00000000; total number of genes: 4158), both reveal a genome length shorter than that of strain DSM 17069T, but also remain still in draft state.
The fraction of shared genes between strain R. mucosus DSM 17069T and the strains R. nubinhibens ISM and Roseovarius sp. TM1035 is shown in a Venn diagram (Figure 4). The numbers of pairwise shared homologous genes were inferred from an analysis with a reimplementation of the TribeMCL algorithm [48], applying an E-value threshold of 10-5 and an MCL inflation parameter of 2.0.

Venn diagram depicting the intersections of sets of homologous proteins of R. mucosus DSM 17069 T , R. nubinhibens ISM and Roseovarius sp. TM1035. Their cardinalities are given in parentheses; the total number of proteins and the resources are listed in the text. The Venn diagram was calculated with the corresponding R package [47].
Many members of the Roseobacter group contain plasmids [49]. Genome sequencing of strain R. mucosus DSM 17069T revealed the presence of four plasmids with sizes between 21 kb and 123 kb (Table 5). The two largest plasmids pRosmuc_A123 and pRosmuc_B85 contain characteristic RepABC replication operons including the repAB partitioning genes. The respective replicases that mediate the initiation of replication are designated according to the established plasmid classification scheme [50]. The different numbering of the replicases RepC-7 and RepC-1 corresponds to specific plasmid compatibility groups that are required for a stable coexistence of the replicons within the same cell. The smallest plasmid pRosmuc_D21 contains a RepAIV type replicase [51], but a typical parAB partitioning operon is lacking.
This distribution may correspond with a higher plasmid copy number within the cell thus assuring the replicon maintenance in the daughter cells after cell division. The plasmid pRosmuc_C27 harbors a ParF-type ATPase (rosmuc_03930) downstream of the replicase RepA, which may ensure successful partitioning. The largest plasmid pRosmuc_A123 contains apart from the RepC-7 replication operon an additional solitary replicase RepA-III and may hence represent a composite replicon resulting from plasmid fusion [52].
The locus tags of all replicases, plasmid stability modules and the large virB4 and virD4 genes of type IV secretion systems are presented in Table 6. The 85 kb plasmid pRosmuc_B85 contains two postsegregational killing systems (PSK) consisting of a typical operon with two small genes encoding a stable toxin and an unstable antitoxin [53]. The RepABC-7/RepA-III-type plasmid pRosmuc_A123 contains a complete type-IV secretion system including the virB operon for the formation of a transmembrane channel. The relaxase VirD2, which is required for the strand-specific DNA nicking at the origin of transfer (oriT), and the coupling protein VirD4 support the presence of functional conjugation systems [49, 54]. Moreover, this plasmid contains an additional MobA-type relaxase (rosmuc_04055) located in close proximity to a second coupling protein VirD4 (rosmuc_04058) and the replicase RepA-III (rosmuc_04060) thus supporting the idea that the replicon represents a composite plasmid.
Commensalism and pathogenic potential
Commensal bacteria colonize a host and benefit from it without having a negative effect on its growth. Several representatives of the Roseobacter group were found to live in a close and stable ecological relationship with phototrophic dinoflagellates [2]. This symbiosis is not obligate, so that both organisms can also proliferate independently from each other. The exact type of the association between dinoflagellates and Roseobacter species is, however, rarely determined and may vary depending on the involved species. In the case of Dinoroseobacter shibae a mutualistic symbiosis was found in which the dinoflagellate assimilates essential B vitamins that are produced by the bacterium [55]. In the following paragraphs some features encoded in the genome of strain DSM 17069T are mentioned that could play a role in the interrelationship of R. mucosus with the dinoflagellate A. ostenfeldii and other marine organisms.
The first step in establishing a stable association of free-living R. mucosus cells with a potential dinoflagellate host is the attachment to its surface. Factors that could control attachment are the production of flagella and pili. Swimming cells were rarely observed in laboratory cultures of strain DSM 17069T (this study), but may play an important role in the marine ecosystem for targeting dinoflagellates for subsequent colonizing. Genes for the synthesis of flagellae, a flagellar motor and a chemotactic response were located dispersed within the genome sequence, but the two largest gene clusters are found at rosmuc_00543-00574 (mainly genes for the flagellar basal body) and rosmuc_02499-02544 (flagellar motor and chemotaxis). After reaching a suitable dinoflagellate, pili or fimbriae could mediate attachment to the surface of the eukaryotic host. Genes encoding the synthesis of Flp pili belonging to the type IV family were located at rosmuc_01726-01737. It is assumed that Flp pili play a role in the attachment of bacteria to eukaryotic cells, and it was reported that in Ralstonia solanacearum Flp pili are required for the virulence on potato [56]. Upon successful attachment bacteria colonizing surfaces often produce extracellular polymeric substances to establish biofilms that stabilize the association and provide a suitable microenvironment for growth. Formation of slime capsules by cells of R. mucosus is suggested by a slimy appearance of colonies on agar plates [3]. In addition, the genome of DSM 17069T encodes two transporters for the export of capsular polysaccharides, one is of the ABC type (rosmuc_00889-00891) and the other belongs to the CPS-E family (rosmuc_01918-01920).
Dimethylsulfoniopropionate (DMSP) is a common metabolite of marine phytoplankton, especially phototrophic dinoflagellates. It is a compatible solute that can reach high intracellular concentrations and probably has various cellular functions, but osmoregulation seems to be the most important one [57]. Intracellular DMSP can be released into the environment by leakage, senescence or lysis and represents therefore a major source of reduced carbon and sulfur for heterotrophic marine bacteria. Due to the rapid dilution of DMSP in the marine surface water, bacteria colonizing DMSP producers have an advantage over free-living bacteria. DMSP can be utilized as substrate either by cleavage or demethylation pathways, and R. mucosus seems to encode key genes of both routes. In the genome of DSM 17069T genes for a dimethylsulfoniopropionate demethylase (DmdA) were located at two different sites (rosmuc_01718 and rosmuc_02463), which were both reliably identified by sharing protein-sequence identity values above 85% with translated dmdA genes of other Roseovarius strains. The DmdA enzyme catalyzes the transfer of a methyl group to tetrahydrofolate (THF) resulting in 5-methyl-THF and methylmercaptopropionate (MMPA) [58]. The DmdA gene at rosmuc_01718 is probably part of an operon that encodes also the genes for acyl-CoA synthetase (DmdB, rosmuc_01719), methylmercaptopropionyl-CoA dehydrogenase (DmdC, rosmuc_01721) and methylthioacryloyl-CoA hydratase (DmdD, rosmuc_01720), which catalyze the further degradation of MMPA to acetaldehyde and methanethiol. Acetaldehyde and 5-methyl-THF can be funneled into the central carbon metabolism, whereas the reduced sulfur of methanethiol is probably assimilated as methionine by the enzyme cystathionine gamma-synthetase (rosmuc_01420). The alternative DMSP cleavage pathway starts in R. mucosus with the dimethylsulfoniopropionate lyase DddP (rosmuc_03355) that was originally identified in R. nubinhibens and produces dimethylsulfide (DMS) and acrylate from DMSP. No genes encoding enzymes acting on DMS were identified in the genome of DSM 17069T, so that this volatile sulfur compound cannot be further utilized by R. mucosus and is released into the environment. Consequently, the DMSP cleavage pathway is probably mainly used if enough energy for the sulfur assimilation by the reduction of sulfate via the APS/PAPS pathway is available.
The genome of R. mucosus encodes a large number of potentially cytotoxic compounds, including more than 20 genes encoding RTX-like toxins. The cytolytic activity of RTX toxins is based on forming cation-selective pores in membranes of target cells which eventually leads to cell death and lysis [59]. Typical RTX-like toxins are post-translationally activated by fatty acylation and exported into the extracellular space by a unique type I secretion system that mediates translocation across the cytoplasmic and outer membrane in a single step. Albeit some of the RTX protein genes in R. mucosus DSM 17069T were annotated as putative hemolysins (e.g., rosmuc_00250, rosmuc_00896, rosmuc_01947, and rosmuc_02953), they were not located in typical RTX operons encoding also genes for a type I secretion system and a specific acyltransferase. Apparently, these genes are dispersed within the genome and likely expressed constitutively. For instance, genes for a complete type-I secretion system comprising an outer membrane protein, ABC transporter and the membrane fusion protein are encoded at rosmuc_03175-03177. Although, RTX-like toxins are secreted from some representatives of the Roseobacter group in large amounts [60], their ecological function in avirulent strains is largely unknown. Potential functions could include the inhibition of competing bacterial species colonizing the dinoflagellate surface, a contribution to the killing or incapacitation of zooplankton species grazing on the dinoflagellate host or an infection of shellfish after filter feeding on dinoflagellates. In this regard it is noteworthy that the related species Roseovarius crassostreae was identified as a putative pathogen of juvenile oysters [61]. Alternative functions of RTX-like proteins could, however, also include various exo-enzymatic activities that may be required for the release of dissolved organic carbon from macromolecules [59].
Many representatives of the Roseobacter group encode and express gene sequences associated with quorum sensing (QS), a process of cell-to-cell communication and interaction between relatives in dependence of a certain detected population threshold value [62]. They produce small signal molecules called autoinducers, which are detected by their conspecific bacteria when excreted in the environment. Consequently, when the concentration of those membrane-diffusible autoinducers reaches a specific threshold value, the population responds with an activation of gene expression to coordinate a population-wide behavior. Especially, for bacteria colonizing or infecting hosts it is important to determine if the cell density of their population is high enough to make colonization or infection successful. Genome analysis of R. mucosus DSM 17069T revealed the presence of genes putatively associated with QS activity, like e.g. a N-acyl-L-homoserine lactone synthetase (luxI homolog; rosmuc_03495) and several regulator proteins belonging to the luxR family (e.g., rosmuc_01010, rosmuc_02138, rosmuc_02881, rosmuc_03496, and rosmuc_03642).
Mixotrophic growth
In the absence of a suitable dinoflagellate host R. mucosus has to survive as free-living cells in seawater. A well-known characteristic of most marine environments is a depletion of organic nutrients, so that the utilization of alternative energy sources is an important part of bacterial survival strategies in marine surface waters. In evolution marine heterotrophic bacteria have developed essentially two different approaches to overcome the lack of organic carbon sources as electron donors. One is the use of reduced inorganic compounds for lithoheterotrophic growth and the other harvesting of light as additional energy source for photoheterotrophy. According to the genome sequence both mechanisms for generation of energy could be operative in R. mucosus DSM 17069T.
A large cluster of genes involved in the production of a [NiFe] uptake-type hydrogenase (hydrogen:quinone oxidoreductase) was detected at rosmuc_03325-03344. A major source of hydrogen in surface seawater may be nitrogen-fixing cyanobacteria, where it is produced as by-product to the formation of ammonia from the nitrogenase enzyme [63].
Another potential electron donor in ocean waters is carbon monoxide (CO) that is formed by photolysis of dissolved organic matter [64]. It has been reported that only representatives of the Roseobacter group that encode both the definitive form I and a putative form II of the carbon monoxide dehydrogenase are able to oxidize CO to CO2 in laboratory experiments [65]. R. mucosus DSM 17069T encodes indeed a carbon monoxide dehydrogenase of form I (coxLSM; rosmuc_00985-00987) and form II (coxSLM; rosmuc_01269-01271), so that this strain most likely can use CO as supplementary electron donor.
Besides the dissolved gasses H2 and CO reduced inorganic sulfur compounds represent main sources for mixotrophic growth in marine environments. Thiosulfate is one of the most common forms of reduced inorganic sulfur in seawater, because it is quite stable under oxic conditions and thus can accumulate to recognizable amounts in open ocean waters. It can be produced abiotically during oxidation of sulfide at oxic-anoxic transition zones or as metabolic by-product during the degradation of organic sulfur compounds. Especially, the latter source seems to be important at niches which are far away from sites of sulfide production but close to phytoplankton producing organic sulfur compounds. Examples of thiosulfate producing marine bacteria are Chromatocurvus halotolerans growing on glutathione [66] and Methylophaga sulfidovorans growing on dimethylsulfide [67]. However, in most aerobic marine bacteria the sulfur moiety of organic compounds seems to be oxidized to sulfate as end product, although it is assumed that thiosulfate may represent an intermediate metabolite in sulfur oxidation in some of these bacteria [66]. In most representatives of the Roseobacter group oxidation of thiosulfate to sulfate is catalyzed by the periplasmic Sox multienzyme complex, which allows the generation of additional energy for mixotrophic growth in Ruegeria (formerly Sillicibacter) pomeroyi [68]. In the genome of R. mucosus DSM 17069T a large set of sox genes (soxRSVWXYZABCDEF) encoding all proteins of a functional Sox multienzyme complex was found at rosmuc_00222-00234. The generation of metabolically useful energy from the oxidation of thiosulfate was however not tested in this strain so far.
Light can be used as additional energy source by aerobic marine bacteria either by using rhodopsins or a photosynthetic apparatus. Genomic analyses of bacteria inhabiting the photic zone of marine environments revealed that harvesting of light using proton-pumping proteorhodopsins is widely distributed and frequently found in members of the Flavobacteria,Alphaproteobacteria and Gammaproteobacteria [69]. In contrast, bacteria expressing a photosynthetic apparatus enabling aerobic photoheterotrophic growth are mainly restricted to representatives of the Roseobacter group [2] and the gammaproteobacterial OM60/NOR5 clade [70]. A large coherent photosynthetic gene cluster comprising genes for bacteriochlorophyll and carotenoid synthesis, a photosynthetic reaction center and a light-harvesting antenna complex was detected in the genome of R. mucosus DSM 17069T at rosmuc_03187-03225 (Figure 5). In addition, two proteins containing a BLUF domain enabling sensing of blue light using FAD were located outside of the photosynthesis gene cluster at rosmuc_00390 and rosmuc_04258. The description of R. mucosus reports low levels of cellular bacteriochlorophyll a [3], which indicates that the photosynthesis genes are expressed and functional in this strain. The regulation of photosynthetic gene expression was not further analyzed, so that it is unknown if higher amounts of bacteriochlorophyll a may be produced in cells of R. mucosus under distinct environmental conditions.

Conclusion
Members of the marine Roseobacter group are widely distributed in the marine environment. In this study we analysed the genome sequence of R. mucosus DSM 17069T, which was isolated from the dinoflagellate Alexandrium ostenfeldii. Genome analysis of this type strain revealed the presence of key functional characteristics. We summarized some of them, such as genes associated with host colonization, DSMP utilization, cytotoxins and quorum sensing that could play a role in a possible interrelationship of R. mucosus with the dinoflagellate A. ostenfeldii and other marine organisms. In addition, genome analysis of R. mucosus DSM 17069T revealed a possible lithotrophic as well as photoheterotrophic lifestyle.
Abbreviations
- HSP:
-
High-scoring Segment Pair
- ML:
-
Maximum Likelihood
- WGS:
-
Whole Genome Shotgun
- MCL:
-
Markov Cluster
- DMSP:
-
Dimethylsulfoniopropionate
- DmdA:
-
Dimethylsulfoniopropionate demethylase
- DmdB:
-
Acyl-CoA synthetase
- DmdC:
-
Methylmercaptopropionyl-CoA dehydrogenase
- DmdD:
-
Methylthioacryloyl-CoA hydratase
- DddP:
-
Dimethylsulfoniopropionate lyase
- THF:
-
Tetrahydrofolate
- MMPA:
-
Methylmercaptopropionate
- DMS:
-
Dimethylsulfide
- QS:
-
Quorum Sensing
- BLUF:
-
Sensors of Blue Light using FAD
- FAD:
-
Flavin Adenine Dinucleotide.
References
Buchan A, González JM, Moran MA: Overview of the Roseobacter lineage. Appl Environ Microbiol 2005, 71:5665–5677. 10.1128/AEM.71.10.5665-5677.2005
Wagner-Döbler I, Biebl H: Environmental biology of the marine Roseobacter lineage. Annu Rev Microbiol 2006, 60:255–280. 10.1146/annurev.micro.60.080805.142115
Biebl H, Allgaier M, Lünsdorf H, Pukall R, Tindall BJ, Wagner-Döbler I: Roseovarius mucosus sp. nov., a member of the Roseobacter clade with trace amounts of bacteriochlorophyll a . Int J Syst Evol Microbiol 2005, 55:2377–2383. 10.1099/ijs.0.63832-0
Euzéby JP: List of prokaryotic names with standing in nomenclature. [http://www.bacterio.net/roseovarius.html]
Parte AC: LPSN-list of prokaryotic names with standing in nomenclature. Nucleic Acids Res 2014, 42:D613-D616. 10.1093/nar/gkt1111
Allgaier M, Uphoff H, Felske A, Wagner-Döbler I: Aerobic anoxygenic photosynthesis in Roseobacter clade bacteria from diverse marine habitats. Appl Environ Microbiol 2003, 69:5051–5059. 10.1128/AEM.69.9.5051-5059.2003
Lee C, Grasso C, Sharlow MF: Multiple sequence alignment using partial order graphs. Bioinformatics 2002, 18:452–464. 10.1093/bioinformatics/18.3.452
Castresana J: Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol 2000, 17:540–552. 10.1093/oxfordjournals.molbev.a026334
Stamatakis A, Hoover P, Rougemont J: A rapid bootstrap algorithm for the RAxML web-servers. Syst Biol 2008, 57:758–771. 10.1080/10635150802429642
Hess PN, De Moraes Russo CA: An empirical test of the midpoint rooting method. Biol J Linn Soc 2007, 92:669–674. 10.1111/j.1095-8312.2007.00864.x
Pattengale ND, Alipour M, Beninda-Emonds ORP, Moret BME, Stamatakis A: How many bootstrap replicates are necessary? Lecture notes in Computer Science 2009, 5541:184–200. 10.1007/978-3-642-02008-7_13
Swofford DL: PAUP*: Phylogenetic Analysis Using Parsimony (*and Other Methods), Version 4.0 b10. Sunderland: Sinauer Associates; 2002.
Pagani I, Liolios K, Jansson J, Chen IM, Smirnova T, Nosrat B, et al.: The Genomes OnLine Database (GOLD) v. 4: status of genomic and metagenomic projects and their associated metadata. Nucleic Acids Res 2012, 40:D571-D579. 10.1093/nar/gkr1100
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ: Basic local alignment search tool. J Mol Biol 1990, 215:403–410. 10.1016/S0022-2836(05)80360-2
DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, et al.: Greengenes, a Chimera-Checked 16S rRNA gene Database and Workbench Compatible with ARB. Appl Env Microbiol 2006, 72:5069–5072. 10.1128/AEM.03006-05
Porter MF: An algoritm for suffix stripping. Program: electronic library and information systems 1980, 14:130–137. 10.1108/eb046814
Field D, Garrity G, Gray T, Morrison N, Selengut J, Sterk P, et al.: The minimum information about a genome sequence (MIGS) specification. Nat Biotechnol 2008, 26:541–547. 10.1038/nbt1360
Field D, Amaral-Zettler L, Cochrane G, Cole JR, Dawyndt P, Garrity GM, et al.: Clarifying Concepts and Terms in Biodiversity Informatics. PLoS Biol 2013, 9:e1001088.
Woese CR, Kandler O, Weelis ML: Towards a natural system of organisms. Proposal for the domains Archaea and Bacteria . Proc Natl Acad Sci U S A 1990, 87:4576–4579. 10.1073/pnas.87.12.4576
Garrity GM, Bell JA, Lilburn T: Phylum XIV. Proteobacteria phyl nov. In Bergey’s Manual of Sytematic Bacteriology. Volume 2 (The Proteobacteria), part B (The Gammaproteobacteria), second edition. Edited by: Brenner DJ, Krieg NR, Stanley JT, Garrity GM. New York: Springer; 2005:1.
Garrity GM, Bell JA, Lilburn T: Class I. Alphaproteobacteria class. nov. In Bergey’s Manual of Sytematic Bacteriology. Volume 2 (The Proteobacteria), part C (The Alpha-, Beta-, Delta-, and Epsilonproteobacteria), second edition. Edited by: Brenner DJ, Krieg NR, Stanley JT, Garrity GM. New York: Springer; 2005:1.
Validation List No. 107. List of new names and new combinations previously effectively, but not validly, published Int J Syst Evol Microbiol 2006, 56:1–6.
Garrity GM, Bell JA, Lilburn T: Order III. Rhodobacterales ord. nov. In Bergey’s Manual of Sytematic Bacteriology. Volume 2 (The Proteobacteria), part C (The Alpha-, Beta-, Delta-, and Epsilonproteobacteria), second edition. Edited by: Brenner DJ, Krieg NR, Stanley JT, Garrity GM. New York: Springer; 2005:161.
Garrity GM, Bell JA, Lilburn T: Family I. Rhodobacteraceae fam. nov. In Bergey’s Manual of Sytematic Bacteriology. Volume 2 (The Proteobacteria), part C (The Alpha-, Beta-, Delta-, and Epsilonproteobacteria), second edition. Edited by: Brenner DJ, Krieg NR, Stanley JT, Garrity GM. New York: Springer; 2005:161.
Labrenz M, Collins MD, Lawson PA, Tindall BJ, Schumann P, Hirsch P: Roseovarius tolerans gen. nov., sp. nov., a budding bacterium with variable bacteriochlorophyll a production from hypersaline Ekho Lake. Int J Syst Bacteriol 1999, 49:137–147. 10.1099/00207713-49-1-137
BAuA: TRBA 466 “Classification of prokaryotes (bacteria and archaea) into risk groups”. [http://www.baua.de/en/Topics-from-A-to-Z/Biological-Agents/TRBA/TRBA-466.html]
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al.: Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 2000, 25:25–29.
Vaas LAI, Sikorski J, Michael V, Göker M, Klenk HP: Visualization and curve-parameter estimation strategies for efficient exploration of phenotype microarray kinetics. PLoS ONE 2012, 7:e34846. 10.1371/journal.pone.0034846
Vaas LAI, Marheine M, Sikorski J, Göker M, Schumacher M: Impacts of pr-10a overexpression at the molecular and the phenotypic level. International Journal of Molecular Sciences 2013, 14:15141–15166. 10.3390/ijms140715141
Göker M, Klenk HP: Phylogeny-driven target selection for large-scale genome-sequencing (and other) projects. Stand Gen Sci 2013, 8:360–374. 10.4056/sigs.3446951
Wu D, Hugenholtz P, Mavromatis K, Pukall R, Dalin E, Ivanova NN, et al.: A phylogeny-driven Genomic Encyclopaedia of Bacteria and Archaea . Nature 2009, 462:1056–1060. 10.1038/nature08656
Mavromatis K, Land M, Brettin TS, Quest DJ, Copeland A, Clum A, et al.: The fast changing landscape of sequencing technologies and their impact on microbial assemblies and annotation. PLoS ONE 2012, 7:e48837. 10.1371/journal.pone.0048837
Markowitz VM, Chen IMA, Palaniappan K, Chu K, Szeto E, Grechkin Y, et al.: IMG: the integrated microbial genomes database and comparitive analysis system. Nucleic Acids Res 2012, 40:D115-D122. 10.1093/nar/gkr1044
List of growth media used at the DSMZ [http://www.dmsz.de/catalogues/cataloque-microorganisms/culture-technology/list-of-media-for-microorganisms.html]
Gemeinholzer B, Dröge G, Zetzsche H, Haszprunar G, Klenk HP, Güntsch A, et al.: The DNA Bank Network: the start from a German initiative. Biopreserv Biobank 2011, 9:51–55. 10.1089/bio.2010.0029
Aronesty E: ea-utils: Command-line tools for processing biological sequencing data. [http://code.google.com/p/ea-utils]
Kelley DR, Schatz MC, Salzberg SL: Quake: quality-aware detection and correction of sequencing errors. Genome Biol 2010, 11:R116. 10.1186/gb-2010-11-11-r116
Zerbino DR, Birney E: Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 2008, 18:821–829. 10.1101/gr.074492.107
Gordon D, Abajian C, Green P: Consed: a graphical tool for sequence finishing. Genome Res 1998, 8:195–202. 10.1101/gr.8.3.195
Hyatt D, Chen GL, LoCascio PF, Land ML, Larimer FW, Hauser LJ: Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 2010, 11:119. 10.1186/1471-2105-11-119
Finn DR, Clements J, Eddy SR: HMMER web server: interactive sequence similarity searching. Nucleic Acids Res 2011, 39:W37-W29. Web Server Issue 10.1093/nar/gkq1294
Lowe TM, Eddy SR: tRNAscan-SE: A Program for Improved Detection of Transfer RNA Genes in Genomic Sequence. Nucleic Acids Res 1997, 25:955–964. 10.1093/nar/25.5.0955
Nawrocki EP, Kolbe DL, Eddy SR: Infernal 1.0: Inference of RNA alignments. Bioinformatics 2009, 25:1335–1337. 10.1093/bioinformatics/btp157
Markowitz VM, Ivanova NN, Chen IMA, Chu K, Kyrpides NC: IMG ER: a system for microbial genome annotation expert review and curation. Bioinformatics 2009, 25:2271–2278. 10.1093/bioinformatics/btp393
Bland C, Ramsey TL, Sabree F, Lowe M, Brown K, Kyrpides NC, et al.: CRISPR recognition tool (CRT): a tool for automatic detection of clustered regularly interspaced palindromic repeats. BMC Bioinformatics 2007, 8:209. 10.1186/1471-2105-8-209
Edgar RC, Myers EW: PILER: identification and classification of genomic repeats. Bioinformatics 2005, 21:i152-i158. 10.1093/bioinformatics/bti1003
Chen H, Boutros PC: VennDiagram: a package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinformatics 2011, 12:35. 10.1186/1471-2105-12-35
Enright AJ, van Dongen SM, Ouzounis CA: An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res 2002, 30:1575–1584. 10.1093/nar/30.7.1575
Petersen J, Frank O, Göker M, Pradella S: Extrachromosomal, extraordinary and essential–the plasmids of the Roseobacter clade. Appl Microbiol Biotechnol 2013, 97:2805–2815. 10.1007/s00253-013-4746-8
Petersen J, Brinkmann H, Pradella S: Diversity and evolution of repABC type plasmids in Rhodobacterales . Environ Microbiol 2009, 11:2627–2638. 10.1111/j.1462-2920.2009.01987.x
Petersen J, Brinkmann H, Berger M, Brinkhoff T, Päuker O, Pradella S: Origin and evolution of a novel DnaA-like plasmid replication type in Rhodobacterales . Mol Biol Evol 2011, 28:1229–1240. 10.1093/molbev/msq310
Zhong Z, Caspi R, Helinski D, Knauf V, Sykes S, O’Byrne C, et al.: Nucleotide sequence based characterizations of two cryptic plasmids from the marine bacterium Ruegeria isolate PR1b. Plasmid 2003, 49:233–252. 10.1016/S0147-619X(03)00014-3
Zielenkiewicz U, Ceglowski P: Mechanisms of plasmid stable maintenance with special focus on plasmid addiction systems. Acta Biochim Pol 2001, 48:1003–1023.
Cascales E, Christie PJ: The versatile bacterial type IV secretion systems. Nat Rev Microbiol 2003, 1:137–149. 10.1038/nrmicro753
Wagner-Döbler I, Ballhausen B, Berger M, Brinkhoff T, Buchholz I, Bunk B, et al.: The complete genome sequence of the algal symbiont Dinoroseobacter shibae : a hitchhiker’s guide to life in the sea. ISME J 2010, 4:61–77. 10.1038/ismej.2009.94
Wairuri CK, van der Waals JE, van Schalkwyk A, Theron J: Ralstonia solanacearum needs Flp pili for virulence on potato. Mol Plant Microbe Interact 2012, 25:546–556. 10.1094/MPMI-06-11-0166
Yoch DC: Dimethylsulfoniopropionate: its sources, role in the marine food web, and biological degradation to dimethylsulfide. Appl Environ Microbiol 2002, 68:5804–5815. 10.1128/AEM.68.12.5804-5815.2002
Reisch CR, Moran MA, Whitman WB: Bacterial catabolism of dimethylsulfoniopropionate (DMSP). Front Microbiol 2011, 2:172.
Linhartova I, Bumba L, Masin J, Basler M, Osicka R, et al.: RTX proteins: a highly diverse family secreted by a common mechanism. FEMS Microbiol Rev 2010, 34:1076–1112.
Durighello E, Christie-Oleza JA, Armengaud J: Assessing the exoproteome of marine bacteria, lesson from a RTX-toxin abundantly secreted by Phaeobacter strain DSM 17395. PLoS ONE 2014, 9:e89691. 10.1371/journal.pone.0089691
Boettcher KJ, Geaghan KK, Maloy AP, Barber BJ: Roseovarius crassostreae sp. nov., a member of the Roseobacter clade and the apparent cause of juvenile oyster disease (JOD) in cultured Eastern oysters. Int J Syst Evol Microbiol 2005, 55:1531–1537. 10.1099/ijs.0.63620-0
Fuqua C, Winans S, Greenberg E: Census and consensus in bacterial ecosystems: the LuxR-LuxI family of quorumsensing transcriptional regulators. Annu Rev Microbiol 1996, 50:727–751. 10.1146/annurev.micro.50.1.727
Scranton MI, Novelli PC, Michaels A, Horrigan SG, Carpenter EJ: Hydrogen production and nitrogen fixation by Oscillatoria thiebautii during in situ incubations. Limnol Oceangr 1987, 32:998–1006. 10.4319/lo.1987.32.4.0998
Zuo Y, Jones RD: Formation of carbon monoxide by photolysis of dissolved marine organic material and its significance in the carbon cycling of the oceans. Naturwiss 1995, 82:472–474. 10.1007/BF01131598
Cunliffe M: Correlating carbon monoxide oxidation with cox genes in the abundant Marine Roseobacter clade. ISME J 2011, 5:685–691. 10.1038/ismej.2010.170
Spring S: Function and evolution of the sox multienzyme complex in the marine gammaproteobacterium Congregibacter litoralis . ISRN Microbiol 2014., 2014: Article ID 597418
De Zwart JMM, Sluis JMR, Kuenen JG: Competition for dimethylsulfide and hydrogen sulfide by Methylophaga sulfidovorans and Thiobacillus thioparus T5 in continuous cultures. Appl Environ Microbiol 1997, 63:3318–3322.
Moran MA, Buchan A, González JM, Heidelberg JF, Whitman WB, Kiene RP, et al.: Genome sequence of Silicibacter pomeroyi reveals adaptations to the marine environment. Nature 2004, 432:910–913. 10.1038/nature03170
Fuhrman JA, Schwalbach MS, Stingl U: Proteorhodopsins: An array of physiological roles? Nat Rev Microbiol 2008, 6:488–494.
Spring S, Riedel T, Spröer C, Yan S, Harder J, Fuchs BM: Taxonomy and evolution of bacteriochlorophyll a -containing members of the OM60/NOR5 clade of marine gammaproteobacteria: Description of Luminiphilus syltensis gen. nov., sp. nov., reclassification of Haliea rubra as Pseudohaliea rubra gen. nov., comb. nov., and emendation of Chromatocurvus halotolerans . BMC Microbiol 2013, 13:118. 10.1186/1471-2180-13-118
Spring S, Lünsdorf H, Fuchs BM, Tindall BJ: The photosynthetic apparatus and its regulation in the aerobic gammaproteobacterium Congregibacter litoralis gen. nov., sp. nov. PLoS One 2009, 4:e4866. 10.1371/journal.pone.0004866
Riedel T, Fiebig A, Göker M, Klenk HP: Complete genome sequence of the bacteriochlorophyll a -containing Roseibacterium elongatum type strain (DSM 19469 T ), a representative of the Roseobacter group from Australian coast sand. Stand Genomic Sci 2014, 9:840–854. 10.4056/sigs.5541028
Acknowledgements
The authors gratefully acknowledge the help of Iljana Schröder for growing cells of R. mucosus, Evelyne Brambilla for DNA extraction and quality control, and Jan Meier-Kolthoff for useful hints regarding Venn diagrams (all at DSMZ). The work was performed under the auspices of the German Research Foundation (DFG) Transregio-SFB 51 Roseobacter grant.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
MG and HPK devised the study. TR, SS, AF, CS, JP, MG, and HPK contributed materials and analyses. TR wrote the paper. All authors read and approved the final manuscript.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Riedel, T., Spring, S., Fiebig, A. et al. Genome sequence of the Roseovarius mucosus type strain (DSM 17069T), a bacteriochlorophyll a-containing representative of the marine Roseobacter group isolated from the dinoflagellate Alexandrium ostenfeldii . Stand in Genomic Sci 10, 17 (2015). https://doi.org/10.1186/1944-3277-10-17
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1944-3277-10-17