
Abstract
Despite advances in characterizing genetic influences on addiction liability and treatment response, clinical applications of these efforts have been slow to evolve. Although challenges to clinical translation remain, stakeholders already face decisions about evidentiary thresholds for the uptake of pharmacogenetic tests in practice. There is optimism about potential pharmacogenetic applications for the treatment of alcohol use disorders, with particular interest in the OPRM1 A118G polymorphism as a moderator of naltrexone response. Findings from human and animal studies suggest preliminary evidence for the clinical validity of this association; on this basis, arguments for clinical implementation can be made in accordance with existing frameworks for the uptake of genomic applications. However, generating evidence-based guidelines requires evaluating the clinical utility of pharmacogenetic tests. This goal will remain challenging, largely due to minimal data to inform clinical utility estimates. The pace of genomic discovery highlights the need for clinical utility and implementation research to inform future translation efforts. Near-term implementation of promising pharmacogenetic tests can help expedite this goal, generating an evidence base to enable efficient translation as additional gene-drug associations are discovered.
Similar content being viewed by others


Introduction
Initial optimism about the prospect of genomic medicine has given way to the realization that clinical applications will be challenging to identify and implement[1, 2]. Efforts to predict disease incidence or severity based on common genetic variants have shown limited success[1], raising questions about the utility of genomic applications in assessing risk for common health conditions. However, advances in pharmacogenetics have already led to treatment innovations in tertiary care contexts. The U.S. Food and Drug Administration currently lists pharmacogenetic content in >100 drug labels[3]. Genetic testing is standard for some therapies, and evidence for potentially actionable pharmacogenetic variants is accumulating quickly[4, 5].
The clinical translation of pharmacogenetics research also faces numerous challenges[6–10]. One challenge is that the rapid pace of genomic sciences has left little time to apply and evaluate emergent knowledge in clinical contexts[10–12], resulting in a translational research gap[12, 13]. This evidence gap is consistent with the observation that funding initiatives place heavy priority on genomic discovery (T1) research, with a comparatively minute focus on translational (T2–T4) research[6, 12]. Further complicating translation efforts are numerous challenges inherent to the clinical implementation of pharmacogenetic tests. At minimum, these challenges include establishing the clinical validity and clinical utility of candidate biomarkers; training health care providers in their application; addressing ethical, legal, and social implications of genetic testing; navigating third-party reimbursement issues; and updating bioinformatics resources to accommodate genomic information[7–9, 14–16].
A key question concerns the evidentiary thresholds necessary to justify the adoption of promising pharmacogenetic tests in practice[6, 7, 9, 17, 18]. Perspectives on this question will differ across disciplines and clinical scenarios, suggesting the need to navigate this issue as it relates to addiction therapeutics. Because most interventions for alcohol use disorders have only moderate efficacy, identifying prognostic markers of treatment response is a topic of significant interest[19–22]. However, existing research has focused predominantly on the identification or replication of gene–drug response associations, with little discussion about preconditions for clinical implementation. This paper provides a brief discussion of considerations for implementation, emphasizing the need for near-term translation research to inform the clinical utility of pharmacogenetic protocols in alcohol treatment contexts. As a case scenario, evidence for the OPRM1 A118G polymorphism as a moderator of naltrexone response is discussed. However, these considerations will be relevant for other gene–drug response associations under study in the context of alcohol treatments, summaries of which are provided elsewhere[23–25].
Evidence for genetic moderation of naltrexone response
Naltrexone is an opioid receptor antagonist and front-line therapy for alcohol dependence. A widely studied single nucleotide polymorphism in exon 1 of the μ-opioid receptor gene (OPRM1 A118G, rs1799971) is generally considered the most promising genetic moderator of alcohol treatment outcomes[21, 22]. Although molecular evidence concerning the functional role of A118G is not conclusive[24, 26, 27], preclinical and human research implies functional relevance of OPRM1 for phenotypes related to pain and analgesia, stress response, and response to psychoactive drugs[26]. Of particular clinical relevance are findings that alcohol-dependent participants with the minor (118G) allele show relatively better clinical outcomes (e.g., lower rates of relapse to heavy drinking) during treatment with naltrexone, but not placebo, compared to those homozygous for the 118A variant (for review see[19, 24, 26, 27]). In weighing this evidence, it is important to note instances of nonreplication[28] and the lack of large-scale prospective trials[29]. Nonetheless, the first meta-analysis on this topic[30] reached conclusions consistent with initial promising findings reported by Oslin and others[31, 32].
Human laboratory evidence also suggested that OPRM1 A118G moderated naltrexone-related reductions in the hedonic effects of alcohol[33]. This finding is notable in that stimulant effects of alcohol relate to the risk for heavy drinking[34] and reflect a potential target of naltrexone. The identification of a functional equivalent to OPRM1 A118G in rhesus monkeys (C77G) has also allowed investigations in primate models[35–37]. Primates with the 77G variant (which is considered analogous to 118G) showed higher levels of alcohol preference and self-administration, and naltrexone preferentially reduced alcohol self-administration in these animals[35, 37], providing evidence for cross-species convergence of this finding[19, 35]. Therefore, while the association of OPRM1 with alcohol use disorder etiology remains controversial[26], findings suggest the relevance of OPRM1 for a phenotype of clinical interest: reduction in alcohol consumption during treatment with naltrexone versus placebo[19, 35]. Overall, these findings offer preliminary evidence for the clinical validity of OPRM1 A118G as a marker of therapeutic response.
Defining sufficient conditions for clinical implementation
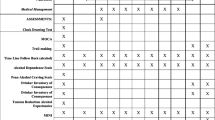
A much-debated issue concerns the degree of evidence necessary to incorporate a promising pharmacogenetic test in practice. This topic has received limited public discussion in the alcohol field, despite that decision-making frameworks for determining the uptake of genomic applications are available. For example, the Centers for Disease Control and Prevention (CDC) endorses the ACCE model[38] for evaluating candidate genomic tests for readiness in practice settings. ACCE emphasizes three performance aspects of genetic tests: analytic validity (availability of a reliable laboratory assay for the genetic variant), clinical validity (prognostic value in relation to clinical phenotypes or intermediate phenotypes of interest), and clinical utility (net benefits minus harms of implementing the test in practice). A fourth category addresses ethical, legal and social implications of implementing the genetic test in a specific clinical scenario. The ACCE model includes 44 standard questions to guide evidence-based evaluation, although adaptations have also been made[39]. Example questions are presented in Table1.
Analytic validity should not pose a concern in the OPRM1-naltrexone scenario, since A118G is a single-point mutation and identifiable with commercial assays. As for clinical validity, the evidence for OPRM1 is by no means conclusive, however, initial findings could be considered promising. Importantly, the ACCE model and similar frameworks do not require conclusive evidence from high-quality randomized trials to evaluate clinical validity. More limited sources of evidence (for example, nonrandomized studies of acceptable quality, systematic reviews of lower-quality studies, or meta-analyses with evidence of heterogeneity) can be considered[5]. In the case of OPRM1, the existence of several retrospective trials and subsequent meta-analyses[30, 40] is likely sufficient for initial evaluations of clinical validity.
Assuming an encouraging clinical validity profile, implementation decisions will hinge largely on clinical utility, defined broadly as the net effect (benefits minus harms) of implementation[7, 41]. Common indices of clinical utility include relevant treatment endpoints (e.g., relapse to heavy drinking), as well as side effects, adverse events, metabolism profiles, or important intermediate phenotypes. Endpoints that can be linked to broader health or economic metrics (e.g., morbidity/mortality, quality-adjusted life-years, treatment costs) are seen as particularly important for determining clinical utility. However, demonstrating that genotype-based treatment protocols improve these outcomes is not necessarily sufficient, because numerous other factors will influence clinical utility in a given context[42]. As one example, a pharmacogenetic protocol that improves clinical outcomes under efficacy scenarios could have zero clinical utility without adequate uptake of the genetic test and/or medication by providers. Similarly, improved treatment endpoints or distal health outcomes do not serve as the only indicators of clinical utility[41]. Proximal or “soft” clinical outcomes can be important in genetic testing scenarios[42]; for instance, personalized treatment protocols could influence treatment motivation, perceived quality of care, and adherence to treatment regimens[42, 43]. Also, perceived utility of a genetic test for therapeutic choice could influence uptake of the test or related medications by providers[41, 42]. Ethical, psychosocial, and legal implications of genetic testing are also important for informing clinical utility[41, 44]. Importantly, numerous parameters of clinical utility exist and most are context-dependent, potentially varying based on the clinical syndrome, population, treatment setting, and other contextual factors[7, 39, 41].
A major barrier to the clinical translation of pharmacogenetics is the shortfall of data to inform estimates of clinical utility, contributing to the so-called “evidence dilemma” in genomic medicine[6, 45]. This gap exists primarily at the T2 stage, such that even the most promising applications are rarely subjected to rigorous research to inform clinical utility profiles and implementation questions[6, 12, 46]. A consequence of this “T2 bottleneck” is that decisions about clinical implementation must be made absent comprehensive evidence for or against clinical utility—or else deferred indefinitely[6]. With few exceptions[47], very little data is available to inform clinical utility estimates for genetic applications in alcohol treatment settings, although somewhat more progress has been made in the area of nicotine addiction[10, 15, 48–53]. Given this evidence shortfall, initial clinical recommendations for pharmacogenetic tests must be made without sufficient evidence for or against clinical utility—a scenario common to most of medicine[6, 54].
Addressing implementation in the context of limited evidence
Most genetic tests are not federally regulated, leaving stakeholders (e.g., the scientific community, clinicians, third-party payers) to navigate implementation decisions[42], usually in the context of insufficient evidence[54]. To facilitate evidence-based recommendations, the CDC sponsored the Evaluation of Genomic Applications in Practice and Prevention (EGAPP) initiative. The chief aim of EGAPP is to develop and test guidelines for the systematic evaluation of candidate genomic applications[5]. Candidate applications are subjected to evidence-based review by an expert panel, following the ACCE framework and resulting in published recommendations[55]. The CDC endorses a three-tier evidentiary framework for assessing candidate genomic applications[54, 56]. Tier 1 denotes applications with sufficient evidence to recommend clinical use (e.g., BRCA testing in the context of family risk for breast and ovarian cancer). Evidence sufficient to discourage clinical use of a test results in assignment to Tier 3. Tier 2 is used for applications with promising evidence for clinical validity, but insufficient evidence to recommend for or against clinical adoption—often reflecting limited data on clinical utility. The vast majority of pharmacogenetic applications will fall under Tier 2[54]. Other Tier 2 examples include genetic risk indicators routinely assessed in practice, but without clear evidence for clinical utility (e.g., assessing family history during depression screening in primary care). The Clinical Pharmacogenetic Implementation Consortium (CPIC) and other expert groups have endorsed similar three-tier models for evaluating genomic applications[4, 57].
The three-tier scheme is noteworthy in that it accommodates clinical implementation in the absence of sufficient evidence for or against clinical utility[54, 57]. To aid these decisions, Tier 2 biomarkers can be further triaged based on anticipated risk-benefit profiles. For example, Tier 2 tests with unfavorable risk-benefit profiles can be subcategorized with a recommendation of “do not use,” whereas those with neutral or marginally favorable profiles may receive recommendations such as “consider use in clinical practice” or “use with evidence-based development”[54, 57]. Importantly, the latter designations can justify clinical implementation in the absence of comprehensive data on clinical utility. No evidence-based guidelines for gene–drug combinations specific to addiction therapeutics have been published by EGAPP or CPIC. In the absence of formal evidence-based reviews—which will emerge slowly, given an enormous number of potential gene–drug associations—stakeholders will largely be tasked with making initial implementation decisions. Clinical annotations for candidate pharmacogenetic tests[58] and models for evidence-based evaluation of genomic applications[59] are increasingly available to guide these decisions.
Noninferiority as a basis for implementation
Placebo-controlled, prospective randomized controlled trials (RCTs) will remain the gold standard for validating pharmacogenetic applications. These designs reduce sources of bias and are necessary for testing genotype-based treatment algorithms[19, 29]. However, predicating clinical adoption on prospective RCTs alone will mean a protracted path from discovery to translation[12, 60]. An alternative view is that noninferiority of pharmacogenetic protocols can be sufficient for initial implementation[60]. From this perspective, tests for which the anticipated risk-benefit profile is neutral or marginally positive could be implemented and evaluated in practice settings prior to establishing clear utility on the basis of RCTs[60]. A noninferiority approach is not a universal solution[61] but can serve several purposes, such as acclimating health care providers and patients to genetic testing; evaluating and refining treatment algorithms; optimizing the design of bioinformatics resources and electronic medical records; and identifying pragmatic barriers to implementation. These steps can generate important information to inform clinical utility profiles and clinical practice protocols[60]. On the other hand, deferring these steps until supportive data from prospective RCTs accumulates will likely delay clinical translation for those tests that ultimately show acceptable clinical utility[13, 60]. As noted above, a reliance on traditional RCTs alone is also insufficient because most trials are not designed to collect comprehensive data on clinical utility.
Because a noninferiority rationale assumes no foreseeable net harms, an important ethical consideration is whether pharmacogenetic protocols could compromise access to best-practice interventions. Ideal scenarios for early implementation might include those where a genetic test informs assignment to one of two empirically supported interventions. For instance, evidence for comparable efficacy of naltrexone and cognitive-behavioral therapy (CBT) for alcohol dependence[62] could justify genotype-based assignment (e.g., assigning naltrexone and CBT as the front-line interventions for 118G carriers and noncarriers, respectively), other considerations being equal. However, contextual factors are important to consider. For instance, the range of available therapeutic options will vary across treatment settings. Assuming noninferiority of a pharmacogenetic protocol and availability of both naltrexone and CBT, a more liberal evidentiary threshold might be used to base treatment selection on OPRM1. However, in the hypothetical scenario that naltrexone is the only evidence-based treatment available, the evidentiary bar should arguably be higher before restricting use of naltrexone to 118G carriers.
Developing an evidence base
Drawing from discussions in other disciplines, several recommendations can be made for near-term efforts to promote translation research in addiction therapeutics. A general goal is to evaluate candidate pharmacogenetic applications with approaches that balance methodological rigor with clinical applicability[8, 13, 54]. To meet this aim, a comprehensive, portfolio-based approach to translation research has been recommended[8, 12]. This approach includes an emphasis on alternative research designs (e.g., feasibility studies, pragmatic clinical trials, adaptive designs, observational studies) that can complement RCTs to generate data on clinical utility and implementation questions[7–9, 57]. Early translation efforts can prioritize the most promising genetic markers as prototypes, allowing for clinical implementation to be studied concurrent with (rather than being predicated on) prospective RCTs[12]. One critique of this approach is its apparent circularity; that is, candidate tests require initial implementation in order to generate evidence that can inform broader implementation. In fact, this observation illustrates the evidence dilemma in genomic medicine[6]. A proposed solution is that, rather than dictating a linear progression in the steps from bench to bedside, translation research proceeds as an ongoing, iterative process, including bidirectional knowledge exchange between basic and clinical researchers beginning at an early stage[12].
These considerations support an argument for provisional implementation of promising pharmacogenetic protocols, if and when feasible[60]. Such an approach assumes neutral or marginally favorable risk-benefit profiles for these protocols relative to standard care, as well as supportive local conditions for implementation. Determining the ideal implementation conditions in addiction treatment settings will take additional work, although some minimal conditions can be inferred (e.g., financial and administrative support; acceptability of genetic testing by staff and patients; affiliation with a laboratory that meets necessary regulatory standards; the ability to educate providers on genetic protocols). Implementing candidate genetic tests in the context of naturalistic, observational, pragmatic, or comparative effectiveness trials can produce data on specific implementation questions, complementing findings from RCTs to inform clinical utility[13, 54]. Ideally, these efforts can be designed to inform specific translation questions, including those relevant to the ACCE criteria. This strategy reflects the chain-of-evidence approach utilized by EGAPP, such that various sources of data are seen as relevant to informing clinical utility[5, 57]. The identification of key “early adopter” sites can help to organize implementation research[8]. Large-scale biobanking and prospective genotyping are commonplace in some hospitals[63, 64], offering particularly good venues. To ensure financial support, funding streams for translational (T2–T4) pharmacogenetics research in alcohol treatment contexts are likely critical to these aims.
Preliminary evidence on OPRM1 and naltrexone response makes this a logical prototype for near-term translation research. At the same time, more research is clearly needed to characterize clinical validity and utility profiles for OPRM1. For example, assessments of clinical validity require specific attention to test prognostics (sensitivity, specificity, positive predictive value, and negative predictive value) in relation to the clinical endpoint in question[65]. Consensus is needed around the primary clinical endpoint(s) for such analyses, with particular attention to what constitutes a “positive” or “negative” treatment response. Relapse to heavy drinking has served as a common outcome in retrospective trials[30], but alternative definitions are likely important[31]. Ideally, these outcomes should be defined a priori and evaluated in prospective studies[29]. Another question concerns the most appropriate comparator condition for sensitivity/specificity estimates[65]. One consideration is that real-world treatment algorithms will not include placebo, and the lack of a single “gold standard” intervention for alcohol dependence further complicates this question. Importantly, clinical validity estimates for most genetic tests will always be imprecise, due to environmental and contextual moderators specific to a given setting[65].
Regarding clinical utility, a key question is whether genotype-based treatment algorithms offer advantages over standard protocols when implemented in clinical practice scenarios. Research protocols that compare genotype-based treatment assignment to conventional methods are needed to address this question. Adaptive treatment designs[21] could be an important element of such studies. In addition to measuring traditional clinical endpoints, such trials should also evaluate proximal outcomes (e.g., medication adherence, physician attitudes) that are ultimately important for informing clinical utility profiles. As noted, such questions can be addressed using prototype scenarios, even if the overall evidence for clinical validity and utility remains limited[43]. As reviewed above, the use of alternative treatment designs, including noninferiority studies and observational studies in naturalistic settings, can also help generate clinical utility data.
Although ethical issues are potentially less obvious in pharmacogenetic scenarios relative to other areas of genetic testing[60], research on these issues is needed. Example issues include privacy considerations related to biobanking[44] and whether ancillary health risk information could be revealed based on test results[66]. A particularly important issue is the potential for racial or ethnic group disparities in access to pharmacogenetic applications or associated treatments[44, 67]. For example, the low prevalence of the 118G allele in people of African descent is one potential explanation for limited efficacy of naltrexone in this population[22]. However, using self-reported race as a basis for treatment decisions can have significant ethical and societal implications, potentially exacerbating disparities in health care access and health outcomes[44].
A key end goal is the development of evidence-based guidelines for candidate pharmacogenetic applications. Published guidelines are seen as critical for promoting awareness of candidate applications, enhancing their uptake[4, 9, 13], and informing third-party reimbursement[6]. Importantly, the process of guideline development is largely stakeholder-driven[54, 59], meaning that those in the alcohol research and treatment communities will be tasked with developing and disseminating these guidelines. In the scenario of OPRM1, a decade of research following the initial report[32] has generated multiple studies, leading to initial meta-analyses[30, 40]. The publication of upcoming prospective trials (and integration of these results in meta-analyses) would provide an ideal time for an updated data synthesis that includes clinical recommendations. Such a process could follow the format of the EGAPP reviews[55]. Reflecting the general lack of clinical utility data, the foreseeable result of such a review is a conclusion of “insufficient evidence” to recommend using OPRM1 for clinical purposes. The question then becomes whether to recommend provisional implementation, for example, in specific clinical scenarios or for purposes of evidence development. Such a decision depends on whether the aggregate evidence, even if limited, is deemed “encouraging” or “discouraging” overall. Importantly, setting the implementation threshold either too high or too low can have adverse implications for future translation efforts[6].
Anticipating barriers
Numerous barriers to the clinical implementation of pharmacogenetic tests have been outlined[9, 10, 12, 15, 44, 50]. Research is needed to evaluate how these barriers apply in alcohol treatment settings, and to identify barriers unique to these settings. A common critique is the potential for adverse responses to genetic testing, including concerns that genetic testing might undermine behavior change efforts. Importantly, evidence for “genetic fatalism” is limited overall[68]. Genetic tests could also have favorable effects on treatment motivation or adherence[43, 69, 70], and initial evidence suggests acceptability of genetic tests among those seeking treatment for alcohol use disorders[47]. Efforts should be made to address these concerns empirically, rather than deferring to “genetic exceptionalism” (i.e., treating genetic tests with greater scrutiny than other diagnostic tools)[71]. One example of a clear barrier in the OPRM1 scenario is the low utilization of naltrexone in both general and specialty settings[72], which ultimately limits clinical utility. Numerous other barriers to implementation can be anticipated, some of which will be evident only as implementation efforts progress[7].
Conclusion
The likelihood that pharmacogenetic protocols will see common use in alcohol treatment scenarios remains unknown. However, there is optimism about the potential for personalized approaches and consensus on the importance of predicting treatment response[19–22, 24, 67], giving reason to support near-term clinical translation research. Ultimately, the potential public health benefits of pharmacogenetics research are contingent on efficient translation—in turn requiring greater commitment to clinical utility and implementation research[12, 45]. Stakeholders in the field will be responsible for setting translational research and funding priorities, navigating implementation issues, and developing consensus guidelines for candidate applications. A clear goal is to generate much-needed evidence on the clinical utility of such applications in treatment settings, in turn leading to evidence-based guidelines for those applications that show the potential for utility. Near-term evaluation of candidate pharmacogenetic protocols in clinical settings can help expedite this goal, generating an evidence base to enable efficient translation as additional gene–drug response associations are discovered.
References
Evans JP, Meslin EM, Marteau TM, Caulfield T: Genomics. Deflating the genomic bubble. Science. 2011, 331: 861-862. 10.1126/science.1198039.
Hall WD, Mathews R, Morley KI: Being more realistic about the public health impact of genomic medicine. PLoS Med. 2010, 7: e1000347-10.1371/journal.pmed.1000347.
U.S. Food and Drug Administration: Table of pharmacogenomic biomarkers in drug labeling. [http://www.fda.gov/Drugs/ScienceResearch/ResearchAreas/Pharmacogenetics/ucm083378.htm]
Relling MV, Klein TE: CPIC: clinical pharmacogenetics implementation consortium of the pharmacogenomics research network. Clin Pharmacol Ther. 2011, 89: 464-467. 10.1038/clpt.2010.279.
Teutsch SM, Bradley LA, Palomaki GE, Haddow JE, Piper M, Calonge N, Dotson WD, Douglas MP, Berg AO, EGAPP Working Group: The evaluation of genomic applications in practice and prevention (EGAPP) initiative: methods of the EGAPP working group. Genet Med. 2009, 11: 3-14. 10.1097/GIM.0b013e318184137c.
Khoury MJ, Berg A, Coates R, Evans J, Teutsch SM, Bradley LA: The evidence dilemma in genomic medicine. Health Aff (Millwood). 2008, 27: 1600-1611. 10.1377/hlthaff.27.6.1600.
Lesko LJ, Zineh I, Huang SM: What is clinical utility and why should we care?. Clin Pharmacol Ther. 2010, 88: 729-733. 10.1038/clpt.2010.229.
Manolio TA, Chisholm RL, Ozenberger B, Roden DM, Williams MS, Wilson R, Bick D, Bottinger EP, Brilliant MH, Eng C, Frazer KA, Korf B, Ledbetter DH, Lupski JR, Marsh C, Mrazek D, Murray MF, O'Donnell PH, Rader DJ, Relling MV, Shuldiner AR, Valle D, Weinshilboum R, Green ED, Ginsburg GS: Implementing genomic medicine in the clinic: the future is here. Genet Med. 2013, 15: 258-267. 10.1038/gim.2012.157.
Pirmohamed M: Acceptance of biomarker-based tests for application in clinical practice: criteria and obstacles. Clin Pharmacol Ther. 2010, 88: 862-866. 10.1038/clpt.2010.245.
Shields AE, Blumenthal D, Weiss KB, Comstock CB, Currivan D, Lerman C: Barriers to translating emerging genetic research on smoking into clinical practice. Perspectives of primary care physicians. J Gen Intern Med. 2005, 20: 131-138. 10.1111/j.1525-1497.2005.30429.x.
Auffray C, Caulfield T, Khoury MJ, Lupski JR, Schwab M, Veenstra T: Genome medicine: past, present and future. Genome Med. 2011, 3: 6-10.1186/gm220.
McBride CM, Bowen D, Brody LC, Condit CM, Croyle RT, Gwinn M, Khoury MJ, Koehly LM, Korf BR, Marteau TM, McLeroy K, Parick K, Valente TW: Future health applications of genomics: priorities for communication, behavioral, and social sciences research. Am J Prev Med. 2010, 38: 556-565. 10.1016/j.amepre.2010.01.027.
Mrazek DA, Lerman C: Facilitating clinical implementation of pharmacogenomics. JAMA. 2011, 306: 304-305.
Roden DM, Tyndale RF: Genomic medicine, precision medicine, personalized medicine: what's in a name?. Clin Pharmacol Ther. 2013, 94: 169-172. 10.1038/clpt.2013.101.
Schnoll RA, Shields AE: Physician barriers to incorporating pharmacogenetic treatment strategies for nicotine dependence into clinical practice. Clin Pharmacol Ther. 2011, 89: 345-347. 10.1038/clpt.2010.267.
Shuldiner AR, Relling MV, Peterson JF, Hicks JK, Freimuth RR, Sadee W, Pereira NL, Roden DM, Johnson JA, Klein TE, Shuldiner AR, Vesely M, Robinson SW, Ambulos N, Stass SA, Kelemen MD, Brown LA, Pollin TI, Beitelshees AL, Zhao RY, Pakyz RE, Palmer K, Alestock T, O'Neill C, Maloney K, Branham A, Sewell D, Relling MV, Crews K, Pharmacogenomics Research Network Translational Pharmacogenetics Program Group: The pharmacogenomics research network translational pharmacogenetics program: overcoming challenges of real-world implementation. Clin Pharmacol Ther. 2013, 94: 207-210. 10.1038/clpt.2013.59.
Mroziewicz M, Tyndale RF: Pharmacogenetics: a tool for identifying genetic factors in drug dependence and response to treatment. Addict Sci Clin Pract. 2010, 5: 17-29.
Schnoll RA, Leone FT: Biomarkers to optimize the treatment of nicotine dependence. Biomark Med. 2011, 5: 745-761. 10.2217/bmm.11.91.
Heilig M, Goldman D, Berrettini W, O'Brien CP: Pharmacogenetic approaches to the treatment of alcohol addiction. Nature Rev Neurosci. 2011, 12: 670-684.
Hutchison KE: Substance use disorders: realizing the promise of pharmacogenomics and personalized medicine. Annu Rev Clin Psychol. 2010, 6: 577-589. 10.1146/annurev.clinpsy.121208.131441.
Kranzler HR, McKay JR: Personalized treatment of alcohol dependence. Curr Psychiatry Rep. 2012, 14: 486-493. 10.1007/s11920-012-0296-5.
Oslin D: Personalized addiction treatment: how close are we?. Alcohol Alcohol. 2011, 46: 231-232. 10.1093/alcalc/agr030.
Ho MK, Goldman D, Heinz A, Kaprio J, Kreek MJ, Li MD, Munafo MR, Tyndale RF: Breaking barriers in the genomics and pharmacogenetics of drug addiction. Clin Pharmacol Ther. 2010, 88: 779-791. 10.1038/clpt.2010.175.
Kranzler HR, Edenberg HJ: Pharmacogenetics of alcohol and alcohol dependence treatment. Curr Pharm Des. 2010, 16: 2141-2148. 10.2174/138161210791516387.
Sturgess JE, George TP, Kennedy JL, Heinz A, Muller DJ: Pharmacogenetics of alcohol, nicotine and drug addiction treatments. Addict Biol. 2011, 16: 357-376. 10.1111/j.1369-1600.2010.00287.x.
Mague SD, Blendy JA: OPRM1 SNP (A118G): involvement in disease development, treatment response, and animal models. Drug Alcohol Depend. 2010, 108: 172-182. 10.1016/j.drugalcdep.2009.12.016.
Ray LA, Barr CS, Blendy JA, Oslin D, Goldman D, Anton RF: The role of the Asn40Asp polymorphism of the mu opioid receptor gene (OPRM1) on alcoholism etiology and treatment: a critical review. Alcohol Clin Exp Res. 2012, 36: 385-394. 10.1111/j.1530-0277.2011.01633.x.
Gelernter J, Gueorguieva R, Kranzler HR, Zhang H, Cramer J, Rosenheck R, Krystal JH, VA Cooperative Study #425 Study Group: Opioid receptor gene (OPRM1, OPRK1, and OPRD1) variants and response to naltrexone treatment for alcohol dependence: results from the VA cooperative study. Alcohol Clin Exp Res. 2007, 31: 555-563.
Kranzler HR: Commentary on Garbutt et al. (2014): Can we predict who benefits from naltrexone in the treatment of alcohol dependence?. Addiction. 2014, 109: 1285-1286. 10.1111/add.12618.
Chamorro AJ, Marcos M, Miron-Canelo JA, Pastor I, Gonzalez-Sarmiento R, Laso FJ: Association of μ-opioid receptor (OPRM1) gene polymorphism with response to naltrexone in alcohol dependence: a systematic review and meta-analysis. Addict Biol. 2012, 17: 505-512. 10.1111/j.1369-1600.2012.00442.x.
Anton RF, Oroszi G, O'Malley S, Couper D, Swift R, Pettinati H, Goldman D: An evaluation of mu-opioid receptor (OPRM1) as a predictor of naltrexone response in the treatment of alcohol dependence: results from the combined pharmacotherapies and behavioral interventions for alcohol dependence (COMBINE) study. Arch Gen Psychiatry. 2008, 65: 135-144. 10.1001/archpsyc.65.2.135.
Oslin DW, Berrettini W, Kranzler HR, Pettinati H, Gelernter J, Volpicelli JR, O'Brien CP: A functional polymorphism of the mu-opioid receptor gene is associated with naltrexone response in alcohol-dependent patients. Neuropsychopharmacology. 2003, 28: 1546-1552. 10.1038/sj.npp.1300219.
Ray LA, Hutchison KE: Effects of naltrexone on alcohol sensitivity and genetic moderators of medication response—a double-blind placebo-controlled study. Arch Gen Psychiatry. 2007, 64: 1069-1077. 10.1001/archpsyc.64.9.1069.
King AC, de Wit H, McNamara PJ, Cao D: Rewarding, stimulant, and sedative alcohol responses and relationship to future binge drinking. Arch Gen Psychiatry. 2011, 68: 389-399. 10.1001/archgenpsychiatry.2011.26.
Barr CS, Chen SA, Schwandt ML, Lindell SG, Sun H, Suomi SJ, Heilig M: Suppression of alcohol preference by naltrexone in the rhesus macaque: a critical role of genetic variation at the micro-opioid receptor gene locus. Biol Psychiatry. 2010, 67: 78-80. 10.1016/j.biopsych.2009.07.026.
Barr CS, Schwandt M, Lindell SG, Chen SA, Goldman D, Suomi SJ, Higley JD, Heilig M: Association of a functional polymorphism in the mu-opioid receptor gene with alcohol response and consumption in male rhesus macaques. Arch Gen Psychiatry. 2007, 64: 369-376. 10.1001/archpsyc.64.3.369.
Vallender EJ, Ruedi-Bettschen D, Miller GM, Platt DM: A pharmacogenetic model of naltrexone-induced attenuation of alcohol consumption in rhesus monkeys. Drug Alcohol Depend. 2010, 109: 252-256. 10.1016/j.drugalcdep.2010.01.005.
Haddow JE, Palomaki PG: ACCE: A Model Process for Evaluating Data on Emerging Genetic Tests. Human Genome Epidemiology: A Scientific Foundation for Using Genetic Information to Improve Health and Prevent Disease. Edited by: Khoury MJ, Little J, Burke W. 2004, New York: Oxford University Press, 217-233.
Burke W, Zimmern R: Moving Beyond ACCE: An Expanded Framework for Genetic Test Evaluation. 2007, PHG Foundation: A paper for the United Kingdom Genetic Testing Registry
Jonas DE, Amick HR, Feltner C, Bobashev G, Thomas K, Wines R, Kim MM, Shanahan E, Gass CE, Rowe CJ, Garbutt JC: (Prepared by the RTI International–University of North Carolina Evidence-based Practice Center under Contract No. 290-2012-00008-I.) AHRQ Publication No. 14-EHC029-EF. Pharmacotherapy for Adults With Alcohol-Use Disorders in Outpatient Settings. Comparative Effectiveness Review No. 134. Rockville, MD: Agency for Healthcare Research and Quality, May 2014. http://www.effectivehealthcare.ahrq.gov/reports/final.cfm
Grosse SD, Khoury MJ: What is the clinical utility of genetic testing?. Genet Med. 2006, 8: 448-450. 10.1097/01.gim.0000227935.26763.c6.
Botkin JR, Teutsch SM, Kaye CI, Hayes M, Haddow JE, Bradley LA, Szegda K, Dotson WD, EGAPP Working Group: Outcomes of interest in evidence-based evaluations of genetic tests. Genet Med. 2010, 12: 228-235. 10.1097/GIM.0b013e3181cdde04.
Marteau TM, Aveyard P, Munafo MR, Prevost AT, Hollands GJ, Armstrong D, Sutton S, Hill C, Johnstone E, Kinmonth AL: Effect on adherence to nicotine replacement therapy of informing smokers their dose is determined by their genotype: a randomised controlled trial. PLoS One. 2012, 7: e35249-10.1371/journal.pone.0035249.
Shields AE: Ethical concerns related to developing pharmacogenomic treatment strategies for addiction. Addict Sci Clin Pract. 2011, 6: 32-43.
Khoury MJ: Dealing with the evidence dilemma in genomics and personalized medicine. Clin Pharmacol Ther. 2010, 87: 635-638. 10.1038/clpt.2010.4.
Khoury MJ, Gwinn M, Yoon PW, Dowling N, Moore CA, Bradley L: The continuum of translation research in genomic medicine: how can we accelerate the appropriate integration of human genome discoveries into health care and disease prevention?. Genet Med. 2007, 9: 665-674. 10.1097/GIM.0b013e31815699d0.
Strobel B, McManus L, Leong S, Blow F, Slaymaker V, Berrettini W, Gordon AJ, O'Brien C, Oslin D: A cross-sectional study of attitudes about the use of genetic testing for clinical care among patients with an alcohol use disorder. Alcohol Alcohol. 2013, 48: 700-703. 10.1093/alcalc/agt130.
McClure JB, Swan GE, St John J, Fauver R, Javitz HS, Bergen AW, Nishita D, Niaura R, Munafo MR, David SP: Pharmacogenetic smoking cessation intervention in a health care setting: a pilot feasibility study. Nicotine Tob Res. 2013, 15: 518-526. 10.1093/ntr/nts173.
Sanderson SC, Humphries SE, Hubbart C, Hughes E, Jarvis MJ, Wardle J: Psychological and behavioural impact of genetic testing smokers for lung cancer risk: a phase II exploratory trial. J Health Psychol. 2008, 13: 481-494. 10.1177/1359105308088519.
Shields AE, Levy DE, Blumenthal D, Currivan D, McGinn-Shapiro M, Weiss KB, Yucel R, Lerman C: Primary care physicians' willingness to offer a new genetic test to tailor smoking treatment, according to test characteristics. Nicotine Tob Res. 2008, 10: 1037-1045. 10.1080/14622200802087580.
Wright AJ, Aveyard P, Guo B, Murphy M, Brown K, Marteau TM: Is attributing smoking to genetic causes associated with a reduced probability of quit attempt success? A cohort study. Addiction. 2007, 102: 1657-1664. 10.1111/j.1360-0443.2007.01937.x.
Wright AJ, French DP, Weinman J, Marteau TM: Can genetic risk information enhance motivation for smoking cessation? An analogue study. Health Psychol. 2006, 25: 740-752.
Wright AJ, Weinman J, Marteau TM: The impact of learning of a genetic predisposition to nicotine dependence: an analogue study. Tob Control. 2003, 12: 227-230. 10.1136/tc.12.2.227.
Khoury MJ, Coates RJ, Evans JP: Evidence-based classification of recommendations on use of genomic tests in clinical practice: dealing with insufficient evidence. Genet Med. 2010, 12: 680-683. 10.1097/GIM.0b013e3181f9ad55.
Evaluation of Genomic Applications in Practice and Prevention (EGAPP) Working Group: The EGAPP initiative: lessons learned. Genet Med. 2014, 16: 217-224.
Dotson WD, Douglas MP, Kolor K, Stewart AC, Bowen MS, Gwinn M, Wulf A, Anders HM, Chang CQ, Clyne M, Lam TK, Schully SD, Marrone M, Feero WG, Khoury MJ: Prioritizing genomic applications for action by level of evidence: a horizon-scanning method. Clin Pharmacol Ther. 2014, 95: 394-402. 10.1038/clpt.2013.226.
Veenstra DL, Roth JA, Garrison LP, Ramsey SD, Burke W: A formal risk-benefit framework for genomic tests: facilitating the appropriate translation of genomics into clinical practice. Genet Med. 2010, 12: 686-693. 10.1097/GIM.0b013e3181eff533.
Thorn CF, Klein TE, Altman RB: PharmGKB: the pharmacogenetics and pharmacogenomics knowledge base. Methods Mol Biol. 2005, 311: 179-191.
Schully SD, Lam TK, Dotson WD, Chang CQ, Aronson N, Birkeland ML, Brewster SJ, Boccia S, Buchanan AH, Calonge N, Calzone K, Djulbegovic B, Goddard KA, Klein RD, Klein TE, Lau J, Long R, Lyman GH, Morgan RL, Palmer CG, Relling MV, Rubinstein WS, Swen JJ, Terry SF, Williams MS, Khoury MJ: Evidence synthesis and guideline development in genomic medicine: current status and future prospects. Genet Med. 2014, doi:10.1038/gim.2014.69
Altman RB: Pharmacogenomics: "noninferiority" is sufficient for initial implementation. Clin Pharmacol Ther. 2011, 89: 348-350. 10.1038/clpt.2010.310.
Khoury MJ, Gwinn M, Dotson WD, Bowen MS: Is there a need for PGxceptionalism?. Genet Med. 2011, 13: 866-867. 10.1097/GIM.0b013e3182265bed.
Anton RF, O'Malley SS, Ciraulo DA, Cisler RA, Couper D, Donovan DM, Gastfriend DR, Hosking JD, Johnson BA, LoCastro JS, Longabaugh R, Mason BJ, Mattson ME, Miller WR, Pettinati HM, Randall CL, Swift R, Weiss RD, Williams LD, Zweben A, COMBINE Study Research Group: Combined pharmacotherapies and behavioral interventions for alcohol dependence: the COMBINE study: a randomized controlled trial. JAMA. 2006, 295: 2003-2017. 10.1001/jama.295.17.2003.
Gottesman O, Scott SA, Ellis SB, Overby CL, Ludtke A, Hulot JS, Hall J, Chatani K, Myers K, Kannry JL, Bottinger EP: The CLIPMERGE PGx program: clinical implementation of personalized medicine through electronic health records and genomics-pharmacogenomics. Clin Pharmacol Ther. 2013, 94: 214-217. 10.1038/clpt.2013.72.
Pulley JM, Denny JC, Peterson JF, Bernard GR, Vnencak-Jones CL, Ramirez AH, Delaney JT, Bowton E, Brothers K, Johnson K, Crawford DC, Schildcrout J, Masys DR, Dilks HH, Wilke RA, Clayton EW, Shultz E, Laposata M, McPherson J, Jirjis JN, Roden DM: Operational implementation of prospective genotyping for personalized medicine: the design of the Vanderbilt PREDICT project. Clin Pharmacol Ther. 2012, 92: 87-95. 10.1038/clpt.2011.371.
Burke W: Genetic tests: clinical validity and clinical utility. Curr Protoc Hum Genet. 2014, 81: 9-15. 1–8
Haga SB, Burke W: Pharmacogenetic testing: not as simple as it seems. Genet Med. 2008, 10: 391-395. 10.1097/GIM.0b013e31817701d4.
Ray LA, Courtney KE, Bujarski S, Squeglia LM: Pharmacogenetics of alcoholism: a clinical neuroscience perspective. Pharmacogenomics. 2012, 13: 129-132. 10.2217/pgs.11.173.
Collins RE, Wright AJ, Marteau TM: Impact of communicating personalized genetic risk information on perceived control over the risk: a systematic review. Genet Med. 2011, 13: 273-277. 10.1097/GIM.0b013e3181f710ca.
Haga SB, LaPointe NM: The potential impact of pharmacogenetic testing on medication adherence. Pharmacogenomics J. 2013, 13: 481-483. 10.1038/tpj.2013.33.
Scheuner MT, Rotter JI: Quantifying the health benefits of genetic tests: a clinical perspective. Genet Med. 2006, 8: 141-142. 10.1097/01.gim.0000206657.19102.a0.
Relling MV, Altman RB, Goetz MP, Evans WE: Clinical implementation of pharmacogenomics: overcoming genetic exceptionalism. Lancet Oncol. 2010, 11: 507-509. 10.1016/S1470-2045(10)70097-8.
Mark TL, Kranzler HR, Song X: Understanding US addiction physicians' low rate of naltrexone prescription. Drug Alcohol Depend. 2003, 71: 219-228. 10.1016/S0376-8716(03)00134-0.
Acknowledgements
This work was supported in part by Canadian Institutes of Health Research (CIHR) grants 288905/307742 and a grant from the McLaughlin Centre, University of Toronto. The author thanks Rachel Tyndale, Caryn Lerman, and Kent Hutchison for their helpful comments on an earlier version of this article.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The author declares no competing interests.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Hendershot, C.S. Pharmacogenetic approaches in the treatment of alcohol use disorders: addressing clinical utility and implementation thresholds. Addict Sci Clin Pract 9, 20 (2014). https://doi.org/10.1186/1940-0640-9-20
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1940-0640-9-20