
Abstract
Lynch syndrome (HNPCC) is a dominantly inherited disorder characterized by germline defects in DNA mismatch repair (MMR) genes and the development of a variety of cancers, predominantly colorectal and endometrial. We present a 44-year-old woman who was shown to carry the truncating MSH2 gene mutation that had previously been identified in her family. Recently, she had been diagnosed with an undifferentiated carcinoma of the thyroid and an adenoma of her coecum. Although the thyroid carcinoma was not MSI-high (1 out of 5 microsatellites instable), it did show complete loss of immunohistochemical expression for the MSH2 protein, suggesting that this tumour was not coincidental. Although the risks for some tumour types, including breast cancer, soft tissue sarcoma and prostate cancer, are not significantly increased in Lynch syndrome, MMR deficiency in the presence of a corresponding germline defect has been demonstrated in incidental cases of a growing range of tumour types, which is reviewed in this paper. Interestingly, the MSH2-associated tumour spectrum appears to be wider than that of MLH1 and generally the risk for most extra-colonic cancers appears to be higher for MSH2 than for MLH1 mutation carriers. Together with a previously reported case, our findings show that anaplastic thyroid carcinoma can develop in the setting of Lynch syndrome. Uncommon Lynch syndrome-associated tumour types might be useful in the genetic analysis of a Lynch syndrome suspected family if samples from typical Lynch syndrome tumours are unavailable.
Similar content being viewed by others


Introduction
Lynch syndrome, also known as hereditary nonpolyposis colorectal cancer (HNPCC), is an autosomal dominant disorder associated with a germline mutation in one of the DNA mismatch repair (MMR) genes, most commonly MLH1 and MSH2 and less frequently MSH6 and PMS2. Lynch syndrome is characterized by a strongly increased risk of developing colorectal cancer and several extra-colonic malignancies including carcinomas of the endometrium, ovary, ureter, stomach and small intestine. Tumours develop at a relatively young age. Although the risks for some common types of cancer, for example breast cancer [1–3], or rarer tumour types, for example malignant fibrous histiocytoma (MFH) [4], do not appear to be significantly increased in Lynch syndrome, MMR deficiency in the presence of a corresponding germline defect has been demonstrated in incidental cases of these tumours.
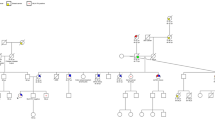
Here we report a 44-year-old woman from a Lynch syndrome, Amsterdam positive family who was referred for DNA testing. She had a recent history of a colorectal adenoma and an undifferentiated carcinoma of her thyroid and was shown to carry the truncating MSH2 mutation that was known to segregate in her family. Traditionally, thyroid cancer is not considered to be part of the Lynch syndrome tumour spectrum. Our findings, however, suggest that this tumour was not coincidental, but likely developed in association with the underlying germline defect in the MSH2 gene. We reviewed the literature on unusual manifestations of inherited mismatch repair gene mutations.
Methods
After genetic counselling, DNA analysis of the MSH2 gene was performed in this 44-year-old woman by extracting DNA from lymphocytes, followed by a PCR amplification of exon 11 of the MSH2 gene. The PCR product was analyzed by denaturing gradient gel electrophoresis (DGGE) and compared with DNA from a family member carrying the mutation [5].
Immunohistochemical staining for MLH1, PMS2, MSH2 and MSH6 protein expression was performed on formalin-fixed, paraffin-embedded sections of tumour as described previously [6].
DNA was extracted from both tumour and normal tissue. Microsatellite instability analysis was performed on formalin-fixed, paraffin-embedded sections of tumour and corresponding normal tissue. Following DNA amplification using fluorescent labelled primers, a panel of five microsatellites recommended by the NCI [7] and consisting of BAT25, BAT26, D2S123, D5S346 and D17S250 was analyzed for allelic shift. The amplified PCR products were analyzed on an ABI Genetic Analyzer.
We searched the English literature through Entrez PubMed http://www.ncbi.nlm.nih.gov/sites/entrez using sets of keywords to identify publications on tumours reported in patients with germline mismatch repair gene mutations. The reference lists of publications found through this approach were searched for additional relevant papers.
Results
Genetic analysis of the MSH2 gene in the patient revealed the c.1704_1705delAG mutation, already known to segregate in her family. Her undifferentiated thyroid carcinoma showed complete loss of immunohi-stochemical expression of the MSH2 and MSH6 protein in the presence of normal positive internal controls, and no loss of the MLH1 and PMS2 protein. Of the five microsatellite markers tested, BAT26 showed instability. Therefore the thyroid tumour was classified as MSI-low.
The acknowledged Lynch syndrome tumour spectrum is shown in Table 1. The cumulative risks and average ages of diagnosis shown in this table were retrieved from papers reporting these data in proven mutation carriers [8–17], rather than from papers that had included untested patients and/or first-degree relatives in their analyses. The reports on 'unusual' tumours in Lynch syndrome patients are presented in Table 2. For comparison, we list the tumour spectrum associated with bi-allelic MMR gene germline mutations in Table 3.
Discussion
Undifferentiated thyroid carcinoma is not commonly associated with Lynch syndrome. In our patient the immunohistochemical loss of expression for the MSH2 and MSH6 protein suggested that this tumour was not coincidental, but due to the underlying mutation in the MSH2 gene. Loss of MSH6 expression in tumours is often observed in case of germline MSH2 mutations and can be explained by loss of its stabilizing partner MSH2. Broaddus et al. [18] contended that for both an adrenal and a thyroid carcinoma an MSH2 gene mutation was causally linked because the tumour showed loss of MSH2 protein with immunohistochemical staining, but retained expression of MLH1. This staining pattern was similar to that seen in the more common Lynch syndrome related malignancies in these families. Although both adrenal and thyroid carcinoma showed loss of MSH2 immunohistochemical expression, neither tumour was microsatellite instable (MSI-high). Loss of protein expression in the absence of MSI has been observed before in Lynch syndrome, most notably in patients with MSH6 mutations [6, 19].
In the past, the Lynch syndrome tumour spectrum has primarily been defined through an epidemiological and statistical approach. From a clinical point of view this approach is of course still very valid as many clinicians will be primarily interested in tumours that have a significantly increased risk of developing in their patients. Cumulative cancer risks for Lynch syndrome were usually based on retrospective cohort analysis of families meeting the Amsterdam criteria, often including families without proven mutations and untested first-degree relatives. More recently studies have focused on proven mutation carriers only. The risk figures listed in Table 1 are based on the latter type of studies [8–17]. Interestingly, the risk for gastric, ovarian, ureter/renal pyelum and brain tumours appears to be higher for carriers of MSH2 mutations than for carriers of MLH1 mutations. In addition to the statistical approach, the tumour spectrum can be broadened through analysis of tumours occurring in MMR gene mutation carriers. Again, patients with atypical Lynch syndrome tumours as listed in Table 2 more often have been reported to carry an MSH2 than an MLH1 mutation. Also a wider range of tumours is observed for MSH2 than for MLH1 in these patients. At this point we can only speculate on the reason for these differences. MLH1 and MSH2 each create a heterodimer with different partners and have different roles in the detection and repair of DNA mismatches. For each of these protein complexes, deficiency might have a different impact on types and quantity of mismatches left unrepaired and the effect deficiency has on different target genes. The absence of MSH6 and PMS2 mutations in Table 2 might simply be caused by the fact that these mutations have been less frequently observed in Lynch syndrome in general. Ascertainment bias, however, cannot be excluded as laboratories did not test MSH6 and PMS2 in their analyses of Lynch syndrome suspected patients until fairly recently. Nevertheless, the absence of MSH6 and PMS2 from the listed reports might also reflect a true difference in associated tumour spectrum.
The tumours listed in Table 2 are not known to develop significantly more frequently in MMR gene mutation carriers than in the general population. Loss of MMR function may or may not have contributed significantly to tumour development in these particular cases. Generally, in these organs loss of the wild type allele in MMR gene mutation carriers and/or subsequently the accumulation of clinically important unrepaired mutations in cancer-associated target genes are apparently relatively rare. It is interesting to look at the types of cancer that develop in patients who have inherited bi-allelic MMR gene mutations (Tabele 3.). These patients are born with a mismatch repair deficiency and can present with tumours that rarely occur in carriers of single allele MMR gene mutations who need to lose their WT allele in their tissues first. Several studies have demonstrated that these bi-allelic mutations can lead to a phenotypically distinct recessive syndrome with predominantly childhood onset brain tumours, leukaemia and lymphoma, bowel tumours and endometrial carcinoma [20–44]. A striking feature of these patients is that nearly all of them display some features, spotty hyperpigmentation of the skin and Lisch nodules of the irides, usually observed in neurofibromatosis type I. Some of the reported tumour types, sarcoma, NHL and early-onset breast cancer match the types incidentally reported in patients with single allele MMR gene mutations, which further supports the notion that these tumour types could be causally linked to inherited MMR gene mutations.
Whether or not MMR deficiency contributed significantly to development of the types of cancer occasionally seen in Lynch syndrome patients remains to be determined. From a practical point of view, we conclude that unusual tumours in Lynch syndrome can show loss of immunohistochemical staining that corresponds to the MMR germline mutation. Therefore these tumours, especially of those types that rarely occur in the general population, could be useful when trying to predict MMR gene mutations in Lynch syndrome suspected families for mutation analysis [6, 19] if the typical Lynch syndrome-associated tumours are unavailable.
References
Blokhuis MM, Goldberg PA, Pietersen GE, Algar U, Vorster AA, Govender D, Ramesar RS: The extracolonic cancer spectrum in females with the common 'South African' hMLH1 c.C1528T mutation. Fam Cancer 2007.
Risinger JI, Barrett JC, Watson P, Lynch HT, Boyd J: Molecular genetic evidence of the occurrence of breast cancer as an integral tumor in patients with the hereditary nonpolyposis colorectal carcinoma syndrome. Cancer 1996, 77: 1836–1843. Publisher Full Text 10.1002/(SICI)1097-0142(19960501)77:9<1836::AID-CNCR12>3.0.CO;2-0
Westenend PJ, Schutte R, Hoogmans MM, Wagner A, Dinjens WN: Breast cancer in an MSH2 gene mutation carrier. Hum Pathol 2005, 36: 1322–1326. 10.1016/j.humpath.2005.08.025
Sijmons R, Hofstra R, Hollema H, Mensink R, Hout A, Hoekstra H, Kleibeuker J, Molenaar W, Wijnen J, Fodde R, Vasen H, Buys C: Inclusion of malignant fibrous histiocytoma in the tumour spectrum associated with hereditary non-polyposis colorectal cancer. Genes Chromosomes Cancer 2000, 29: 353–355. 10.1002/1098-2264(2000)9999:9999<::AID-GCC1042>3.0.CO;2-T
Wu Y, Nyström-Lahti M, Osinga J, Looman MW, Peltomäki P, Aaltonen LA, de la Chapelle A, Hofstra RM, Buys CH: MSH2 and MLH1 mutations in sporadic replication error-positive colorectal carcinoma as assessed by two-dimensional DNA electrophoresis. Genes Chromosomes Cancer 1997, 18: 269–278. 10.1002/(SICI)1098-2264(199704)18:4<269::AID-GCC4>3.0.CO;2-Z
Berends MJ, Wu Y, Sijmons RH, Mensink RG, Sluis T, Hordijk-Hos JM, de Vries EG, Hollema H, Karrenbeld A, Buys CH, Zee AG, Hofstra RM, Kleibeuker JH: Molecular and clinical characteristics of MSH6 variants: an analysis of 25 index carriers of a germline variant. Am J Hum Genet 2002, 70: 26–37. 10.1086/337944
Umar A, Boland CR, Terdiman JP, Syngal S, de la Chapelle A, Rüschoff J, Fishel R, Lindor NM, Burgart LJ, Hamelin R, Hamilton SR, Hiatt RA, Jass J, Lindblom A, Lynch HT, Peltomaki P, Ramsey SD, Rodriguez-Bigas MA, Vasen HF, Hawk ET, Barrett JC, Freedman AN, Srivastava S: Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst 2004, 96: 261–268.
Aarnio M, Sankila R, Pukkala E, Salovaara R, Aaltonen LA, de la Chapelle A, Peltomäki P, Mecklin JP, Järvinen HJ: Cancer risk in mutation carriers of DNA-mismatch-repair genes. Int J Cancer 1999, 81: 214–8. 10.1002/(SICI)1097-0215(19990412)81:2<214::AID-IJC8>3.0.CO;2-L
Dunlop MG, Farrington SM, Carothers AD, Wyllie AH, Sharp L, Burn J, Liu B, Kinzler KW, Vogelstein B: Cancer risk associated with germline DNA mismatch repair gene mutations. Hum Mol Genet 1997, 6: 105–110. 10.1093/hmg/6.1.105
Hendriks YM, Wagner A, Morreau H, Menko F, Stormorken A, Quehenberger F, Sandkuijl L, Møller P, Genuardi M, Van Houwelingen H, Tops C, Van Puijenbroek M, Verkuijlen P, Kenter G, Van Mil A, Meijers-Heijboer H, Tan GB, Breuning MH, Fodde R, Wijnen JT, Bröcker-Vriends AH, Vasen H: Cancer risk in hereditary nonpolyposis colorectal cancer due to MSH6 mutations: impact on counseling and surveillance. Gastroenterology 2004, 127: 17–25. 10.1053/j.gastro.2004.03.068
Plaschke J, Engel C, Krüger S, Holinski-Feder E, Pagenstecher C, Mangold E, Moeslein G, Schulmann K, Gebert J, von Knebel Doeberitz M, Rüschoff J, Loeffler M, Schackert HK: Lower incidence of colorectal cancer and later age of disease onset in 27 families with pathogenic MSH6 germline mutations compared with families with MLH1 or MSH2 mutations: the German Hereditary Nonpolyposis Colorectal Cancer Consortium. J Clin Oncol 2004, 22: 4486–4494. 10.1200/JCO.2004.02.033
Quehenberger F, Vasen HF, van Houwelingen HC: Risk of colorectal and endometrial cancer for carriers of mutations of the hMLH1 and hMSH2 gene: correction for ascertainment. J Med Genet 2005, 42: 491–496. 10.1136/jmg.2004.024299
Vasen HF, Wijnen JT, Menko FH, Kleibeuker JH, Taal BG, Griffioen G, Nagengast FM, Meijers-Heijboer EH, Bertario L, Varesco L, Bisgaard ML, Mohr J, Fodde R, Khan PM: Cancer risk in families with hereditary nonpolyposis colorectal cancer diagnosed by mutation analysis. Gastroenterology 1996, 110: 1020–1027. 10.1053/gast.1996.v110.pm8612988
Vasen HF, Stormorken A, Menko FH, Nagengast FM, Kleibeuker JH, Griffioen G, Taal BG, Moller P, Wijnen JT: MSH2 mutation carriers are at higher risk of cancer than MLH1 mutation carriers: a study of hereditary nonpolyposis colorectal cancer families. J Clin Oncol 2001, 19: 4074–4080.
Hampel H, Stephens JA, Pukkala E, Sankila R, Aaltonen LA, Mecklin JP, de la Chapelle A: Cancer risk in hereditary nonpolyposis colorectal cancer syndrome: later age of onset. Gastroenterology 2005, 129: 415–421.
Carayol J, Khlat M, Maccario J, Bonaïti-Pellié C: Hereditary non-polyposis colorectal cancer: current risks of colorectal cancer largely overestimated. J Med Genet 2002, 39: 335–339. 10.1136/jmg.39.5.335
Buttin BM, Powell MA, Mutch DG, Babb SA, Huettner PC, Edmonston TB, Herzog TJ, Rader JS, Gibb RK, Whelan AJ, Goodfellow PJ: Penetrance and expressivity of MSH6 germline mutations in seven kindreds not ascertained by family history. Am J Hum Genet 2004, 74: 1262–1269. 10.1086/421332
Broaddus RR, Lynch PM, Lu KH, Luthra R, Michelson SJ: Unusual tumors associated with the hereditary nonpolyposis colorectal cancer syndrome. Mod Pathol 2004, 17: 981–989. 10.1038/modpathol.3800150
Niessen RC, Berends MJ, Wu Y, Sijmons RH, Hollema H, Ligtenberg MJ, de Walle HE, de Vries EG, Karrenbeld A, Buys CH, Zee AG, Hofstra RM, Kleibeuker JH: Identification of mismatch repair gene mutations in young patients with colorectal cancer and in patients with multiple tumours associated with hereditary non-polyposis colorectal cancer. Gut 2006, 55: 1781–1788. 10.1136/gut.2005.090159
Agostini M, Tibiletti MG, Lucci-Cordisco E, Chiaravalli A, Morreau H, Furlan D, Boccuto L, Pucciarelli S, Capella C, Boiocchi M, Viel A: Two PMS2 mutations in a Turcot syndrome family with small bowel cancers. Am J Gastroenterol 2005, 100: 1886–1891. 10.1111/j.1572-0241.2005.50441.x
Auclair J, Leroux D, Desseigne F, Lasset C, Saurin JC, Joly MO, Pinson S, Xu XL, Montmain G, Ruano E, Navarro C, Puisieux A, Wang Q: Novel biallelic mutations in MSH6 and PMS2 genes: gene conversion as a likely cause of PMS2 gene inactivation. Hum Mutat 2007, 28: 1084–1090. 10.1002/humu.20569
Bougeard G, Charbonnier F, Moerman A, Martin C, Ruchoux MM, Drouot N, Frébourg T: Early onset brain tumor and lymphoma in MSH2-deficient children. Am J Hum Genet 2003, 72: 213–216. 10.1086/345297
De Rosa M, Fasano C, Panariello L, Scarano MI, Belli G, Iannelli A, Ciciliano F, Izzo P: Evidence for a recessive inheritance of Turcot's syndrome caused by compound heterozygous mutations within the PMS2 gene. Oncogene 2000, 19: 1719–1723. 10.1038/sj.onc.1203447
De Vos M, Hayward BE, Picton S, Sheridan E, Bonthron DT: Novel PMS2 pseudogenes can conceal recessive mutations causing a distinctive childhood cancer syndrome. Am J Hum Genet 2004, 74: 954–964. 10.1086/420796
De Vos M, Hayward BE, Charlton R, Taylor GR, Glaser AW, Picton S, Cole TR, Maher ER, McKeown CM, Mann JR, Yates JR, Baralle D, Rankin J, Bonthron DT, Sheridan E: PMS2 mutations in childhood cancer. J Natl Cancer Inst 2006, 98: 358–361.
Gallinger S, Aronson M, Shayan K, Ratcliffe EM, Gerstle JT, Parkin PC, Rothenmund H, Croitoru M, Baumann E, Durie PR, Weksberg R, Pollett A, Riddell RH, Ngan BY, Cutz E, Lagarde AE, Chan HS: Gastrointestinal cancers and neurofibromatosis type 1 features in children with a germline homozygous MLH1 mutation. Gastroenterology 2004, 126: 576–585. 10.1053/j.gastro.2003.11.008
Hackman P, Tannergård P, Osei-Mensa S, Chen J, Kane MF, Kolodner R, Lambert B, Hellgren D, Lindblom A: A human compound heterozygote for two MLH1 missense mutations. Nat Genet 1997, 17: 135–136. 10.1038/ng1097-135
Hamilton SR, Liu B, Parsons RE, Papadopoulos N, Jen J, Powell SM, Krush AJ, Berk T, Cohen Z, Tetu B, et al.: The molecular basis of Turcot's syndrome. N Engl J Med 1995, 332: 839–847. 10.1056/NEJM199503303321302
Hegde MR, Chong B, Blazo ME, Chin LH, Ward PA, Chintagumpala MM, Kim JY, Plon SE, Richards CS: A homozygous mutation in MSH6 causes Turcot syndrome. Clin Cancer Res 2005, 11: 4689–4693. 10.1158/1078-0432.CCR-04-2025
Krüger S, Kinzel M, Walldorf C, Gottschling S, Bier A, Tinschert S, von Stackelberg A, Henn W, Görgens H, Boue S, Kölble K, Büttner R, Schackert HK: Homozygous PMS2 germline mutations in two families with early-onset haematological malignancy, brain tumours, HNPCC-associated tumours, and signs of neurofibromatosis type 1. Eur J Hum Genet 2008, 16: 62–72. 10.1038/sj.ejhg.5201923
Liu T, Tannergård P, Hackman P, Rubio C, Kressner U, Lindmark G, Hellgren D, Lambert B, Lindblom A: Missense mutations in hMLH1 associated with colorectal cancer. Hum Genet 1999, 105: 437–441. 10.1007/s004390051127
Menko FH, Kaspers GL, Meijer GA, Claes K, van Hagen JM, Gille JJ: A homozygous MSH6 mutation in a child with cafe-au-lait spots, oligodendroglioma and rectal cancer. Fam Cancer 2004, 3: 123–127. 10.1023/B:FAME.0000039893.19289.18
Müller A, Schackert HK, Lange B, Rüschoff J, Füzesi L, Willert J, Burfeind P, Shah P, Becker H, Epplen JT, Stemmler S: A novel MSH2 germline mutation in homozygous state in two brothers with colorectal cancers diagnosed at the age of 11 and 12 years. Am J Med Genet A 2006, 140: 195–199.
Ostergaard JR, Sunde L, Okkels H: Neurofibromatosis von Recklinghausen type I phenotype and early onset of cancers in siblings compound heterozygous for mutations in MSH6. Am J Med Genet A 2005, 139: 96–105.
Plaschke J, Linnebacher M, Kloor M, Gebert J, Cremer FW, Tinschert S, Aust DE, von Knebel Doeberitz M, Schackert HK: Compound heterozygosity for two MSH6 mutations in a patient with early onset of HNPCC-associated cancers, but without hematological malignancy and brain tumor. Eur J Hum Genet 2006, 14: 561–566. 10.1038/sj.ejhg.5201568
Poley JW, Wagner A, Hoogmans MM, Menko FH, Tops C, Kros JM, Reddingius RE, Meijers-Heijboer H, Kuipers EJ, Dinjens WN, Rotterdam Initiative on Gastrointestinal Hereditary Tumors: Biallelic germline mutations of mismatch-repair genes: a possible cause for multiple pediatric malignancies. Cancer 2007, 109: 2349–2356. 10.1002/cncr.22697
Rey JM, Noruzinia M, Brouillet JP, Sarda P, Maudelonde T, Pujol P: Six novel heterozygous MLH1, MSH2, and MSH6 and one homozygous MLH1 germline mutations in hereditary nonpolyposis colorectal cancer. Cancer Genet Cytogenet 2004, 155: 149–151. 10.1016/j.cancergencyto.2004.03.012
Ricciardone MD, Ozçelik T, Cevher B, Ozdag H, Tuncer M, Gürgey A, Uzunalimoglu O, Cetinkaya H, Tanyeli A, Erken E, Oztürk M: Human MLH1 deficiency predisposes to hematological malignancy and neurofibromatosis type 1. Cancer Res 1999, 59: 290–293.
Scott RH, Mansour S, Pritchard-Jones K, Kumar D, MacSweeney F, Rahman N: Medulloblastoma, acute myelocytic leukemia and colonic carcinomas in a child with biallelic MSH6 mutations. Nat Clin Pract Oncol 2007, 4: 130–134. 10.1038/ncponc0719
Scott RH, Homfray T, Huxter NL, Mitton SG, Nash R, Potter MN, Lancaster D, Rahman N: Familial T-cell non-Hodgkin lymphoma caused by biallelic MSH2 mutations. J Med Genet 2007, 44: e83. 10.1136/jmg.2007.048942
Trimbath JD, Petersen GM, Erdman SH, Ferre M, Luce MC, Giardiello FM: Cafe-au-lait spots and early onset colorectal neoplasia: a variant of HNPCC? Fam Cancer 2001, 1: 101–105. 10.1023/A:1013881832014
Vilkki S, Tsao JL, Loukola A, Poyhonen M, Vierimaa O, Herva R, Aaltonen LA, Shibata D: Extensive somatic microsatellite mutations in normal human tissue. Cancer Res 2001, 61: 4541–4544.
Wang Q, Lasset C, Desseigne F, Frappaz D, Bergeron C, Navarro C, Ruano E, Puisieux A: Neurofibromatosis and early onset of cancers in hMLH1-deficient children. Cancer Res 1999, 59: 294–297.
Whiteside D, McLeod R, Graham G, Steckley JL, Booth K, Somerville MJ, Andrew SE: A homozygous germ-line mutation in the human MSH2 gene predisposes to hematological malignancy and multiple cafe-au-lait spots. Cancer Res 2002, 62: 359–362.
Pineda M, Castellsague E, Musulen E, Llort G, Frebourg T, Baert-Desurmont S, Gonzalez S, Capella G, Blanco I: Non-Hodgkin lymphoma related to hereditary nonpolyposis colorectal cancer in a patient with a novel heterozygous complex deletion in the MSH2 gene. Genes Chromosomes Cancer 2008, 47: 326–332. 10.1002/gcc.20536
den Bakker MA, Seynaeve C, Kliffen M, Dinjens WN: Microsatellite instability in a pleomorphic rhabdomyosarcoma in a patient with hereditary non-polyposis colorectal cancer. Histopathology 2003, 43: 297–299. 10.1046/j.1365-2559.2003.01681.x
Banville N, Geraghty R, Fox E, Leahy DT, Green A, Keegan D, Geoghegan J, O'Donoghue D, Hyland J, Sheahan K: Medullary carcinoma of the pancreas in a man with hereditary nonpolyposis colorectal cancer due to a mutation of the MSH2 mismatch repair gene. Hum Pathol 2006, 37: 1498–1502. 10.1016/j.humpath.2006.06.024
Soravia C, Klift H, Bründler MA, Blouin JL, Wijnen J, Hutter P, Fodde R, Delozier-Blanchet C: Prostate cancer is part of the hereditary non-polyposis colorectal cancer (HNPCC) tumor spectrum. Am J Med Genet A 2003, 121: 159–162. 10.1002/ajmg.a.20106
Hirata K, Kanemitsu S, Nakayama Y, Nagata N, Itoh H, Ohnishi H, Ishikawa H, Furukawa Y, HNPCC registry and genetic testing project of the Japanese Society for Cancer of the Colon and Rectum (JSCCR): A novel germline mutation of MSH2 in a hereditary nonpolyposis colorectal cancer patient with liposarcoma. Am J Gastroenterol 2006, 101: 193–196. 10.1111/j.1572-0241.2005.00308.x
Vernez M, Hutter P, Monnerat C, Halkic N, Gugerli O, Bouzourene H: A case of Muir-Torre syndrome associated with mucinous hepatic cholangiocarcinoma and a novel germline mutation of the MSH2 gene. Fam Cancer 2007, 6: 141–145. 10.1007/s10689-006-9105-9
South SA, Hutton M, Farrell C, Mhawech-Fauceglia P, Rodabaugh KJ: Uterine carcinosarcoma associated with hereditary nonpolyposis colorectal cancer. Obstet Gynecol 2007, 110: 543–545.
Boyd J, Rhei E, Federici MG, Borgen PI, Watson P, Franklin B, Karr B, Lynch J, Lemon SJ, Lynch HT: Male breast cancer in the hereditary nonpolyposis colorectal cancer syndrome. Breast Cancer Res Treat 1999, 53: 87–91. 10.1023/A:1006030116357
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution 2.0 International License (https://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Stulp, R.P., Herkert, J.C., Karrenbeld, A. et al. Thyroid cancer in a patient with a germline MSH2 mutation. Case report and review of the Lynch syndrome expanding tumour spectrum. Hered Cancer Clin Pract 6, 15 (2008). https://doi.org/10.1186/1897-4287-6-1-15
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1897-4287-6-1-15