
Abstract
Background
The occurrence of asthma is weakly explained by known genetic variants. Epigenetic marks, DNA methylation (DNA-M) in particular, are considered to add to the explanation of asthma. However, no etiological model has yet been developed that integrates genetic variants and DNA-M. To explore a new model, we focused on one asthma candidate gene, the IL-4 receptor (IL4R). We hypothesized that genetic variants of IL4R in interaction with DNA-M at cytosine-phosphate-guanine (CpG) sites jointly alter the risk of asthma during adolescence. Blood samples were collected at age 18 years from 245 female cohort participants randomly selected for methylation analysis from a birth cohort (n = 1,456, Isle of Wight, UK). Genome-wide DNA-M was assessed using the Illumina Infinium HumanMethylation450 BeadChip.
Results
Thirteen single nucleotide polymorphisms (SNPs) and twelve CpG sites of IL4R gene were analyzed. Based on linkage disequilibrium and association with asthma, eight SNPs and one CpG site were selected for further analyses. Of the twelve CpG sites in the IL4R gene, only methylation levels of cg09791102 showed an association with asthma at age 18 years (Wilcoxon test: P = 0.01). Log-linear models were used to estimate risk ratios (RRs) for asthma adjusting for uncorrelated SNPs within the IL4R gene and covariates. Testing for interaction between the eight SNPs and the methylation levels of cg09791102 on the risk for asthma at age 18 years, we identified the statistically significant interaction term of SNP rs3024685 × methylation levels of cg09791102 (P = 0.002; after adjusting for false discovery rate). A total of 84 participants had methylation levels ≤0.88, 112 participants between 0.89 and 0.90, and 35 between 0.91 and 0.92. For the SNP rs3024685 (‘CC’ vs. ‘TT’) at methylation levels of ≤0.85, 0.86, 0.90, 0.91, and 0.92, the RRs were 0.01, 0.04, 4.65, 14.76, 14.90, respectively (interaction effect, P = 0.0003).
Conclusions
Adjusting for multiple testing, our results suggest that DNA-M modulates the risk of asthma related to genetic variants in the IL4R gene. The strong interaction of one SNP and DNA-M is encouraging and provides a novel model of how a joint effect of genetic variants and DNA-M can explain occurrence of asthma.
Similar content being viewed by others

Background
Asthma is a common chronic disease that affects around 235 million people around the world and 5.4 million in the United Kingdom (UK) [1]. The burden of disease affects 1.1 million children between ages 0 to 17 years in the UK. Asthma is characterized clinically by shortness of breath, wheezing episodes, chest tightness, and acute episodes of coughing [2]. The disease etiology is poorly understood and the postnatal development is not well established. Genetic susceptibility, environmental factors, and gene × environment interaction are believed to play a critical role in the development of asthma. Over 200 genes have been suggested to contribute to asthma occurrence [3–5]. The high heritability (35% to 95%) and the co-occurrence of asthma within families highlight the importance of a genetic component in disease pathogenesis [1]. In this work we focus on the interleukin receptor (IL4R) gene which has been clearly established as an asthma susceptibility gene in multiple candidate gene association studies [3–5].
There is evidence that interleukin-4 (IL-4) and its receptor (IL-4R) are involved in the pathogenesis of asthma [6–8]. A recent meta-analysis indicated a modest risk associated with IL4R single nucleotide polymorphisms (SNPs) on occurrence of asthma, but other investigators found conflicting results [7]. Analysis of asthma candidate genes in a genome-wide association study population showed that SNPs in IL4R were significant related to asthma with significance level between P = 0.05 and P = 0.0035 [3] despite IL4R not being identified in genome-wide association study (GWAS) analysis suggesting that IL4R variation is not well captured in current GWAS platforms. Other genetic regulatory mechanisms beyond DNA sequence variation may aid in explaining the role of IL4R in asthma. It has been suggested that epigenetic mechanisms play a role in T-cell differentiation and regulation, a crucial event in the onset of atopic diseases such as asthma [9]. Epigenetic regulatory mechanisms, such as DNA-methylation (DNA-M), may alter gene expression and protein production without changing the DNA sequence. No etiological model has yet been developed that integrates genetic variants and DNA-M. We will explore the idea that an increase of DNA-M may silence or a decrease of DNA-M may activate the effect of specific SNPs. To test this new model, we focus on one asthma candidate gene, the IL4R gene. We hypothesized that SNPs in interaction with cytosine-phosphate-guanine (CpG) sites jointly predispose to asthma at age 18 years. To test vertical transmission of DNA-M to offspring in future steps, this work focuses on women.
Methods
Study design and population
A whole-population birth cohort was established on the Isle of Wight in 1989 to prospectively study the natural history of asthma and allergic conditions. After exclusion of adoptions, perinatal deaths and refusal, 1,456 children (95%) were enrolled. The local research ethics committee approved the study and informed written parental consent was obtained for all participants at recruitment and subsequently at follow-ups, which were conducted at ages 1, 2, 4, 10, and 18 years of age. The birth cohort has been described in detail elsewhere [10, 11]. In this study we focused on blood samples collected at 18 years of age from 245 female cohort participants who were randomly selected for genomic sequencing and DNA-M.
Clinical data collection and outcome
Maternal history of asthma and smoking during pregnancy was ascertained at birth. Birth weight was obtained from birth records. At ages 1, 2, 4, 10, and 18 years, the original questionnaire-based information was updated, and weight and height of the child were measured. Breastfeeding duration was assessed at follow-up visits at ages 1 and 2 years. At age 18 years, the questionnaire-based information was updated using the International Study of Asthma and Allergies in Childhood (ISAAC) questionnaire [12]. Asthma at age 18 years was defined as subjects with a physician diagnosis of asthma plus current symptoms and/or asthma medication.
SNP selection for the IL4R gene
An efficient genotype tagging scheme was developed that gave priority to variants that 1) showed strong association with asthma in the Isle of Wight birth cohort, and/or 2) have been reported by others to be associated with asthma/allergy, and/or 3) have functional importance. A literature search for IL4R gene plus asthma and allergy was used to identify associated variants (SNPs, indels). Functional variants included those that were non-synonymous, located in conserved DNA, and/or present in DNA regions with gene regulatory potential. Tagger implemented in Haploview 3.2 using Caucasian Hapmap data was used to develop a tagging scheme for the IL4R gene region, including 10 kb upstream and downstream of the gene [13]. An r2 value of 0.85 was the threshold for tagging and one, two and three SNP marker combination tests were used. The result was an efficient number of genotyped variants (n = 13) that would provide the needed information to statistically support or exclude the gene in its association with asthma outcomes.
DNA methylation protocol
DNA was extracted from whole blood using a standard salting out procedure [14]. DNA concentration was determined by PicoGreen quantitation. One microgram DNA was bisulfite-treated for cytosine to thymine conversion using the EZ 96-DNA methylation kit (Zymo Research, Irvine, CA, USA), following the manufacturer’s standard protocol. Genome-wide DNA methylation was assessed using the Illumina Infinium HumanMethylation450 BeadChip (Illumina, Inc., San Diego, CA, USA), which interrogates >484,000 CpG sites associated with approximately 24,000 genes. Arrays were processed using a standard protocol as described elsewhere [15], with multiple identical control samples assigned to each bisulphite conversion batch to assess assay variability and samples randomly distributed on microarrays to control against batch effects. The BeadChips were scanned using a BeadStation, and the methylation level (beta value) calculated for each queried CpG locus using the Methylation Module of BeadStudio software.
Exposures
The main exposures are SNPs and the methylation levels at CpG sites in the IL4R gene (Table 1). The following SNPs were included in the analysis: rs3024622, rs3024685, rs6498012, rs12102586, rs16976728, rs4787423, rs3024676, and rs2057768.
Statistical analysis
To assess whether our analytic sample (245 DNA samples) was representative of the total cohort available at age 18 years, we compared the characteristics of these two subsets by using the chi-square test. After cleaning the DNA-M data, beta (β) values presented as the proportion of intensity of methylated (M) over the sum of methylated (M) and unmethylated (U) sites (β = M/[c + M + U] with c being a constant to prevent dividing by zero) were used to estimate the effect of DNA methylation [16]. The methylation levels of 12 CpG sites spanning the genomic region of the IL4R gene (Table 1) were tested for association with asthma at age 18 years using Wilcoxon tests. Of these CpG sites, only methylation levels of cg09791102 showed a statistically significant association with asthma at age 18 years (Wilcoxon test: P = 0.01).
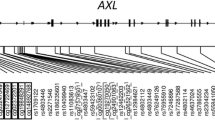
The 13 SNPs shown in Figure 1 were tested for Hardy-Weinberg equilibrium using Haploview 3.2 software [13] and estimates of linkage disequilibrium (LD) between SNPs were calculated using D’ and r2[17], to select one SNP that represents each LD block or an unlinked area.

IL4R LD plot; standard (D’/LOD) color scheme; D’ LD values displayed.
After identifying eight uncorrelated IL4R SNPs (Figure 1; Table 1) and identifying which CpG site was significantly associated with asthma, we ran eight independent models to estimate statistical interactions between these SNPs and the methylation level of cg09791102 on the risk for asthma at age 18 years. We assessed the interaction on a multiplicative scale in log-linear models using an overall chi-square test as a cutoff P value = 0.05 for each. Only one interaction (SNP rs3024685 × cg09791102) showed a significant effect on asthma at age 18. This interaction and those SNPs and four covariates that confounded the association between the SNP and CpG interaction with asthma at age 18 years were included in the final log-linear model. We then inspected which genotype (CC, CT, or TT) explains the overall effect. Confounders include child’s BMI at age 18 (kg/m2), maternal history of asthma, maternal smoking during pregnancy, and breastfeeding duration (weeks). All confounders were simultaneously entered as indicator variables into the log-linear model. A backward elimination process was used to identify confounders, those that changed the association of interest by 10% or more were retained in the final model. For the reduced model, we estimated risk ratios (RR) and their 95% confidence intervals (CI).
Since we tested a total of eight crude SNP × methylation interactions before selecting the full model, we adjusted for multiple testing by applying false discovery rate (P = 0.05) [18]. All statistical analyses were performed using the SAS statistical package, Version 9.2 (SAS Institute, Cary, NC, USA), except for cleaning the DNA methylation data, which was done using R statistical computing package [19].
Results
Blood samples from a subset of 245 of 750 female birth cohort participants were used to determine DNA-M at CpG sites. There were no substantial differences in prevalence of low birth weight, asthma at 18, BMI at 18, breastfeeding duration, maternal BMI, maternal history of asthma, nor maternal smoking between the female participants of the cohort and the subset included in this analysis (Table 2). For the subgroup with available methylation data 12% had maternal history of asthma, 19% had mothers that smoked during pregnancy, and 14.3% (35/245) had asthma at age 18 years.
Of the thirteen SNPs genotyped in the IL4R gene, eight SNPs were analyzed since they were uncorrelated (D’ <0.95) (Figure 1, Table 1). A total of 12 CpG sites spanning the genomic region of the IL4R gene were analyzed for association with asthma at age 18 years. Only methylation levels of cg09791102 showed an association with asthma at age 18 years (Wilcoxon test: P = 0.01). Testing for interaction between the eight SNPs and the methylation levels of cg09791102 on the risk for asthma at age 18 years, we identified that the interaction term of SNP rs3024685 × methylation levels of cg09791102 was statistically significant (P = 0.0003; FDR adjusted P value = 0.002; Table 3). In other words, the genetic risk of asthma associated with rs3024685 increases as the methylation level of cg09791102 rises (Figure 2).

Risk Ratio of asthma at age 18 years versus methylation score at different genotypes of IL4R rs3024685. The blue bars present the relative frequency of the DNA methylation levels. For instance, 87% methylation is found in 10% of the participants. The reference genotype is ‘TT’. The solid horizontal line that indicates a risk ratio value of ‘1’ shows the risk ratio of the reference ‘TT’ genotype. The black dot represents the ‘CC’ genotype, and the diamond is ‘CT’ genotype.
The DNA-M level range for cg09791102 was 0.48 to 0.92 (blue bars in Figure 2). Since the number of participants at methylation levels of 0.85 or less were low, we grouped these methylation levels into ≤0.85 (n = 9). For descriptive purposes, 84 participants had methylation levels of 0.88 and less, 112 participants of 0.89 to 0.90, and 35 of 0.91 to 0.92. Since the mode of inheritance is additive, we compared participants who had the ‘CC’ and ‘CT’ genotypes with those who were ‘TT’ genotype at rs3024685. For the genotype ‘CC’, compared to ‘TT’, we found that at methylation levels of 0.85, 0.86, 0.90, 0.91, and 0.92, the RRs of asthma were 0.01, 0.04, 4.65, 14.76, and 46.90 (Figure 2; FDR adjusted P value = 0.002), respectively. Similar results were found with ‘CT’ genotype, however the interaction term did not achieve statistical significance (P = 0.06).
Descriptively, 13.2% and 14.3% of the participants had asthma at a methylation level of 0.88 at the genotype ‘CT’ and ‘TT’, respectively; and none of the ‘CC’ genotype had asthma. Between 0.89 and 0.90 methylation levels, 15.0% of the ‘CC’, 16.7% of the ‘CT’, and 7.9% of the ‘TT’ genotype had asthma. At methylation levels larger than 0.90, 54.6% of the ‘CC’ and 16.7% of the ‘CT’ genotype had asthma, and none of the ‘TT’ genotype had asthma.
Discussion
This is the first study to determine the role of both genetic and epigenetic factors within the genomic region of the IL4R gene on the risk for asthma. Although the CpG site cg09791102 is located 23,496 base pairs away from SNP rs3024685 in the intragenic region of the IL4R gene, we found that the risk of asthma is modulated by this CpG site even after adjusting for multiple testing. The distance between the SNP and the CpG sites is large. However, Bell et al. have demonstrated that for a regulation in cis even larger distances can show statistically significant effects [20]. Hence, these two factors (SNP and CpG site) may jointly contribute to gene expression or alternative splicing.
The SNP rs3024685 in the 3′UTR region has no independent effect on asthma at age 18 years; however in interaction with the CpG site cg09791102 (gene body, Table 1) it is strongly associated with asthma in female participants. At 92% methylation level, rs3024685 (‘CC’ genotype compared to ‘TT’) showed a 46.9-fold increase risk for asthma. Our observation of a role of gene-body methylation is further supported by the emerging evidence, which shows that methylation in intragenic regions can be positively correlated with gene expression levels and phenotype variation [21, 22]. Intragenic DNA methylation has been linked to ‘exon definition’ through interaction with auxiliary proteins, by which DNA methylation in the body may result in alternative pre-mRNA splicing regulation (for example, inclusion or exclusion of exons) [23–25]. We assume that a higher DNA-M may mask an otherwise protective effect of rs3024685 and thus increases the risk of asthma [26]. Our results indicate that considering both genetic variants and DNA methylation will significantly improve the explanation of asthma. Replication of these findings in an independent study population is needed to validate the interplay of DNA methylation with genetic polymorphism, which results in an increased asthma risk. However, currently there are only few studies that can provide both genetic and DNA methylation data.
A limitation of our study is that the RRs at methylation levels larger than 90% are high, which is due to the limited number of individuals (n = 36) with methylation levels larger than 90%. Evidence of selection bias is absent since prevalence of asthma and IL4R SNPs is comparable between those analyzed in this study and those from the original cohort. Multiple testing was a concern since we tested the joint effect of differential DNA methylation of cg09791102 and eight IL4R SNPs separately (a total of eight tests). Nevertheless, the observed increased risk remained statistically significant after penalizing its P value for false discovery rate. Regarding reliability and specificity of methylation status of CpG sites, a recent report demonstrated that the Infinium HumanMethylation450 array, which was used to obtained DNA methylation profiles in this study, had strong reproducibility and high validity [27]. The extent to which DNA methylation measured in blood relate to other tissues and whether can be used as a biomarker for phenotype variation is unclear and is an area of current scientific dispute [28–30].
Conclusions
The strong interaction of one SNP and DNA-M is encouraging and provides a novel model how a joint effect of genetic variants and DNA-M can explain asthma. Although the sample size is limited and focused on female participants, our results should generally motivate other studies to replicate the interaction we found, while also searching for new interactions between genetic variants and DNA methylation, in particular for the IL4R gene and asthma.
Abbreviations
- CI:
-
Confidence interval
- CpG:
-
Cytosine-phosphate-guanine
- DNA-M:
-
DNA methylation
- GWAS:
-
Genome-wide association study
- IL4R:
-
Interleukin-4 receptor
- ISAAC:
-
International Study of Asthma and Allergies in Childhood
- LD:
-
Linkage disequilibrium
- RR:
-
Risk ratio
- SNP:
-
Single nucleotide polymorphisms
- UTR:
-
Untranslated region.
References
Ober C, Yao TC: The genetics of asthma and allergic disease: a 21st century perspective. Immunol Rev. 2011, 242: 10-30. 10.1111/j.1600-065X.2011.01029.x.
Bjornson CL, Mitchell I: Gender differences in asthma in childhood and adolescence. J Gend Specif Med. 2000, 3: 57-61.
Michel S, Liang L, Depner M, Klopp N, Ruether A, Kumar A, Schedel M, Vogelberg C, von Mutius E, von Berg A, Bufe A, Rietschel E, Heinzmann A, Laub O, Simma B, Frischer T, Genuneit J, Gut IG, Schreiber S, Lathrop M, Illig T, Kabesch M: Unifying candidate gene and GWAS approaches in asthma. PLoS One. 2010, 5: e13894-10.1371/journal.pone.0013894.
Wu H, Romieu I, Shi M, Hancock DB, Li H, Sienra-Monge JJ, Chiu GY, Xu H, del Rio-Navarro BE, London SJ: Evaluation of candidate genes in a genome-wide association study of childhood asthma in Mexicans. J Allergy Clin Immunol. 2010, 125: e313-e327.
Vercelli D: Discovering susceptibility genes for asthma and allergy. Nat Rev Immunol. 2008, 8: 169-182. 10.1038/nri2257.
Perkins C, Yanase N, Smulian G, Gildea L, Orekov T, Potter C, Brombacher F, Aronow B, Wills-Karp M, Finkelman FD: Selective stimulation of IL-4 receptor on smooth muscle induces airway hyperresponsiveness in mice. J Exp Med. 2011, 208: 853-867. 10.1084/jem.20100023.
Loza MJ, Chang BL: Association between Q551R IL4R genetic variants and atopic asthma risk demonstrated by meta-analysis. J Allergy Clin Immunol. 2007, 120: 578-585. 10.1016/j.jaci.2007.05.019.
Rosa-Rosa L, Zimmermann N, Bernstein JA, Rothenberg ME, Khurana Hershey GK: The R576 IL-4 receptor alpha allele correlates with asthma severity. J Allergy Clin Immunol. 1999, 104: 1008-1014. 10.1016/S0091-6749(99)70082-5.
Kabesch M, Michel S, Tost J: Epigenetic mechanisms and the relationship to childhood asthma. Eur Respir J. 2010, 36: 950-961. 10.1183/09031936.00019310.
Kurukulaaratchy RJ, Fenn MH, Waterhouse LM, Matthews SM, Holgate ST, Arshad SH: Characterization of wheezing phenotypes in the first 10 years of life. Clin Exp Allergy. 2003, 33: 573-578. 10.1046/j.1365-2222.2003.01657.x.
Arshad SH, Hide DW: Effect of environmental factors on the development pf allergic disorders in infancy. J Allergy Clin Immunol. 1992, 90: 235-241. 10.1016/0091-6749(92)90077-F.
Asher MI, Keil U, Anderson HR, Beasley R, Crane J, Martinez F, Mitchell EA, Pearce N, Sibbald B, Stewart AW: International Study of Asthma and Allergies in Childhood (ISAAC): rationale and methods. Eur Respir J. 1995, 8: 483-491. 10.1183/09031936.95.08030483.
Barrett JC, Fry B, Maller J, Daly MJ: Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005, 21: 263-265. 10.1093/bioinformatics/bth457.
Miller SA, Dykes DD, Polesky HF: A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988, 16: 1215-10.1093/nar/16.3.1215.
Bibikova M, Fan JB: GoldenGate assay for DNA methylation profiling. Methods Mol Biol. 2009, 507: 149-163. 10.1007/978-1-59745-522-0_12.
Kuan PF, Wang S, Zhou X, Chu H: A statistical framework for Illumina DNA methylation arrays. Bioinformatics. 2010, 26: 2849-2855. 10.1093/bioinformatics/btq553.
Hill WG, Robertson A: The effect of linkage on limits to artificial selection. Genet Res. 1966, 8: 269-294. 10.1017/S0016672300010156.
Benjamini Y, Hochberg Y: Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Roy Stat Soc Ser B Meth. 1995, 57: 289-300.
R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing: R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing. 2012, R Development Core Team, Vienna,http://www.R-project.org,
Bell JT, Pai AA, Pickrell JK, Gaffney DJ, Pique-Regi R, Degner JF, Gilad Y, Pritchard JK: DNA methylation patterns associate with genetic and gene expression variation in HapMap cell lines. Genome Biol. 2011, 12: R10-10.1186/gb-2011-12-1-r10.
Jjingo D, Conley AB, Yi SV, Lunyak VV, Jordan IK: On the presence and role of human gene-body DNA methylation. Oncotarget. 2012, 3: 462-474.
Shenker N, Flanagan JM: Intragenic DNA methylation: implications of this epigenetic mechanism for cancer research. Br J Cancer. 2012, 106: 248-253. 10.1038/bjc.2011.550.
Oberdoerffer S: A conserved role for intragenic DNA methylation in alternative pre-mRNA splicing. Transcription. 2012, 3: 106-109. 10.4161/trns.19816.
Lyko F, Foret S, Kucharski R, Wolf S, Falckenhayn C, Maleszka R: The honey bee epigenomes: differential methylation of brain DNA in queens and workers. PLoS Biol. 2010, 8: e1000506-10.1371/journal.pbio.1000506.
Shukla S, Kavak E, Gregory M, Imashimizu M, Shutinoski B, Kashlev M, Oberdoerffer P, Sandberg R, Oberdoerffer S: CTCF-promoted RNA polymerase II pausing links DNA methylation to splicing. Nature. 2011, 479: 74-79. 10.1038/nature10442.
Berlivet S, Moussette S, Ouimet M, Verlaan DJ, Koka V, Al Tuwaijri A, Kwan T, Sinnett D, Pastinen T, Naumova AK: Interaction between genetic and epigenetic variation defines gene expression patterns at the asthma-associated locus 17q12-q21 in lymphoblastoid cell lines. Hum Genet. 2012, 131: 1161-1171. 10.1007/s00439-012-1142-x.
Bibikova M, Barnes B, Tsan C, Ho V, Klotzle B, Le JM, Delano D, Zhang L, Schroth GP, Gunderson KL, Fan JB, Shen R: High density DNA methylation array with single CpG site resolution. Genomics. 2011, 98: 288-295. 10.1016/j.ygeno.2011.07.007.
Terry MB, Delgado-Cruzata L, Vin-Raviv N, Wu HC, Santella RM: DNA methylation in white blood cells: association with risk factors in epidemiologic studies. Epigenetics. 2011, 6: 828-837. 10.4161/epi.6.7.16500.
Heijmans BT, Mill J: Commentary: the seven plagues of epigenetic epidemiology. Int J Epidemiol. 2012, 41: 74-78. 10.1093/ije/dyr225.
Talens RP, Boomsma DI, Tobi EW, Kremer D, Jukema JW, Willemsen G, Putter H, Slagboom PE, Heijmans BT: Variation, patterns, and temporal stability of DNA methylation: considerations for epigenetic epidemiology. FASEB J. 2010, 24: 3135-3144. 10.1096/fj.09-150490.
Acknowledgements
Research reported in this publication was supported by the National Institute of Allergy and Infectious Diseases under Award Number R01 AI091905-01 (PI: Wilfried Karmaus) and R01 AI061471 (PI: Susan Ewart). The 10-year follow-up of this study was funded by National Asthma Campaign, UK (Grant No 364) and the 18-year follow-up by NIH/NHLBI R01 HL082925-01 (PI: S. Hasan Arshad). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The authors gratefully acknowledge the cooperation of the children and parents who participated in this study, and appreciate the hard work of Mrs. Sharon Matthews and the Isle of Wight research team in collecting data and Nikki Graham for technical support. We thank the High-Throughput Genomics Group at the Wellcome Trust Centre for Human Genetics (funded by Wellcome Trust grant reference 090532/Z/09/Z and MRC Hub grant G0900747 91070) for the generation of the methylation data.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
NSR conducted the statistical analysis, interpreted the data, and drafted the manuscript. SHA and VP contributed to the clinical interpretation and helped with the manuscript. JH supervised the assessment of the DNA methylation and revised the manuscript. HZ directed the statistical analysis and aided in their interpretation and the final editing. SE and ES selected and measured the single nucleotide polymorphisms. SE and SHA contributed to funding acquisition and the manuscript. WK designed the study, reviewed the data quality, helped with statistical analyses, and revised the manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Soto-Ramírez, N., Arshad, S.H., Holloway, J.W. et al. The interaction of genetic variants and DNA methylation of the interleukin-4 receptor gene increase the risk of asthma at age 18 years. Clin Epigenet 5, 1 (2013). https://doi.org/10.1186/1868-7083-5-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1868-7083-5-1