
Abstract
Genomic instability is a common feature of cancer etiology. This provides an avenue for therapeutic intervention, since cancer cells are more susceptible than normal cells to DNA damaging agents. However, there is growing evidence that the epigenetic mechanisms that impact DNA methylation and histone status also contribute to genomic instability. The DNA damage response, for example, is modulated by the acetylation status of histone and non-histone proteins, and by the opposing activities of histone acetyltransferase and histone deacetylase (HDAC) enzymes. Many HDACs overexpressed in cancer cells have been implicated in protecting such cells from genotoxic insults. Thus, HDAC inhibitors, in addition to unsilencing tumor suppressor genes, also can silence DNA repair pathways, inactivate non-histone proteins that are required for DNA stability, and induce reactive oxygen species and DNA double-strand breaks. This review summarizes how dietary phytochemicals that affect the epigenome also can trigger DNA damage and repair mechanisms. Where such data is available, examples are cited from studies in vitro and in vivo of polyphenols, organosulfur/organoselenium compounds, indoles, sesquiterpene lactones, and miscellaneous agents such as anacardic acid. Finally, by virtue of their genetic and epigenetic mechanisms, cancer chemopreventive agents are being redefined as chemo- or radio-sensitizers. A sustained DNA damage response coupled with insufficient repair may be a pivotal mechanism for apoptosis induction in cancer cells exposed to dietary phytochemicals. Future research, including appropriate clinical investigation, should clarify these emerging concepts in the context of both genetic and epigenetic mechanisms dysregulated in cancer, and the pros and cons of specific dietary intervention strategies.
Similar content being viewed by others

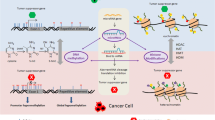
Introduction
Genomic instability is a key feature of cancer development, often associated with the acquisition of mutations in oncogenes, tumor suppressor genes, and DNA repair genes [1]. Thus, DNA repair pathways and cell cycle checkpoint controls have important consequences for genome stability, and have come under much scrutiny [2]. Defects in genome stability increase the sensitivity of cells to DNA damaging agents, and provide an "Achilles heel" for cancer therapeutics [3]. Indeed, there are numerous efforts to manipulate the DNA damage response so as to selectively induce tumor cell death through catastrophic genomic instability [4, 5]. Differences in the DNA damage response between normal cells and cancer cells often underlie the utility of DNA damaging agents in cancer treatment. Radiotherapy and chemotherapeutic drugs are known to function by DNA damage-induced tumor cell death, and there are ongoing efforts to improve sensitivity while overcoming resistance to these agents. Poly(ADP-ribose)polymerase (PARP) inhibitors that target defects in double-strand break repair in women with hereditary breast cancer [6] illustrate the concept of selective "synthetic lethality". Other examples include inhibitors of apurinic/apyrimidinic endonuclease-1 (APE1), DNA repair protein RecA homolog (RAD51), ataxia-telangiectasia mutated (ATM), and DNA-dependent protein kinase (DNAPK), some of which have entered clinical trials. As we learn more about the DNA damage response pathways dysregulated in cancer cells, new combinations of agents are being developed with enhanced therapeutic efficacy [7].
Epigenetic mechanisms influence DNA damage and repair pathways; the reader is referred to related reviews in the current journal [8–10]. In eukaryotic cells, DNA damage repair occurs in the context of chromatin, and it is now clear that DNA damage response impacts specific aspects of chromatin structure and chromatin remodeling. Post-translational histone modifications, histone variants, and chromatin-binding proteins provide a regulatory platform for controlling DNA template-directed processes, including gene transcription, DNA replication, and repair of DNA damage [11, 12]. Such responses may be mediated by chromatin modifiers involved in histone methylation, acetylation and biotinylation [13–15].
Recently, it was reported that histone deacetylase (HDAC) inhibitors have the potential to interfere with DNA repair mechanisms [16]. A recent review summarized the ways in which HDAC inhibitors trigger apoptosis by taking advantage of genomic instability in cancer cells [14]. The latter review highlighted the ways in which HDAC inhibitors lead to impaired mitotic progression, defects in kinetochore assembly, and aberrations in spindle assembly checkpoint controls, resulting in premature exit from mitosis. HDAC inhibitors regulate chromatin structure and activate the DNA damage checkpoint pathway involving ATM [17]. Histone acetyltransferase (HAT) inhibition also has been shown to impair double-strand break repair [18]. Damage signaling involves phosphorylation of H2AX(S139) (γH2AX) by ATM/ATR kinases. This is followed by chromatin opening and the involvement of H3/H4 acetylation, via HATs such as Tip60, GCN5 and CBP/p300. Chromatin restoration after repair involves γH2AX dephosphorylation by phosphatases PP4 and PP2A and deacetylation of H3/H4 lysines by HDACs. Additional histone modifications such as ubiquitination and sumoylation of histones also contribute to this process. Details of this process have been extensively reviewed elsewhere [19] and dealt with in more detail in the next section.
Similarly, acetylation of non-histone proteins can influence chromatin dynamics, protein turnover, and the DNA damage response. Robert et al [20] have recently shown in yeast that depletion of class I and II HDACs by mutation, or via HDAC inhibition with valproic acid (VPA), prevented DNA damage signaling and interfered with DNA break repair. The DNA resection and recombination protein Sae2 (human C-terminal binding protein interacting protein, CtIP) was acetylated, resulting in increased protein turnover and degradation by autophagy. Deacetylation by HDACs stabilized Sae2, but VPA inhibited this process [20]. Consistent with these observations, a recent study showed that a class III HDAC (SIRT6) positively regulated the repair of double-strand breaks (DSBs) through deacetylation of CtIP [21].
The investigation of genome stability and epigenetics dovetails with mechanistic studies on diet and nutrition. Based on epidemiological studies, diets rich in fruits and vegetables can offer protection against cancer development [22–25]. Recent reviews have covered the mechanisms of dietary agents impacting DNA methyltransferases, HDAC or HAT enzymes, and microRNAs [26–29]. In the context of DNA damage, a folate/methyl deficient diet has been conclusively shown to cause genomic instability [30]. Although dietary anticancer compounds modulate drug metabolizing enzymes and scavenge free radicals, under some conditions they have been shown to generate reactive oxygen species (ROS) and cause oxidative DNA damage [31, 32].
Given this background, the present review summarizes recent advances in our understanding of HDACs involved in the DNA damage response, and the possible implications for cancer therapy. Targeting genome integrity in rapidly cycling cells has been a central feature of cancer therapeutics. However, a growing area of interest is the dietary agents that can trigger a DNA damage response via epigenetic mechanisms, involving altered HDAC/HAT activities.
Changes in chromatin structure during DNA damage
DNA wraps around an octameric complex of core histones H2A, H2B, H3 and H4 to form nucleosomes. DSBs induced by ROS, replication stress, or by exogenous agents like UV, radiation, radiotherapy, or other genotoxic agents are thought to be the most dangerous lesions for genomic integrity [33]. Although the exact sequence of events following DSB is still poorly understood, one of the earliest events in the response to DNA breakage involves phosphorylation of H2AX (γH2AX) that surrounds ~2 Mb of each DSB, which marks the sites of breakage [34, 35]. Thus, a common biomarker for DNA-damage nuclear foci is γH2AX, typically assayed by immunofluorescence-based approaches. H2AX is phosphorylated by phosphatidylinositol-3 kinase (PI3K)-like kinases, including ATM, ATM-and Rad3-related (ATR), ATM related kinase (ATX), and DNAPK [36, 37]. Histone H3 acetylation at tail residues K9, K14, K18, K23 and K27, and histone H4 acetylation at tail residues K5, K8, K12, K16 [38] reduces their affinity for negatively-charged DNA. This in turn promotes relaxation of chromatin, and facilitates access of repair proteins. The HAT complex TAT-interacting protein 60 (TIP60) acetylates histones H2A, H3, and H4 [39, 40], whereas HDACs participate in histone deacetylation during repair and chromatin reassembly [41–43], as shown in Figure 1.

Model of DNA damage signaling, histone acetylation and chromatin remodeling. Recognition and signaling of a DSB is followed by opening of chromatin to repair the break, terminating in chromatin restoration after DNA break repair. HDAC inhibition, as indicated by HDACI, has been shown to affect key steps in this process (as illustrated) by virtue of deacetylating histone and non-histone proteins involved in signaling and repair.
H2AX phosphorylation and core histone acetylation assist in the recruitment to DSB sites of chromatin remodeling complexes of the SWI2/SNF2 superfamily [44–46]. This is followed by the accumulation of other PI3K-like members, mediator of DNA damage checkpoint protein 1 (MDC1) or p53-binding protein (53BP1), which play a pivotal role in signaling DSBs [47]. DSB signaling is further amplified by transducer checkpoint kinases, CHK1 and CHK2, which, together with ATM and ATR, phosphorylate breast cancer 1 (BRCA1), RAD51, p53, and its negative regulator, murine double minute (Mdm2) [48]. Phosphorylation of p53 leads to its stabilization, causing cell cycle arrest through induction of cyclin-dependent kinase inhibitor p21 or in the event of severe DNA damage, apoptosis.
DNA damage is sensed and the repair machinery is employed, consisting of MRE11-RAD50-Nbs1 (MRN) mediator complexes or RAD51 enzymes [49, 50] that recruit ATM to the site of DSBs [51]. Histone ubiquitylation, through ubiquitin ligases RNF8 and 168, is an important route for recruitment of additional repair complexes involving BRCA1/Abraxas/Rap80 [52, 53].
Other events include mobilization of high-mobility group N1 (HMGN1) protein for ATM recruitment, and heterochromatin protein 1β (HP1β) [54]. The histone trimethylation mark, H3K9me3, is recognized by chromodomain regions of HP1 and casein kinase 2 (CK2) that mediate the removal of HP1 protein [55]. The recruitment and activation of ATM at DSB sites affects chromatin structure by phosphorylating the KRAB associated protein (KAP-1), thus further relaxing the chromatin structure [56]. Acetylation of histone H3K56 drives chromatin assembly after repair, and signals the completion of repair [57].
The mechanisms that restore chromatin architecture after repair of DSBs involve deacetylation by HDACs [58], proteasomal degradation of MDC1 foci [59, 60], and turnover of the repair machinery. Chromatin assembly factors, including the histone chaperones chromatin assembly factor I (CAF-1) and anti-silencing function 1(Asf1), play essential roles in restoring chromatin structure and cell cycle progression after DNA repair [57].
Role of HDACs in DNA damage response
Acetylation is a reversible process in which histone and non-histone protein acetyltransferases transfer the acetyl moiety from acetyl co-enzyme A to lysine residues, and HDACs remove the acetyl groups. HDACs play major roles in modulating chromatin accessibility during transcription, replication, recombination, and repair [61, 62]; however, the role of individual HDACs in these processes is still unclear.
At the present time, 18 HDACs have been identified in humans that fall into four classes: class I HDACs (HDAC1, 2, 3 and 8) share sequence similarity with the yeast RPD3 deacetylase, are ubiquitously expressed, and they are localized mainly in the nucleus. Class II HDACs (HDAC4, 5, 6, 7, 9 and 10) are homologous to the yeast Hda1 deacetylase, are nuclear and cytoplasmic, and restricted to certain tissues. Class II HDACs are further subdivided into class IIa (HDAC4, 5, 7 and 9) and class IIb (HDAC6 and 10). Class III HDACs are represented by sirtuins (SIRT1 to SIRT7), a family of seven HDACs sharing homology with yeast silent information regulator 2 (Sir2). Class IV has only one member, HDAC11, which shares conserved residues with both class I and II HDACs [63]. Class I, II, and III HDACs have been implicated in the DNA damage response, homologous recombination (HR), and chromatin integrity. This is explained below, and summarized in Table 1.
An important substrate of HDAC1 is the tumor suppressor protein p53. Recruitment of HDAC1 by MDM2 promotes p53 degradation by deacetylation. Thus, HDAC1 decreases DNA damage-induced p53 acetylation, and inhibits the induction of p21 and MDM2 [64]. HDAC1 also regulates several other proteins involved in the DNA damage response, such as proliferating cell nuclear antigen (PCNA) [65], BRCA1 [66], ATM [67], ATR [68], inhibitor of growth 1a (ING1a) [69], replication factor C (RFC) [70], apurinic apyrimidinic endonuclease redox effector factor-1 (APE1/Ref1) [71], and proteins that facilitate non-homologous end-joining (NHEJ) by altering histone H3K56 acetylation [41].
Miller et al. [41] showed that HDAC1 and HDAC2 cooperate in the DNA damage response. Specifically, HDAC1 and HDAC2 were recruited to DNA damage sites and regulated the deacetylation of H3K56 and H4K16, a requirement for DNA repair, particularly through NHEJ. HDAC2 also regulates ATR [68], and alters histone H3K56 acetylation status during the DNA damage response. Based on their findings, the authors suggested that HDAC1 and HDAC2 might repress transcription at sites of DNA damage, thereby preventing transcription from interfering with repair processes, as well as remodeling chromatin into a state that promotes NHEJ. They found that Class I/II HDAC inhibitors, such as butyrate and trichostatin A (TSA), caused defects in the DNA damage response, including hyperacetylation of H3K56 and H4K16, and impairment of NHEJ. Furthermore, HDAC1- and 2-depleted cells were hypersensitive to DNA-damaging agents and showed sustained DNA-damage signaling, phenotypes that reflect defective DSB repair. The authors discussed the potential implications of their findings for HDAC1- and HDAC2-specific therapy [41].
Bhaskara et al. [42, 72] showed that HDAC3 is important for DSB repair. HDAC3 associates with nuclear receptor corepressor (NCOR) and silencing mediator for retinoic and thyroid receptor (SMRT) [73], and is considered a locus-specific corepressor that is recruited to promoters to repress genes regulated by nuclear hormone receptors and other transcription factors [74]. Conditional deletion demonstrated the absolute requirement for cell viability of HDAC3 in murine embryonic fibroblasts (MEFs) [72]. The latter MEFs underwent apoptosis due to impaired S phase progression and formation of DSBs, rather than altered transcriptional programs. The DNA damage was blocked when cells were taken out of the cell cycle by serum starvation, suggesting that HDAC3 acted during S phase. In another study [42], HDAC3-null MEFs increased histone acetylation (H3K9, H3K14, H4K5 and H4K12) in late S phase. Knockdown of NCOR1 and SMRT increased acetylated H4K5 and caused DNA damage, indicating that the HDAC3/NCOR/SMRT axis may be critical for maintaining chromatin structure and genomic stability. Furthermore, two studies have linked HDAC3 to maintenance of the mitotic spindle assembly [75, 76]. Ishii et al. [75] reported on the localization of HDAC3 to the mitotic spindle, and showed that HDAC3 knockdown led to chromosome misalignment, impaired kinetochore-microtubule attachment, and mitotic spindle collapse. Eot-Houllier et al. [76] showed that HDAC3 knockdown induced spindle assembly checkpoint activation and sister chromatid dissociation. Further, down-regulation of HDAC3 mimics actions of the HDAC inhibitor suberoylanilide hydroxamic acid (SAHA, vorinostat) in reducing replication fork velocity and increasing origin firing at sites of replication, likely due to chromatin changes [77].
Among the class II HDACs, HDAC4, and more recently HDAC6, HDAC9 and HDAC10, have been implicated in DNA damage signaling, transcription factor binding, and DNA repair processes [78–80, 43]. Kao et al. [78] showed that HDAC4 co-localized with 53BP1, a PI3K-like member with a pivotal role in signaling DSBs. HDAC4-containing foci gradually disappeared in repair-proficient cells, but persisted in repair-deficient cell lines, suggesting that resolution of HDAC4 foci is linked to successful DNA repair. Silencing of HDAC4 via RNA interference surprisingly also decreased levels of 53BP1 protein, abrogated the DNA damage-induced G2 delay, and radiosensitized HeLa cells. These observations showed that HDAC4 is a critical component of the DNA damage response pathway that acts through 53BP1, and perhaps contributes in maintaining the G2 cell cycle checkpoint. Basile et al. [79] demonstrated that HDAC4 shuttles from the cytoplasm to the nucleus following DNA damage, independent of p53 activation, and becomes associated with gene promoters via a p53-dependent mechanism. Thus, HDAC4 is clearly implicated as a component of the DNA damage response.
Namdar et al. [80] reported that HDAC6 inhibition with tubacin or shRNA activated the intrinsic apoptosis pathway in cancer cells; this led to accumulation of γH2AX, and the expression of growth arrest and DNA damage 153 (GADD153/DDIT3), a transcription factor upregulated in response to cellular stress. Tubacin treatment enhanced cell death induced by topoisomerase II inhibitors etoposide and doxorubicin, and by the pan-HDAC inhibitor SAHA, in transformed cells (LNCaP, MCF-7), an effect not observed in normal cells (human foreskin fibroblast cells). Further, tubacin increased the accumulation of γH2AX and activated Chk2. GADD153/DDIT3 induction was augmented when tubacin was combined with SAHA. The authors suggested that HDAC6-selective inhibition enhances the efficacy of certain anticancer agents in transformed cells [80].
Recently, Kotian et al. [43] showed that depletion of HDAC9 or HDAC10 inhibited HR in a tissue-culture based homology-directed repair assay. The authors showed that HDAC9 and HDAC10 were directly involved in the HR process, and this was not through indirect blocking of the cell cycle. Further, depletion of HDAC9 or HDAC10 resulted in increased sensitivity to mitomycin C [43].
Among the NAD+-dependent class III HDACs (sirtuins), SIRT1, SIRT3 and SIRT6 have definite roles in genome stability and repair [21, 81–84]. SIRT1 plays crucial roles in multiple biological processes affecting gene transcription, cellular metabolism, stress response, and tumorigenesis. SIRT1 is overexpressed in several p53-deficient tumor cell lines, and the transient knockdown of SIRT1 leads to increased apoptosis after DNA damage or oxidative stress [85]. Moreover, several proteins involved in the DNA damage response are deacetylated and inactivated by SIRT1. These targets include p53 [86, 87], forkhead box transcription factor (FoxO) [88, 89], the nonhomologous end joining (NHEJ) factor, Ku70 [90], Tip60 [91], the histone modification H3K56 acetylation [92], and MRN repair complex [93]. Thus, these studies support the idea that SIRT1 can act as an oncogenic protein when overexpressed in cancer cells.
SIRT1 also is thought to act as a tumor suppressor in some scenarios, through its role in deacetylating p53 [94] and Ku70 [90]. CK2 phosphorylates and activates SIRT1, and partly protects cells from ionizing radiation-induced apoptosis [95], whereas Set7/9 methylates SIRT1 and disrupts it's binding to p53, leading to p53 acetylation and activation in response to DNA damage [96]. Several recent reports have shown the importance of SIRT1 in enhancing DNA repair [81–83, 97]. Palacios et al. [81] examined the effects of SIRT1 on telomere maintenance and DNA repair. Using SIRT1-deficient and gain-of-function mouse models, SIRT1 was identified as a positive regulator of telomere length in vivo, and attenuated telomere shortening associated with aging. The authors showed that SIRT1 interacted with telomeric repeats in vivo. In addition, SIRT1 overexpression increased HR throughout the entire genome, including telomeres, centromeres, and chromosome arms. These findings link SIRT1 to telomere biology and global DNA repair, and provide new mechanistic insights into the known functions of SIRT1 in the protection from DNA damage [81]. Uhl et al. [82] showed that Werner helicase (WRN) was required for SIRT1-mediated HR. WRN, in its mutated form, causes premature aging and cancer, and has been linked to Rad51-independent single-strand annealing (SSA) DSB repair pathway. SIRT1 also regulates other DNA repair pathways, viz. base-excision repair (BER) and nucleotide-excision repair (NER) [83, 97, 98] through the transcription of xeroderma pigmentosum (XPA, XPC) group proteins [97, 98]. Yamamori et al. [83] showed that SIRT1 plays a vital role in maintaining genomic integrity by deacetylating APE1, which is an essential component of the BER pathway. Increased association of SIRT1 with APE1 during genotoxic stress facilitated SIRT1-mediated deacetylation of APE1 in vitro and in vivo, thereby reducing genotoxic insult-stimulated lysine acetylation of APE1. Fan and Luo [97] showed that SIRT1 plays an important role in the regulation of NER. Thus, downregulation of SIRT1 significantly sensitized cells to UV irradiation through interaction with xeroderma pigmentosum group A (XPA), a core factor essential for NER. SIRT1 has been shown to deacetylate XPA both in vitro and in vivo[97].
SIRT3 is transported from the nucleus to the mitochondria upon cellular stress, as in the case of DNA damaging agents, and deacetylates histone H4K16 [99]. SIRT3 has been shown to deacetylate and activate mitochondrial isocitrate dehydrogenase 2 (Idh2), leading to increased NADPH levels and an increased glutathione GSH:GSSG ratio in mitochondria, thereby protecting cells from oxidative stress-induced cell death. SIRT3 is thus an essential player in the mitochondrial glutathione antioxidant defense system [100].
Kaidi et al. [21] have shown that human SIRT6 has a role in promoting DNA end-resection, a crucial step in DSB repair by HR. SIRT6 depletion impaired the accumulation of replication protein A (RPA) and single-stranded DNA at damage sites, reduced the rate of HR, and sensitized cells to DSB-inducing agents. The authors identified CtIP as a SIRT6 interaction partner, and showed that SIRT6-dependent CtIP deacetylation promotes DSB resection. Schwer et al. [101] have shown that SIRT6 deletion causes hyperacetylated histone H3K9 and H3K56, two chromatin marks implicated in the regulation of gene activity and chromatin structure, in various brain regions. McCord et al. [102] observed that SIRT6 forms a complex with DNAPK and promotes DSB repair. In addition, the role of SIRT6 in genomic stability has been demonstrated in aging mouse models [84].
Collectively, these studies highlight the roles of multiple HDACs in the DNA damage response and chromatin stability. As a corollary, the question arises as to how such events might be impacted by HDAC inhibitors.
HDAC inhibitors and the DNA damage response
HDAC inhibitors are being developed as anticancer agents, as well as therapies for non-oncologic disorders [63, 103]. Inhibitors of the zinc-dependent HDACs belong to several chemical classes, including hydroxamic acids, cyclic peptides, electrophilic ketones, short-chain fatty acids, and benzamides. Some of these inhibitors affect the interactions of HDACs with protein partners, independent of the deacetylase activity [63]. Thus, HDAC inhibitor mechanisms now include competitive binding in the active site [104], turnover of the HDAC protein by proteasomal degradation [105], and HDAC protein inactivation by alkylation/carbonylation [106, 107]. These HDAC regulatory mechanisms are not necessarily mutually exclusive.
HDAC inhibitors can induce growth arrest of neoplastically-transformed cells and trigger apoptosis via one or more pathways. These events are associated with altered patterns of acetylation in histone and non-histone proteins, including key players involved in the regulation of gene expression, apoptosis, cell cycle progression, redox signaling, mitotic division, DNA repair, cell migration, and angiogenesis [63]. Subsequent to the role of HDACs in maintaining genome stability, as discussed above, histone hyperacetylation induced by HDAC inhibitors causes structural alterations in chromatin. This can open up regions of DNA that are normally protected by heterochromatin, enabling DNA-damaging agents to gain access to the exposed template. Importantly, HDAC inhibitors have been shown to decrease the expression of DNA repair proteins such as Ku70 [108], BRCA1 [109], RAD51 [110] and CtIP [20]. It is not clear whether transcription mediates HDAC inhibitor actions in these circumstances [111], and indeed non-transcriptional targets of HDAC inhibitors have been proposed [112, 113]. Thus, HDAC inhibitors have the potential to target multiple signaling and repair mechanisms in the DNA damage pathway by targeting histones and non-histone proteins, as illustrated in Figure 1.
Several pharmacologic HDAC inhibitors are undergoing clinical trials as monotherapies, or in combination therapies with other anticancer agents. Two of these HDAC inhibitors, vorinostat and romidepsin (depsipeptide), have been approved for the treatment of cutaneous T-cell lymphoma [114]. Apart from effects on gene transcription, evidence is accumulating that HDAC inhibitors influence chromatin stability, mitosis, and DNA repair mechanisms. For example, vorinostat acts at replication origins [77], downregulates the DNA repair gene Rad52[115], and suppresses HR repair genes such as Brca1, Rad51, Chk1, and Bubr1 (a checkpoint kinase), via downregulation of E2F1 transcription factor [16]. These effects also have been reported for other HDAC inhibitors, such as PCI-24781 [110] and VPA [16]. Romidepsin downregulated thioredoxin reductase (TrxR), generated ROS accumulation, and augmented DNA damage and apoptosis [116]. The HDAC inhibitor LAQ-824 also triggered ROS production, with increased γH2AX and Ku70 acetylation [117]. Many other HDAC inhibitors, including TSA, SAHA and MS-275, augment the acetylation of Ku70 [108] and alter genes encoding HR components, such as ATR, Bloom syndrome gene (BLM), BRCA1, BRCA2, and nijmegen breakage syndrome 1(NBS1) [109].
Dietary agents and their effects on chromatin, DNA damage, and repair in cancer cells
In addition to the potent HDAC inhibitor drugs being developed as cancer therapeutic agents, there is growing interest in dietary phytochemicals that also possess HDAC inhibitor activity [118, 119, 26]. A synopsis of dietary chemopreventive agents in the context of DNA damage and repair pathways is shown in Figure 2, and is summarized below for specific chemical classes. As discussed next, some dietary compounds have shown DNA-damaging effects in cancer cells associated with HDAC inhibition. The order in which the compounds are presented below corresponds with the approximate extent of supportive evidence from the literature for HDAC/HAT modulation and DNA damage end-points.

The role of dietary factors in altering histone acetylation and DNA damage signaling.
Isothiocyanates
Brassica or cruciferous vegetables are a rich source of glucosinolates [120]. The hydrolysis of these glucosinolates by the plant enzyme myrosinase generates biologically active isothiocyanates (ITC) and indoles [121]. For example, ITC precursors of sulforaphane (SFN) and phenethyl isothiocyanate (PEITC) are found at high levels in broccoli and watercress, respectively. Epigenetic effects of ITCs have been linked to the inhibition of HDAC activity and histone hyperacetylation, as reported for SFN [122], allyl isothiocyanate (allyl-ITC) [123], benzyl isothiocyanate (BITC) [124], phenylhexyl isothiocyanate (PHITC) [125], PEITC [126], and other longer-chain isothiocyanates [118]. In addition to altering HDAC expression and causing histone acetylation, other histone marks altered by ITCs include histone methylation [127]. BITC [124] and SFN [128, 129] have also been shown to decrease HDAC protein expression in cancer cells.
We know from previous studies that methyl isothiocyanate [130], BITC [131], allyl-ITC and PEITC [132] exert genotoxic effects. For example, BITC (10 μM) increased γH2AX and triggered apoptosis in Capan-2 pancreatic cells [133]. It caused a significant decrease in the expression and activity of HDAC1 and HDAC3, as well as NFκB inactivation, in pancreatic cancer cells but not normal cells. Interestingly, overexpression of HDAC1 or HDAC3 blocked these effects [124].
SFN has been shown to cause both DSBs and single-strand breaks (SSBs) in cancer cells. Sekine-Suzuki et al. [134] observed that 20 μM SFN triggered cell cycle arrest, induced DSB, and elevated γH2AX levels in cervical cancer (HeLa) cells. DSBs generated by SFN were comparable to that triggered with 12 Gy of X-rays. These DSBs were repaired mainly by HR through Rad51 foci formation and not by NHEJ [134]. Sestili et al. [135] reported that a short exposure of cells with SFN (10-30 μM for 1-3 h) triggered SSBs in Jurkat lymphoma and HUVEC cells. They found that DNA damage was causally linked to ROS generation and GSH depletion [135]. DSBs also were triggered in colon cancer cell lines SW620 at 10-50 μM [136] and HCT116 cells at 15 μM SFN, resulting in sustained γH2AX expression (our unpublished data). In prostate cancer cells, SFN-induced DNA damage involved the Chk2-mediated phosphorylation of protein phosphatase Cdc25C [137].
We recently reported SFN-induced loss of HDAC3 and HDAC6 protein expression in a time-dependent manner in HCT116 colon cancer cells, leading to acetylation of histone H4 and tubulin, respectively. By 6 h, SFN was shown to enhance CK2/HDAC3 binding, leading to HDAC3 phosphorylation and nuclear export by 14-3-3 and Pin1 [128]. As noted earlier, this has the potential to affect chromatin structure and DNA repair since HDAC3 is critical for chromatin integrity, mitotic spindle assembly, and DNA replication [72, 75, 76]. We also found that overexpression of HDAC3 or HDAC6 blocked SFN-induced acetylation of respective substrates [128]. It is interesting to note that prostate [129] and colon [138] cancer cells were more sensitive to SFN as compared to normal cells. Clarke et al. [129] demonstrated differential effects of SFN in normal prostate cells versus hyperplastic and cancerous prostate cells based, at least in part, on altered HDAC expression levels.
Further, ITC-induced oxidative DNA damage has been attributed to ROS generation [139–142], inhibition of telomerase [143], lipid peroxidation [144], and covalent binding to protein targets such as tubulin [145]. Thus, it appears that SFN preferentially targets cancer cells over normal cells possibly via a sustained DNA damage response.
Indole-3-carbinol (I3C) and 3,3'-diindolylmethane (DIM)
Cruciferous vegetables contain glucosinolates such as glucoraphanin, the precursor of SFN, and glucobrassicin, the precursor of indole-3-carbinol (I3C). The latter compound and its acid condensation products, such as 3,3'-diindolylmethane (DIM), have been examined extensively for their cancer chemoprotective properties [146]. I3C has been shown to increase ATM signaling and p53 phosphorylation leading to p21 induction and G1 arrest in breast cancer cells [147]. I3C-induced activation of ATM-Chk2 was further shown to degrade the protein phosphatase Cdc25A [148]. Bhatnagar et al. [149] reported that DIM inhibited expression of HDAC1, HDAC2 and HDAC3 in colon cancer cells, which was associated with inhibition of survivin. Li et al. [150] demonstrated that DIM-induced HDAC depletion involved proteasome-mediated HDAC protein degradation. Although the authors found negligible increases in the acetylation of gene promoters, a reduction in the levels of repressive HDACs bound to the p21 and p27 promoters coincided with cell cycle arrest. Further, DIM caused significant increases in γH2AX and chromatin relaxation, with phosphorylation of KAP-1 prior to DNA damage-triggered apoptosis. Interestingly, decreased HDAC expression appeared 24 h prior to DNA damage signaling, suggesting HDAC inhibition/loss as a possible causative mechanism [150].
Other mechanisms related to the DIM-induced DNA damage response include activation of BRCA1 in breast and prostate cancer cells. BRCA1/2 signaling by DIM led to endoplasmic reticulum stress and activation of the GADD45 promoter [151]. Similarly, another study demonstrated that I3C, in combination with genistein, induced GADD gene expression in MCF-7 breast cancer cells and decreased expression of ER-α, thereby triggering apoptosis [152].
Parthenolide
Parthenolide (PN) is a sesquiterpene lactone isolated from Tanacetum parthenium. It has been shown to cause cell cycle arrest, promote cell differentiation, and induce apoptosis [153]. In addition to its other actions, PN was found to specifically deplete HDAC1 protein without affecting other class I/II HDACs. HDAC1 depletion was found to occur via proteasomal degradation that was activated through the DNA-damage-transducer ATM [154]. HDAC1 depletion by PN led to ubiquitination of MDM2 leading to p53 activation and sustained DNA damage response [155].
Anacardic acid
A phytochemical that modifies DNA damage via HAT inhibition is anacardic acid. The anacardic acid 6-pentadecyl salicylic acid (6-PDSA), from cashew nut shell liquid, is a potent HAT inhibitor. It inhibits p300 and p300/CBP-associated HAT activities [156]. In addition, 6-PDSA was shown to inhibit the HAT function of Tip60 and sensitize cancer cells to ionizing radiation [157]. Interestingly, a structural analog of 6-PDSA was reported to reduce histone H3K56 acetylation [158]. On the contrary, in normal human dermal fibroblasts, inhibition of HAT activity by 6-PDSA prevented UV-induced increases in γH2AX, p53, and acetyl-H3 [159], suggesting that histone acetylation is a prerequisite for efficient DNA damage signaling in normal cells.
Allium compounds
Garlic, onions, shallots and other members of the allium family contain an interesting and complex range of water-soluble and fat-soluble organosulfur compounds, some of which have been implicated as cancer chemopreventive agents [160, 161]. Allyl derivatives from garlic were among the first compounds described to impact histone acetylation status. Allyl mercaptan (AM), diallyl disulfide (DADS), S-allylcysteine (SAC), S-allylmercaptocysteine (SAMC) and allicin increased histone acetylation (H3/H4) in human cancer cells [123, 162–164], implicating HDACs as possible targets. AM was the most effective HDAC inhibitor among several garlic-derived organosulfur compounds and their metabolites, including SAMC, SAC, diallyl sulfide (DAS), DADS, diallyl trisulfide (DATS) and allyl methyl sulfide (AMS). In human colon cancer cells, AM caused histone H3 hyperacetylation, and facilitated Sp3 and p53 binding on the P21WAF1 promoter [165].
Recently, DADS and DATS were shown to directly induce the DNA damage response in cancer cells [166, 167]. In skin cancer cells, 25 μM DATS increased γH2AX levels as early as 3 h and produced a 10-fold increase in γH2AX by 24 h. Furthermore, DATS increased the phosphorylation of p53 by 12 h, and induced p21 expression at 24 h. Importantly, such effects were noted in cancer cells but not in normal keratinocytes [166]. The authors suggested that DATS might increase ROS levels and inflict DNA damage. A prior study showed that DATS activated the Chk1-Chk2-Cdc25C pathway, causing cell cycle arrest in prostate cancer cells [168]. Ling et al. [167] reported that DADS induced G2/M arrest through a similar pathway involving Chk1-Cdc25c-cyclin B1, and the DNA damage signaling kinase ATR. The specific role of histone acetylation in DNA damage signaling has not been elucidated in these studies. However, the ATR signaling pathway, known to be activated by allium compounds, is known to initiate a p53 phosphorylation-acetylation cascade leading to p21 expression [169]. In fact, the DNA damage-mediated phosphorylation of p53 promotes acetylation by increasing interaction between p53 and HATs [169]. Whether allium compounds affect DNA repair mechanisms in addition to DNA damage is not clear, since one study shows that DADS does not affect DNA repair genes in a microarray-based study using cancer cells [170].
Numerous studies have implicated ROS, NOS, and hydrogen peroxide (H2O2) in the actions of DADS and DATS, with evidence for anti-cancer activities being blocked by ROS scavengers such as N-acetyl cysteine (NAC) and other anti-oxidants [166, 171–174].
Selenium
Selenium is an essential trace element found as inorganic forms in soil, but also bioaccumulated as organic forms in foods such as Brazil nuts and seafood. Anticarcinogenic effects have been attributed to selenoproteins, and more recently to organoselenium metabolites [175–177]. Selenium may be an effective chemopreventive and anticancer agent in a broad spectrum of human cancers, viz. prostate, colon, bladder, lung, liver, ovarian, and leukemia [178]. Some forms of selenium exert epigenetic effects via histone modifications. HDAC activity was decreased, and histone acetylation increased, by sodium selenite [179], keto-methylselenobutyrate (KMSB), methyl selenocysteine (MSC), and methyl selenopyruvate (MSP) [180, 181]. Histone phosphorylation also was increased by selenomethionine (SM) on the promoters of GJB2 (connexin 26) and serum glucocorticoid kinase genes [182].
Selenium compounds have been reported to cause DNA damage-mediated apoptosis in cancer cells [183]. Recently, two papers described the mechanisms by which selenium compounds trigger DNA damage-induced cell death in cancer cells but not in normal cells [184, 185]. Qi et al. [184] examined methylseleninic acid (MSA, 0-10 μM), methyl selenocysteine (MSC, 0-500 μM), and sodium selenite (0-20 μM) in mismatch repair (MMR)-deficient HCT116 colorectal cancer cells and MMR-proficient HCT116 cells with MutL homolog 1 (MLH1) complementation. The authors found that compared with MMR-deficient HCT116 cells, HCT116+hMLH1 cells were significantly more sensitive to oxidative DNA lesions and γH2AX induction. Further, response to selenium compounds was dependent on ATM kinase and ROS, and required hMLH1-hPMS2. Addition of the ATM kinase inhibitor KU55933, the antioxidant NAC, or the superoxide dismutase mimic Tempo, suppressed the selenium-induced effects. The authors suggested that the hMLH1-hPMS2 complex senses and processes selenium-induced oxidative DNA damage and transmits the signal to ATM kinase, leading to the activation of G2/M checkpoint and death pathways [184]. Hence, in this case, a DNA repair complex acts via genomic instability and mutation to induce cell death. Wu et al. [185] showed that selenium compounds activated similar responses in normal MRC5 cells; however, rather than apoptosis induction they activated cell senescence, as evidenced by the expression of senescence-associated β-galactosidase and BrdU incorporation. In view of the HDAC inhibition, as noted previously for these compounds, it will be interesting to probe whether histone modifications have a role to play in the observed DNA damage signaling. In this regard, we know that MSA and MSC activate ATM [184], which is known to control the transcription of DNA damage genes in response to HDAC inhibition [186]. SM, another selenium compound, also decreased cell proliferation and induced cell-cycle arrest by increasing GADD34 and GADD153 expression [187]. However, such effects were not seen in mammary and prostate cancer cells [188]. Selenocystine, a nutritionally available selenoamino acid, was shown to induce ROS formation leading to DNA strand breaks in cancer cells, but not in normal human fibroblasts [189]. In fact, in normal fibroblast cells, selenium was identified as an important cofactor for various antioxidant enzymes that enhance DNA repair in cells [190].
Polyphenols
Polyphenols occur naturally in many foods and beverages consumed by humans. Promising cancer chemopreventive polyphenols include those in green tea, curry spices, grapes, soy, and berries.
(-)-Epigallocatechin-3-gallate (EGCG)
EGCG, the most abundant polyphenolic catechin in green tea, was identified as an antioxidant in vitro[191], although the possible relevance of this activity to its anticancer properties in vivo is far from established [29]. EGCG was reported to inhibit enzymes involved in DNA methylation, and was subsequently identified as a histone modifier [192–194]. EGCG inhibited HDAC activity and increased histone acetylation in prostate [192], skin [193], and breast cancer cells [194]. Pandey et al. [192] demonstrated that EGCG reduced mRNA expression of HDAC1, HDAC2, and HDAC3, leading to re-expression of GSTP1 in prostate cancer cells. Li et al. [194] showed that EGCG reactivated estrogen receptor (ERα) in breast cancer cells, due to decreased binding of the transcription repressor complex Rb/p130-E2F4/5-HDAC1-SUV39H1-DNMT1. Interestingly, Choi et al. [195] identified EGCG as a HAT inhibitor that suppressed transcription factor p65 (RelA) acetylation, thereby inhibiting nuclear factor kappa B (NFκB), interleukin 6 (IL6), and downstream target genes. In addition to the HAT and HDAC activities, EGCG inhibited polycomb group (PcG) proteins [196] that are key epigenetic regulators [197]. Treatment of skin cancer cells with EGCG reduced expression of PcG proteins BMI-1 and EZH2, leading to global reduction of histone H3K27me3 and reduced cell survival [196].
Although EGCG exhibits antioxidant activity in some in vitro assays, it can induce oxidative DNA damage and generate intracellular and mitochondrial ROS in lung cancer cells [198]. EGCG treatment triggered GADD153 gene expression in combination with celecoxib, via MAPK signaling [199]. Although GADD153 activity is known to be modulated by HDACs [80, 200], it is not clear whether HDAC inhibition played a role on GADD153 gene activation by EGCG. In this regard, it is worth mentioning that LBH589, a well known HDAC inhibitor, activates GADD genes by augmenting histone acetylation at the corresponding gene promoters [201]. More studies need to be carried out to determine if the effects of EGCG on HDACs contribute to its DNA damage effects. Another aspect of EGCG in this pathway is inhibition of CK2 [202], which is an important enzyme in the DNA damage response [55]. In addition, tea catechins are reported to exert DNA demethylating effects in vitro[203, 204], and trigger oxidative degradation of cellular DNA in the presence of copper Cu(II) ions [205].
Curcumin
Curcuminoid polyphenols in Indian spices have antioxidant, anti-inflammatory, and cancer chemoprotective properties [206–209]. There is growing interest in these compounds and their potential to modulate epigenetic endpoints [210–212]. Curcumin, for example, inhibited HAT activity by inducing proteasome-dependent degradation of p300 [213] in multiple cancers at a concentration of 20 μM or higher [214–216]. Curcumin also was shown to inhibit HDAC1 and to upregulate p21 mRNA and protein in a dose- and time-dependent manner in HepG2 hepatoma cells [217]. Another study showed that curcumin inhibited the expression levels of p300, HDAC1, HDAC3, and HDAC8 proteins, repressed NFκB and Notch1, and decreased cell proliferation in Raji lymphoma cells [218]. A more recent report on curcumin also supported its HDAC inhibitory effects [219]. However, curcumin also was found to stabilize HDAC2 protein expression and increase HDAC activity in lung, a beneficial outcome in the context of chronic oxidative stress [220].
Rowe et al. [221] reported that curcumin caused DNA damage in cancer cells, associated with phosphorylation, increased expression, and cytoplasmic retention of the BRCA1 protein. These effects were not seen in normal mammary epithelial cells [221]. Further, the induction of γH2AX and DNA damage by curcumin required ATM/Chk1 signaling [222]. Curcumin induced expression of GADD153 and increased ROS-mediated apoptosis induction in lung cancer cells. Treatment with GADD45- and GADD153-siRNAs inhibited apoptotic induction in these cells [223, 224]. As noted earlier, GADD genes are known to be modified through HAT/HDAC balance [201], as well as ATM kinase activity [186, 225].
Curcumin also inhibits DNA repair pathways in cancer cells, like the fanconi anemia/BRCA (FA/BRCA) pathway [226], or downregulates DNA repair proteins MGMT (O6-methylguanine-DNA methyltransferase), DNAPK, Ku70, Ku80, and ERCC-1 [227]. Other studies have shown that curcumin induces damage to both mitochondrial and nuclear DNA [228], triggers ROS generation [229] and glutathione (GSH) depletion [230], resulting in apoptosis induction in cancer cells.
Resveratrol
Resveratrol, a stilbene found in grapes and wine, has been implicated in anti-aging and cancer prevention mechanisms [231]. Resveratrol was linked with activation of SIRT1 and the acetyl transferase, p300 [94, 232, 233]. There is a debate as to whether these mechanisms are directly or indirectly involved in the protective effects of resveratrol in vitro and in vivo[234, 235]. A recent study concluded that tumor suppressive effects of resveratrol in Apcmin mice were dependent on SIRT expression [236]. Resveratrol can delay cell cycle progression and induce apoptosis in several cancer cell lines; some of these effects have been attributed to the activity of SIRT1.
Several proteins that have a role in the DNA damage response, such as p53, FoxO, and Ku70, are deacetylated and inactivated by SIRT1. Consistent with this role of SIRT1, recent evidence indicates that resveratrol inhibits DNA repair in cancer cells [83, 94, 237–239]. Studies by Wang et al. [94] using SIRT1 mutant mice showed that impaired SIRT1 function resulted in tumor formation in a p53-null background, and that activation of SIRT1 by resveratrol reduced tumorigenesis. Further, SIRT1 activation by resveratrol negatively regulated survivin expression by histone deacetylation in the promoter of the survivin gene [237]. Resveratrol enhanced p53 acetylation and induced apoptosis in prostate cancer cells by inhibiting MTA1/NuRD, an integral component of the nucleosome remodeling and deacetylase complex [238]. Furthermore, resveratrol inhibited both HR and NHEJ via ATM-p53 and ATM/ATR-Nbs1-dependent pathways, respectively [239]. On the contrary, activation of SIRT1 by resveratrol was reported to promote APE1 activity and binding to X-ray cross-complementing-1 (XRCC1) protein, facilitating the BER DNA repair pathway [83].
Several studies support resveratrol inducing a sustained DNA damage response via BRCA1 and activation of the ATM/ATR-Chk1/2-Cdc25C pathway in cancer cells [240–242]. Notably, Tyagi et al [242] observed only marginal effects of resveratrol in normal human foreskin fibroblasts. In addition, a recent study demonstrated that resveratrol caused telomere instability in osteosarcoma cells [241], which resulted in genetic instability, activation of DNA damage response, and cell senescence.
There is increasing evidence that resveratrol exhibits "pro-oxidant" activity in some circumstances [231]. Resveratrol catalyzed oxidative DNA degradation in the presence of transition metal ions, such as copper [243], generated ROS [244, 245], and triggered GSH efflux associated with Bax translocation to the mitochondria [246].
Isoflavones
Soy isoflavones have been implicated in reducing the overall incidence of breast and prostate cancers in Asian countries. Genistein (4',5,7-trihydroxyisoflavone) is the major isoflavone present in soybeans. Genistein is known to inhibit human cancer cell growth, mediated via genes controlling cell cycle progression and apoptosis [247]. One mechanism that has recently received considerable attention is the epigenetic modulation of DNA methylation and/or chromatin marks [248]. Genistein possesses high histone modifying activity compared with other isoflavones, such as biochanin A and diadzein. Genistein impacted histone acetylation and demethylation, leading to activation of tumor suppressors such as p21, p16, FoxO3a, and phosphatase and tensin homolog (PTEN) [249]. Genistein also caused androgen receptor (AR) downregulation through inhibition of HDAC6-Hsp90 co-chaperone functions in prostate cancer cells [250].
Genistein activated stress signaling pathways that phosphorylated p53 and ATM, leading to p21 induction and γH2AX formation [251, 252]. Further, genistein modulated cyclin-dependent kinase Cdc2 activity through the protein phosphatase Cdc25C, thereby activating ATM and causing G2/M arrest in hepatoma cells [253]. Other recent studies confirmed similar effects in lung and prostate cancer cells [254–256]. An in vivo metabolite of genistein, 5,7,3',4'-tetrahydroxyisoflavone, was shown to act via ATR kinase signaling to cause DNA breaks and induce cell cycle arrest [257]. Genistein induced GADD45, p53, and p38 in embryonic cancer cells [258], and enhanced expression of BRCA1 [259] and MDC1 in neuroblastoma cells [260].
Other reported mechanisms for genistein in cancer cells include oxidative DNA damage by ROS generation in the presence of copper [261], and inhibition of topoisomerase II in an ATM-dependant manner [262]. Interestingly, in the non-cancerous MCF-10A breast cell line, genistein protected against polycyclic aromatic hydrocarbon (PAH)-induced oxidative DNA damage [263].
Quercetin
Quercetin is a flavonoid found in foods such as citrus fruit, buckwheat, and onions. Recently, quercetin was shown to increase histone H3 acetylation by both HAT activation and HDAC inhibition in leukemia HL60 cells. The result was FasL-dependent apoptosis, and activation of the extracellular signal-regulated kinase (ERK) and jun N-terminus kinase (JNK) signaling pathways [264]. Quercetin also induced the phosphorylation of ATM and H2AX [251]. Despite its anti-inflammatory and anti-oxidant properties, low concentrations of quercetin induced extensive DNA damage by reacting with Cu(II) in cancer cells [265]. This was confirmed recently, with evidence that a quercetin-copper(II) complex promoted cleavage of plasmid DNA, producing single and double DNA strand breaks in lung cancer A549 cancer cells [266]. In addition, quercetin inhibited DNA repair via competitive inhibition of DNAPK, a repair protein involved in NHEJ [267].
Dietary compounds as chemo- and radio-sensitizers for cancer therapy
In addition to the aspects discussed above, histone modifiers exert synergistic actions when combined with ionizing irradiation (IR) or DNA-damaging drugs [268–270]. HDAC inhibitors can stabilize and enhance γH2AX and interfere with the DNA repair machinery in cancer cells [271]. From the evidence provided above, many dietary compounds can influence the DNA damage response and inhibit specific repair mechanisms. Importantly, histone modifications augment DNA damage in a manner that goes essentially unrepaired in many cancer cells, but is repaired effectively in normal cells. Some illustrative examples from the recent literature are provided below.
ITCs, HDAC inhibitor drugs, and radiotherapies
Radiosensitivity of HeLa cells was reportedly enhanced by SFN pretreatment. Pre-treatment with SFN was found to inhibit DSB repair in irradiated cells leading to apoptosis. This was associated with decreased expression of repair proteins, Rad51 and DNAPK [272]. The authors showed that the combination was also effective in vivo[272]. In PC3 prostate cancer cells, the lipid peroxidation end-product 4-hydroxynonenal resulting from SFN treatment potentiated the anti-tumor effects of the HDAC inhibitor LBH589. Combined SFN+LBH589 treatment induced dephosphorylation of Cdc2 and sustained expression of γH2AX [273]. BITC and other ITCs sensitized pancreatic cancer cells to γ-irradiation. Specifically, BxPC-3 pancreatic cancer cells pretreated with 2.5 μM BITC for 24 h followed by exposure to 5 Gy γ-irradiation had reduced survival and enhanced G2/M arrest as compared to cells exposed to γ-irradiation alone. Cell cycle arrest was associated with DNA damage, phosphorylation of ATR, Chk2, Cdc25C, and Cdk-1, and induction of p21 [274]. Similarly, PEITC significantly enhanced cytotoxicity in a vorinostat-resistant leukemia cell line, HL60/LR, by inhibiting the cytoprotective antioxidant response involving depleted cellular GSH [275].
Anacardic acid enhances radiosensitivity
HAT inhibitors in the anacardic acid family (see above) exert antiproliferative and cytotoxic effects on pituitary adenoma cells associated with an increase in PARP, sub-G1 arrest, and apoptosis. These compounds radiosensitized pituitary adenoma cells by reducing the expression of survivin and X-linked inhibitor of apoptosis protein (XIAP), which are known to be associated with cell survival and radioresistance [276].
Curcumin synergizes with chemo- and radiotherapy
Curcumin has been shown to enhance the toxicity of cyclophosphamide (CTX) in a drug-resistant human lymphoma cell line HT/CTX, via inhibition of the FA/BRCA pathway. The combination of curcumin and CTX produced synergistic effects and reversed multiple drug resistance. Blockade of cell cycle progression and downregulation of fanconi anemia group D2 (FANCD2) were implicated in the anti-tumor mechanism of curcumin [277]. Similarly, curcumin reversed multidrug resistance in multiple myeloma cell line MOLP-2/R through inhibition of FA/BRCA, suggesting beneficial outcomes when used with low-dose DNA cross-linking agents [278]. In a variety of human cancer cells, synergistic inhibition of cell proliferation also was seen for curcumin combined with cisplatin, 5-fluorouracil (5-FU), or celecoxib, via inhibition of DNA repair pathways [226, 279–281]. Curcumin sensitized glioma cells to clinically used chemotherapeutic agents or radiation, which correlated with reduced Bcl-2 and inhibitor of apoptosis (IAP) family members as well as DNA repair enzymes MGMT, DNAPK, Ku70, Ku80, and the excision repair cross-complementary-1 (ERCC-1) [227]. Recently, Lin et al [282] have shown that curcumin downregulates the expression levels of thymidine phosphorylase (TP), an enzyme of the pyrimidine salvage pathway and ERCC1, a protein involved in the process of nucleotide excision repair which helps in overcoming platinum resistance in cancer cells. Interestingly, curcumin also synergized with HDAC inhibitors vorinostat and LBH589, via persistent depletion of Hsp90 client proteins EGFR, Raf-1, Akt, and survivin [283].
Resveratrol and purine analogs
In chronic lymphocytic leukemia (CLL) cells from patients, clinically-used purine analogs fludarabine or cladribine caused a higher rate of apoptosis when combined with resveratrol. Apoptosis was related to the presence of cytogenetic abnormalities and increased DNA damage markers γH2AX and ATM. The authors suggested that resveratrol might provide a new therapeutic approach for CLL due to acceptable safety, lowering the dose of purine analogs, resulting in activation of DNA damage specifically in cancer cells and not in normal cells [284].
Catechins and COX-2 inhibitors
EGCG, in combination with COX-2 inhibitors enhanced apoptosis by increasing the expression of DNA damage- inducible GADD153, GADD45A, and CDKN1A (p21/WAF1/CIP1) genes. Synergistic enhancements of apoptosis and GADD153 gene expression in human non-small cell lung cancer cells by the combination of EGCG and celecoxib were mediated through the activation of the MAPK signaling pathway [199].
In vivo studies and clinical translation: Future perspectives
In vivo studies that demonstrate the functional relevance of epigenetic mechanisms for anti-tumor efficacy are still relatively scarce. At present, the best evidence to demonstrate that nutrition modulates epigenetic status and health outcomes in mammals comes from studies with mice carrying the agouti (Avy) gene [285]. Dietary methyl deficiency (folate, choline, and methionine) in animal models alters hepatic DNA methylation patterns and induces liver cancer in the absence of a carcinogen [286]. Similarly, selenium-deficient diets have been shown to hypomethylate DNA in liver and colon, as compared to rats fed either selenite or selenomethionine [287]. In a very interesting study, high levels of grooming and nursing by rat mothers modified the levels of DNA methylation at a glucocorticoid receptor (GR) gene promoter in the hippocampus of the offspring, leading to altered histone acetylation and binding of a transcription factor (NGFI-A) to the GR promoter [288]. Further, it was observed that a proportion of these changes could be modified by treatment with an HDAC inhibitor or a methyl donor [289].
Dietary HDAC inhibition also is an emerging field, with some evidence for epigenetic modulation in vivo. For example, polyphenon B, a tea polyphenol preparation, decreased HDAC1 levels and modulated the expression of markers of invasion and angiogenesis in dimethylaminoazobenzene-induced liver cancer in rats [290]. Theophylline, also present in tea, was associated with downregulation of the inflammatory response through increased HDAC activity in epithelial cells and macrophages in smokers and chronic obstructive pulmonary disease (COPD) patients, a situation associated with diminished HDAC activity [291–294]. It is noteworthy that the mechanism occurred at therapeutic concentrations [294]. Another polyphenol, quercetin, inhibited HDAC1 and DNA methyl transferase 1 (DNMT1) in carcinogen-treated hamsters and reduced tumor incidence and burden [295].
In the ApcMin/+ mouse model, SFN-containing diet (300 and 600 ppm for 3 weeks) was optimal for achieving SFN tissue concentrations in the 3-30 μM range [296]. In the same animal model, we reported that SFN-containing diet suppressed tumor development via increased global H3/H4 histone acetylation, with a concomitant upregulation of p21 and Bax gene expression [297]. In another study, Myzak et al. [298] demonstrated that oral administration of 7.5 μM SFN per animal per day for 21 days significantly reduced growth of prostate cancer (PC-3) tumor xenografts and decreased HDAC activity in the xenografts, prostates, and mononuclear blood cells. There was a trend towards increased global histone acetylation in these tissues. The study was also extended to human volunteers wherein consumption of 68 g broccoli sprouts resulted in a significant inhibition of HDAC activity in peripheral blood mononuclear cells 3 h following intake [298]. Recently, it was demonstrated that SFN is highly metabolized in mice, achieving micromolar concentrations in plasma, with tissue concentrations in the range 0.003 - 0.35 nmole/mg. Thus, SFN metabolites may play an important role in HDAC and tumor inhibition [299]. In vivo data with BITC, another ITC, clearly indicate that oral administration of 12 μmol BITC significantly suppressed the growth of pancreatic (BxPC-3) tumor xenografts, and that tumor suppression was associated with the reduced NF-κB, cyclin D1, HDAC1, and HDAC3, complementing observations made in vitro. The authors suggested inhibition of HDAC1/HDAC3 by BITC as a plausible mechanism of NF-κB inactivation [124]. Other studies have shown that ITCs can achieve therapeutic serum concentrations both in vivo[300] and in humans [301].
In rats, treatment with AM and/or DADS increased acetylation of histones and caused up-regulation of p21 expression in normal liver and hepatoma cells and in rat colonocytes [163, 302, 303]. However, there is concern about the high concentration of allyl-derivatives used in these studies, which may be associated with toxicity in various tissues.
Li et al. [150] provided direct in vivo evidence on the role of dietary HDAC inhibition and DNA damage for the anti-cancer effects of DIM, an I3C metabolite. Previous reports by Bhatnagar et al. [149] showed DIM significantly inhibited expression of HDAC1, HDAC2 and HDAC3 in colon cancer cells and in APCmin/+ mice. Li et al. [150] demonstrated using colon cancer (HT29) xenografts that DIM downregulates HDAC1 and HDAC2 and this was associated with induction of γH2AX and p21 expression in the xenografts. Importantly, these effects were seen at non-toxic DIM concentrations. An oral dose of 250 mg/kg of DIM produced a plasma concentration of 18 μg/ml in mice, equivalent to ~77 μM. The authors suggested DNA damage as a possible mechanism of cancer cell death induced by DIM [150].
In vivo studies with dietary polyphenols have shown encouraging results on DNA damage and tumor inhibition. Tyagi et al [304] showed that resveratrol (50 mg/kg bw) treatment inhibited head and neck squamous cell carcinoma (FaDu) tumor growth in nude mice, and γH2AX and cleaved caspase-3 were strongly increased in xenografts from resveratrol-treated mice compared to controls. Vanhees et al. [305] have shown that prenatal exposure to both genistein and quercetin supplements in mice induced DSBs and DNA rearrangements in the mixed-lineage leukemia (MLL) gene, especially in the presence of compromised DNA repair. Toyoizumi et al. [306] reported that co-administration of isoflavones and NaNO2 caused DNA damage in mouse stomach via the formation of radicals. Amin et al. [307] observed that EGCG, in combination with luteolin, increased apoptosis in head and neck and lung cancer xenografted tumors in nude mice, possibly by ATM-dependent Ser(15) phosphorylation of p53 resulting from DNA damage.
Pharmacological HDAC inhibitors also have shown promise when acting in vivo, alone or in combination with radiotherapy/chemotherapy. Vorinostat, as a single agent, was shown to induce DSBs associated with the downregulation of DNA repair gene Rad52, thus preventing brain metastasis of triple-negative breast cancer [115]. In a murine metastatic neuroblastoma model, vorinostat was found effective possibly by modulating DNA repair enzyme Ku-86 [308]. Treatment with LBH589, another HDAC inhibitor, led to a dramatic reduction of tumor growth in a colon (HCT116) cancer model. Analysis of the residual tumor revealed that HDAC inhibitor treatment increased histone acetylation, γH2AX accumulation, and apoptosis. The treatment had no obvious detrimental effects on the mice, only acting on the xenografts [309]. In addition, various HDAC inhibitors appear to sensitize tumors to IR in vivo, as demonstrated by vorinostat [308, 310], MS-275 [311], valproic acid [312], LBH589 [313], LAQ824 [314], AN-9 [315] and PCI-24781 [316]. Treatment with these HDAC inhibitors led to greater delay in tumor growth by enhancing IR-induced γH2AX in xenografts, suggesting that HDAC inhibitors interfere with DSB repair and/or render DNA more susceptible to IR-induced damage. However, there is evidence that radiosensitization is not limited to cancer cells, but also occurs in healthy normal cells. Vorinostat, MS-275, sodium-butyrate and valproic acid treatments have been shown to increase radiosensitivity and reduce DSB repair capacity in normal cells too, leading to potential genotoxic effects of HDAC inhibitor treatment [317].
In addition to the growing list of studies in vivo, there are over 300 human clinical trials of HDAC inhibitors, tested either alone or in combination with radiation, chemotherapy, and/or molecular therapy. In particular, some of the human trials with vorinostat and valproic acid in combination with radiation are evaluating the effects on DNA damage and repair factors http://clinicaltrials.gov/. While in vitro models have contributed enormously to our mechanistic understanding of the epigenetic network and its regulation, there remains a paucity of preclinical and clinical data for the majority of dietary compounds. Until this situation is rectified, one must exercise caution when interpreting and extrapolating the significance of current evidence in the literature. Ongoing clinical trials are moving in the right direction, as for example in the evaluation of broccoli sprouts and broccoli sprout extract for modulating epigenetic marks in breast and prostate cancer. In addition to testing for epigenetic biomarkers in blood and tumor biopsies, DNA damage markers (e.g. γH2AX) could be analyzed in tumor samples and adjacent normal tissue to provide insights on the DNA damage response. Monitoring γH2AX levels in a patient's circulating tumor cells [318], PBMCs, or hair samples might prove useful in the clinical setting [319, 320]. Studies in (normal) human volunteers could certainly benefit from such non-invasive techniques. However, it will be important to note that tumor cells also have genetic alterations that impact responses to DNA damage, which differ from normal replicating cells. Notably, normal cells typically respond in a facile manner to "correct' DNA damage responses once the test agent has been removed.
Conclusions
Genomic instability provides a means for selective targeting of cancer cells over normal cells, via epigenetic players with important roles in DNA repair. The active recruitment and de-recruitment of HAT and HDAC enzymes and their binding partners at sites of DNA damage produces localized sites of open chromatin, increasing the genotoxic effectiveness of agents such as UV, IR, and chemotherapeutic agents. The literature supports the role of multiple HDACs in genome surveillance, and HDAC inhibitors appear to facilitate cancer cell death by enhancing the DNA damage response and inhibiting DNA repair. Among the various cancer chemopreventive agents reviewed herein, many cause changes in chromatin conformation, disrupt the intracellular redox balance, and deregulate DNA repair proteins. Thus, these compounds might activate the DNA damage response with particular effectiveness in cancer cells as compared to normal cells as depicted in Figure 3. In vivo therapeutic efficacy of these compounds, as reviewed here and elsewhere [29], suggests that effective concentrations are achievable and modulate DNA damage and repair responses in tumors. Dietary compounds with pleiotropic effects in cancer cells also likely impact DNA damage and repair via other epigenetic mechanisms, such as through DNA methylation and microRNAs. Improved understanding of these various epigenetic mechanisms will, it is hoped, provide a more rational basis for combining specific dietary compounds and standard radiation or chemotherapy approaches, thereby enhancing efficacy in the clinical setting.

The differential effect of DNA damaging agents in cancer and normal cells. HDAC inhibitors are known to cause DSBs through chromatin remodeling and oxidative damage due to ROS generation. Normal cells counteract this by check point activation leading to cell cycle arrest; anti-oxidant mechanisms and effective DNA repair whereas cancer cells known to be defective in some of these mechanisms, for e.g. check point kinases and repair genes, fail to repair the DNA damage leading to cell death.
Abbreviations
- 53BP1:
-
p53-binding protein
- 5-FU:
-
5-fluorouracil
- 6-PDSA:
-
6-pentadecyl salicylic acid
- allyl-ITC:
-
allyl isothiocyanate
- AM:
-
allyl mercaptan
- AMS:
-
allyl methyl sulfide
- APE1:
-
apurinic/apyrimidinic endonuclease-1
- APE1/Ref-1:
-
apurinic apyrimidinic endonuclease redox effector factor-1
- Asf1:
-
anti-silencing function 1
- ATM:
-
ataxia-telangiectasia mutated
- ATR:
-
ATM-and Rad3-related
- ATX:
-
ATM related kinase
- BER:
-
base-excision repair
- BITC:
-
benzyl isothiocyanate
- BLM:
-
bloom syndrome gene
- BRCA1:
-
breast cancer 1
- BRCA2:
-
breast cancer 2
- CAF1:
-
chromatin assembly factor I
- CHK1/CHK2:
-
checkpoint kinase1/2
- CK2:
-
casein kinase 2
- CLL:
-
chronic lymphocytic leukemia
- CtIP:
-
c-terminal binding protein interacting protein
- CTX:
-
cyclophosphamide
- DADS:
-
diallyl disulfide
- DAS:
-
diallyl sulfide
- DATS:
-
diallyl trisulfide
- DIM:
-
3,3'-diindolyl methane
- DMBA:
-
7: 12-dimethylbenz[a]anthracene
- DNAPK:
-
DNA-dependent protein kinase
- DSB:
-
double-strand break
- EGCG:
-
(-)-epigallocatechin-3-gallate
- ERK:
-
extracellular signal-regulated kinase
- FA/BRCA:
-
fanconi anemia/BRCA pathway
- FANCD2:
-
fanconi anemia group D2
- FoxO:
-
forkhead box O
- GADD153:
-
growth arrest and DNA damage 153
- GADD45:
-
growth arrest and DNA damage 45
- GSH:
-
glutathione
- H2O2:
-
hydrogen peroxide
- HAT:
-
histone acetyl transferase
- HDAC:
-
histone deacetylase
- HMGN1:
-
high-mobility group N1
- HP1β:
-
heterochromatin Protein 1β
- HR:
-
homologous recombination
- I3C:
-
indole-3-carbinol
- IAP:
-
inhibitor of apoptosis
- Idh2:
-
isocitrate dehydrogenase 2
- IL6:
-
interleukin 6
- ING1a:
-
inhibitor of growth 1a
- IR:
-
ionizing irradiation
- ITC:
-
isothiocyanate
- JNK:
-
jun N-terminus kinase
- KAP-1:
-
KRAB associated protein
- KMSB:
-
keto-methylselenobutyrate
- Ku70:
-
nonhomologous end joining (NHEJ) factor
- LBH589:
-
panobinostat
- MCL1:
-
induced myeloid leukemia cell differentiation protein
- MDC1:
-
mediator of DNA damage checkpoint protein-1
- Mdm2:
-
murine double minute
- MEFs:
-
murine embryonic fibroblasts
- MGMT:
-
O6-methylguanine-DNA methyl transferase
- MLH1:
-
MutL homolog 1
- MMR:
-
mismatch repair
- MRE11:
-
meiotic recombination 11
- MRN:
-
MRE11-RAD50-Nbs1 complex
- MSA:
-
methylseleninic acid
- MSC:
-
methyl selenocysteine
- MSP:
-
methyl selenopyruvate
- NAC:
-
N-acetyl cysteine
- NADPH:
-
nicotinamide adenine dinucleotide phosphate
- Nbs:
-
nijmegen breakage syndrome
- NCOR:
-
nuclear receptor corepressor
- NER:
-
nucleotide-excision repair
- NFκB:
-
nuclear factor kappa B
- NHEJ:
-
non-homologous end-joining
- PARP:
-
poly(ADP-ribose)polymerase
- PcG:
-
polycomb group protein
- PCNA:
-
proliferating cell nuclear antigen
- PEITC:
-
phenethyl isothiocyanate
- PHITC:
-
phenylhexyl isothiocyanate
- PI3K:
-
phosphatidylinositol-3 kinase
- PN:
-
parthenolide
- PTEN:
-
phosphatase and tensin homolog
- RAD51:
-
DNA repair protein RecA homolog
- RelA:
-
transcription factor p65
- RFC:
-
replication factor C
- RNF8/168:
-
RING finger E3 ubiquitin-protein ligase
- ROS:
-
reactive oxygen species
- RPA:
-
replication protein A
- SAC:
-
S-allylcysteine
- SAHA:
-
suberoylanilide hydroxamic acid
- SAMC:
-
S-allylmercaptocysteine
- SFN:
-
sulforaphane
- Sir2:
-
silent information regulator 2
- SIRT:
-
sirtuin
- SM:
-
selenomethionine
- SMRT:
-
silencing mediator for retinoic and thyroid receptor
- SSA:
-
single-strand annealing
- SSB:
-
single-strand break
- SWI2/SNF2:
-
SWItch/Sucrose NonFermentable
- Tip60:
-
TAT-interacting protein 60
- TP:
-
thymidine phosphorylase
- TrxR:
-
thioredoxin reductase
- VPA:
-
valproic acid
- WRN:
-
werner helicase
- XIAP:
-
X-linked inhibitor of apoptosis protein
- XPA:
-
xeroderma pigmentosum group A
- XPC:
-
xeroderma pigmentosum group C
- XRCC1:
-
X-ray cross-complementing-1
- γH2AX:
-
phosphorylated histone H2AX.
References
Heinen CD, Schmutte C, Fishel R: DNA repair and tumorigenesis: lessons from hereditary cancer syndromes. Cancer Biol Ther. 2002, 1: 477-485.
Cazaux C: Genetic instability as a driver for oncogenesis. Bull Cancer. 2010, 97: 1241-1251.
Barbie DA, Hahn WC, Pellman DS: Destabilization of the cancer genome. Cancer: Principles & Practice of Oncology. Edited by: DeVita VT, Lawrence TS, Rosenberg SA. 2008, Lippincott Williams & Wilkins, Philadelphia, 35-51. 8
Mangerich A, Bürkle A: How to kill tumor cells with inhibitors of poly(ADP-ribosyl)ation. Int J Cancer. 2011, 128: 251-265.
Rassool FV, Tomkinson AE: Targeting abnormal DNA double strand break repair in cancer. Cell Mol Life Sci. 2010, 67: 3699-3710.
Lord CJ, Ashworth A: Targeted therapy for cancer using PARP inhibitors. Curr Opin Pharmacol. 2008, 8: 363-369.
Plummer R: Perspective on the pipeline of drugs being developed with modulation of DNA damage as a target. Clin Cancer Res. 2010, 16: 4527-4531.
Putiri EL, Robertson KD: Epigenetic mechanisms and genome stability. Clin Epigenet. 2010, 2: 299-314.
Purrucker JC, Mahlknecht U: Targeting the epigenome: effects of epigenetic treatment strategies on genomic stability in healthy human cells. Clin Epigenet. 2010, 1: 45-54.
Lichtenstein AV: Cancer: evolutionary, genetic and epigenetic aspects. Clin Epigenet. 2010, 1: 85-100.
Chi P, Allis CD, Wang GG: Covalent histone modifications--miswritten, misinterpreted and mis-erased in human cancers. Nat Rev Cancer. 2010, 10: 457-469.
Zhu Q, Wani AA: Histone modifications: crucial elements for damage response and chromatin restoration. J Cell Physiol. 2010, 223: 283-288.
Vaissière T, Herceg Z: Histone code in the cross-talk during DNA damage signaling. Cell Res. 2010, 20: 113-115.
Eot-Houllier G, Fulcrand G, Magnaghi-Jaulin L, Jaulin C: Histone deacetylase inhibitors and genomic instability. Cancer Lett. 2009, 274: 169-176.
Kothapalli N, Sarath G, Zempleni J: Biotinylation of K12 in histone H4 decreases in response to DNA double-strand breaks in human JAr choriocarcinoma cells. J Nutr. 2005, 135: 2337-2342.
Kachhap SK, Rosmus N, Collis SJ, Kortenhorst MS, Wissing MD, Hedayati M: Downregulation of homologous recombination DNA repair genes by HDAC inhibition in prostate cancer is mediated through the E2F1 transcription factor. PLoS One. 2010, 5: e11208-
Jang ER, Choi JD, Park MA, Jeong G, Cho H, Lee JS: ATM modulates transcription in response to histone deacetylase inhibition as part of its DNA damage response. Exp Mol Med. 2010, 42: 195-204.
Ogiwara H, Ui A, Otsuka A, Satoh H, Yokomi I, Nakajima S, Yasui A, Yokota J, Kohno T: Histone acetylation by CBP and p300 at double-strand break sites facilitates SWI/SNF chromatin remodeling and the recruitment of non-homologous end joining factors. Oncogene. 2011, 30: 2135-2146.
Rossetto D, Truman AW, Kron SJ, Côté J: Epigenetic modifications in double-strand break DNA damage signaling and repair. Clin Cancer Res. 2010, 16: 4543-4552.
Robert T, Vanoli F, Chiolo I, Shubassi G, Bernstein KA, Rothstein R, Botrugno OA: HDACs link the DNA damage response, processing of double-strand breaks and autophagy. Nature. 2011, 471: 74-79.
Kaidi A, Weinert BT, Choudhary C, Jackson SP: Human SIRT6 promotes DNA end resection through CtIP deacetylation. Science. 2010, 329: 1348-1353.
Soerjomataram I, Oomen D, Lemmens V, Oenema A, Benetou V, Trichopoulou A, Coebergh JW, Barendregt J, de Vries E: Increased consumption of fruit and vegetables and future cancer incidence in selected European countries. Eur J Cancer. 2010, 46: 2563-2580.
Miller PE, Lesko SM, Muscat JE, Lazarus P, Hartman TJ: Dietary patterns and colorectal adenoma and cancer risk: a review of the epidemiological evidence. Nutr Cancer. 2010, 62: 413-424.
Steevens J, Schouten LJ, Goldbohm RA, van den Brandt PA: Vegetables and fruits consumption and risk of esophageal and gastric cancer subtypes in the Netherlands cohort study. Int J Cancer. 2011
Büchner FL, Bueno-de-Mesquita HB, Linseisen J, Boshuizen HC: Fruits and vegetables consumption and the risk of histological subtypes of lung cancer in the European Prospective Investigation into Cancer and Nutrition (EPIC). Cancer Causes Control. 2010, 21: 357-371.
Rajendran P, Williams DE, Ho E, Dashwood RH: Metabolism as a key to histone deacetylase inhibition. Crit Rev Biochem Mol Biol. 2011, 46: 181-199.
Meeran SM, Ahmed A, Tollefsbol TO: Epigenetic targets of bioactive dietary components for cancer prevention and therapy. Clin Epigenetics. 2010, 1: 101-116.
Huang J, Plass C, Gerhäuser C: Cancer chemoprevention by targeting the epigenome. Curr Drug Targets. 2010
Link A, Balaguer F, Goel A: Cancer chemoprevention by dietary polyphenols: promising role for epigenetics. Biochem Pharmacol. 2010, 80: 1771-1792.
Pogribny IP, Basnakian AG, Miller BJ, Lopatina NG, Poirier LA, James SJ: Breaks in genomic DNA and within the p53 gene are associated with hypomethylation in livers of folate/methyl-deficient rats. Cancer Res. 1995, 55: 1894-1901.
Bonnesen C, Eggleston IM, Hayes JD: Dietary indoles and isothiocyanates that are generated from cruciferous vegetables can both stimulate apoptosis and confer protection against DNA damage in human colon cell lines. Cancer Res. 2001, 61: 6120-6130.
Antosiewicz J, Ziolkowski W, Kar S, Powolny AA, Singh SV: Role of reactive oxygen intermediates in cellular responses to dietary cancer chemopreventive agents. Planta Med. 2008, 74: 1570-1579.
Jeggo PA: DNA breakage and repair. Adv Genet. 1998, 38: 185-218.
Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM: DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998, 273: 5858-5868.
Rogakou EP, Boon C, Redon C, Bonner WM: Megabase chromatin domains involved in DNA double-strand breaks in vivo. J Cell Biol. 1999, 146: 905-916.
Burma S, Chen BP, Murphy M, Kurimasa A, Chen DJ: ATM phosphorylates histone H2AX in response to DNA doublestrand breaks. J Biol Chem. 2001, 276: 42462-42467.
Ward IM, Chen J: Histone H2AX is phosphorylated in an ATR-dependent manner in response to replicational stress. J Biol Chem. 2001, 276: 47759-47762.
Tamburini BA, Tyler JK: Localized histone acetylation and deacetylation triggered by the homologous recombination pathway of double-strand DNA repair. Mol Cell Biol. 2005, 25: 4903-4913.
Yamamoto T, Horikoshi M: Novel substrate specificity of the histone acetyltransferase activity of HIV-1-Tat interactive protein Tip60. J Biol Chem. 1997, 272: 30595-30598.
Kimura A, Horikoshi M: Tip60 acetylates six lysines of a specific class in core histones in vitro. Genes Cells. 1998, 3: 789-800.
Miller KM, Tjeertes JV, Coates J, Legube G, Polo SE, Britton S, Jackson SP: Human HDAC1 and HDAC2 function in the DNA-damage response to promote DNA nonhomologous end-joining. Nat Struct Mol Biol. 2010, 17: 1144-1151.
Bhaskara S, Knutson SK, Jiang G, Chandrasekharan MB, Wilson AJ, Zheng S: HDAC3 is essential for the maintenance of chromatin structure and genome stability. Cancer Cell. 2010, 18: 436-447.
Kotian S, Liyanarachchi S, Zelent A, Parvin JD: Histone deacetylases 9 and 10 are required for homologous recombination. J Biol Chem. 2011, 286: 7722-7726.
Shen X, Mizuguchi G, Hamiche A, Wu C: A chromatin remodelling complex involved in transcription and DNA processing. Nature. 2000, 406: 541-544.
Krogan NJ, Keogh MC, Datta N, Sawa C, Ryan OW, Ding H: A Snf2 family ATPase complex required for recruitment of the histone H2A variant Htz1. Mol Cell. 2003, 12: 1565-1576.
Krogan NJ, Baetz K, Keogh MC, Datta N, Sawa C: Regulation of chromosome stability by the histone H2A variant Htz1, the Swr1 chromatin remodeling complex, and the histone acetyltransferase NuA4. Proc Natl Acad Sci USA. 2004, 101: 13513-13518.
Paul TT, Rogakou EP, Yamazaki V, Kirchgessner CU, Gellert M, Bonner WM: A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Curr Biol. 2000, 10: 886-895.
Petrini JH: Cell signaling. A touching response to damage. Science. 2007, 316: 1138-1139.
Essers J, Houtsmuller AB, van Veelen L, Paulusma C, Nigg AL, Pastink A, Vermeulen W, H Hoeijmakers, Kanaar R: Nuclear dynamics of RAD52 group homologous recombination proteins in response to DNA damage. Embo J. 2002, 21: 2030-2037.
Yu DS, Sonoda E, Takeda S, Huang CL, Pellegrini L, Blundell TL, Venkitaraman AR: Dynamic control of Rad51 recombinase by self-association and interaction with BRCA2. Mol Cell. 2003, 12: 1029-1041.
Lee JH, Paull TT: ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science. 2005, 308: 551-554.
Sobhian B, Shao G, Lilli DR, Culhane AC, Moreau LA, Xia B, Livingston DM, Greenberg RA: RAP80 targets BRCA1 to specific ubiquitin structures at DNA damage sites. Science. 2007, 316: 1198-1202.
Wang B, Matsuoka S, Ballif BA, Zhang D, Smogorzewska A, Gygi SP, Elledge SJ: Abraxas and RAP80 form a BRCA1 protein complex required for the DNA damage response. Science. 2007, 316: 1194-1198.
Ayoub N, Jeyasekharan AD, Bernal JA, Venkitaraman AR: HP1-beta mobilization promotes chromatin changes that initiate the DNA damage response. Nature. 2008, 453: 682-686.
Sun Y, Jiang X, Xu Y, Ayrapetov MK, Moreau LA, Whetstine JR, Price BD: Histone H3 methylation links DNA damage detection to activation of the tumour suppressor Tip60. Nat Cell Biol. 2009, 11: 1376-1382.
Ziv Y, Bielopolski D, Galanty Y, Lukas C, Taya Y, Schultz DC, Lukas J, Bekker-Jensen S, Bartek J, Shiloh Y: Chromatin relaxation in response to DNA double-strand breaks is modulated by a novel ATM- and KAP-1 dependent pathway. Nat Cell Biol. 2006, 8: 870-876.
Chen CC, Carson JJ, Feser J, Tamburini B, Zabaronick S, Linger J, Tyler JK: Acetylated lysine 56 on histone H3 drives chromatin assembly after repair and signals for the completion of repair. Cell. 2008, 134: 231-243.
Utley RT, Lacoste N, Jobin-Robitaille O, Allard S, Côté J: Regulation of NuA4 histone acetyltransferase activity in transcription and DNA repair by phosphorylation of histone H4. Mol Cell Biol. 2005, 25: 8179-8190.
Ayoub N, Rajendra E, Su X, Jeyasekharan AD, Mahen R, Venkitaraman AR: The carboxyl terminus of Brca2 links the disassembly of Rad51 complexes to mitotic entry. Curr Biol. 2009, 19: 1075-1085.
Shi W, Ma Z, Willers H, Akhtar K, Scott SP, Zhang J, Powell S: Disassembly of MDC1 foci is controlled by ubiquitin-proteasome-dependent degradation. J Biol Chem. 2008, 283: 31608-31616.
Gallinari P, Di Marco S, Jones P, Pallaoro M, Steinkuhler C: HDACs, histone deacetylation and gene transcription: from molecular biology to cancer therapeutics. Cell Res. 2007, 17: 195-211.
Goodarzi AA, Noon AT, Jeggo PA: The impact of heterochromatin on DSB repair. Biochem Soc Trans. 2009, 37: 569-576.
Marks PA, Xu WS: Histone deacetylase inhibitors: Potential in cancer therapy. J Cell Biochem. 2009, 107: 600-608.
Ito A, Kawaguchi Y, Lai CH, Kovacs JJ, Higashimoto Y, Appella E, Yao TP: MDM2-HDAC1-mediated deacetylation of p53 is required for its degradation. EMBO J. 2002, 21: 6236-6245.
Milutinovic S, Zhuang Q, Szyf M: Proliferating cell nuclear antigen associates with histone deacetylase activity, integrating DNA replication and chromatin modification. J Biol Chem. 2002, 277: 20974-20978.
Yarden RI, Brody LC: BRCA1 interacts with components of the histone deacetylase complex. Proc Natl Acad Sci USA. 1999, 96: 4983-4988.
Kim GD, Choi YH, Dimtchev A, Jeong SJ, Dritschilo A, Jung M: Sensing of ionizing radiation-induced DNA damage by ATM through interaction with histone deacetylase. J Biol Chem. 1999, 274: 31127-31130.
Schmidt DR, Schreiber SL: Molecular association between ATR and two components of the nucleosome remodeling and deacetylating complex, HDAC2 and CHD4. Biochemistry. 1999, 38: 14711-14717.
Vieyra D, Loewith R, Scott M, Bonnefin P, Boisvert FM, Cheema P, Pastyryeva S: Human ING1 proteins differentially regulate histone acetylation. J Biol Chem. 2002, 277: 29832-29839.
Anderson LA, Perkins ND: The large subunit of replication factor C interacts with the histone deacetylase, HDAC1. J Biol Chem. 2002, 277: 29550-29554.
Bhakat KK, Mantha AK, Mitra S: Transcriptional regulatory functions of mammalian AP-endonuclease (APE1/Ref-1), an essential multifunctional protein. Antioxid Redox Signal. 2009, 11: 621-638.
Bhaskara S, Chyla BJ, Amann JM, Knutson SK, Cortez D, Sun ZW, Hiebert SW: Deletion of histone deacetylase 3 reveals critical roles in S phase progression and DNA damage control. Mol Cell. 2008, 30: 61-72.
Codina A, Love JD, Li Y, Lazar MA, Neuhaus D, Schwabe JW: Structural insights into the interaction and activation of histone deacetylase 3 by nuclear receptor corepressors. Proc Natl Acad Sci USA. 2005, 102: 6009-6014.
Jones PL, Shi YB: N-CoR-HDAC corepressor complexes: roles in transcriptional regulation by nuclear hormone receptors. Curr Top Microbiol Immunol. 2003, 274: 237-268.
Ishii S, Kurasawa Y, Wong J, Yu-Lee LY: Histone deacetylase 3 localizes to the mitotic spindle and is required for kinetochore-microtubule attachment. Proc Natl Acad Sci USA. 2008, 105: 4179-4184.
Eot-Houllier G, Fulcrand G, Watanabe Y, Magnaghi-Jaulin L, Jaulin C: Histone deacetylase 3 is required for centromeric H3K4 deacetylation and sister chromatid cohesion. Genes Dev. 2008, 22: 2639-2644.
Conti C, Leo E, Eichler GS, Sordet O, Martin MM, Fan A, Aladjem MI, Pommier Y: Inhibition of histone deacetylase in cancer cells slows down replication forks, activates dormant origins, and induces DNA damage. Cancer Res. 2010, 70: 4470-4480.
Kao GD, McKenna WG, Guenther MG, Muschel RJ, Lazar MA, Yen TJ: Histone deacetylase 4 interacts with 53BP1 to mediate the DNA damage response. J Cell Biol. 2003, 160: 1017-1027.
Basile V, Mantovani R, Imbriano C: DNA damage promotes histone deacetylase 4 nuclear localization and repression of G2/M promoters, via p53 C-terminal lysines. J Biol Chem. 2006, 281: 2347-2357.
Namdar M, Perez G, Ngo L, Marks PA: Selective inhibition of histone deacetylase 6 (HDAC6) induces DNA damage and sensitizes transformed cells to anticancer agents. Proc Natl Acad Sci USA. 2010, 107: 20003-20008.
Palacios JA, Herranz D, De Bonis ML, Velasco S, Serrano M, Blasco MA: SIRT1 contributes to telomere maintenance and augments global homologous recombination. J Cell Biol. 2010, 191: 1299-1313.
Uhl M, Csernok A, Aydin S, Kreienberg R, Wiesmüller L, Gatz SA: Role of SIRT1 in homologous recombination. DNA repair. 2010, 9: 383-393.
Yamamori T, DeRicco J, Naqvi A, Hoffman TA, Mattagajasingh I, Kasuno K, Jung SB, Kim CS, Irani K: SIRT1 deacetylates APE1 and regulates cellular base excision repair. Nucleic Acids Res. 2010, 38: 832-845.
Tennen RI, Chua KF: Chromatin regulation and genome maintenance by mammalian SIRT6. Trends Biochem Sci. 2011, 36: 39-46.
Ford J, Jiang M, Milner J: Cancer-specific functions of SIRT1 enable human epithelial cancer cell growth and survival. Cancer Res. 2005, 65: 10457-10463.
Luo J, Nikolaev AY, Imai S, Chen D, Su F, Shiloh A, Guarente L, Gu W: Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell. 2001, 107: 137-148.
Vaziri H, Dessain SK, Ng Eaton E, Imai SI, Frye RA, Pandita TK, Guarente L, Weinberg RA: hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell. 2001, 107: 149-159.
Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, Lin Y, Tran H: Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. 2004, 303: 2011-2015.
Motta MC, Divecha N, Lemieux M, Kamel C, Chen D, Gu W, Bultsma Y, McBurney M, Guarente L: Mammalian SIRT1 represses forkhead transcription factors. Cell. 2004, 116: 551-563.
Jeong J, Juhn K, Lee H, Kim SH, Min BH, Lee KM, Cho MH, Park GH, Lee KH: SIRT1 promotes DNA repair activity and deacetylation of Ku70. Exp Mol Med. 2007, 39: 8-13.
Wang J, Chen J: SIRT1 regulates autoacetylation and histone acetyltransferase activity of TIP60. J Biol Chem. 2010, 285: 11458-11464.
Yuan J, Pu M, Zhang Z, Lou Z: Histone H3-K56 acetylation is important for genomic stability in mammals. Cell Cycle. 2009, 8: 1747-1753.
Yuan Z, Zhang X, Sengupta N, Lane WS, Seto E: SIRT1 regulates the function of the Nijmegen breakage syndrome protein. Mol Cell. 2007, 27: 149-162.
Wang RH, Sengupta K, Li C, Kim HS, Cao L, Xiao C: Impaired DNA damage response, genome instability, and tumorigenesis in SIRT1 mutant mice. Cancer Cell. 2008, 14: 312-323.
Kang H, Jung JW, Kim MK, Chung JH: CK2 is the regulator of SIRT1 substrate-binding affinity, deacetylase activity and cellular response to DNA-damage. PLoS One. 2009, 4: e6611-
Liu X, Wang D, Zhao Y, Tu B, Zheng Z, Wang L, Wang H, Gu W, Roeder RG, Zhu WG: Methyltransferase Set7/9 regulates p53 activity by interacting with Sirtuin 1 (SIRT1). Proc Natl Acad Sci USA. 2011, 108: 1925-1923.
Fan W, Luo J: SIRT1 regulates UV-induced DNA repair through deacetylating XPA. Mol Cell. 2010, 39: 247-258.
Ming M, Shea CR, Guo X, Li X, Soltani K, Han W, He YY: Regulation of global genome nucleotide excision repair by SIRT1 through xeroderma pigmentosum C. Proc Natl Acad Sci USA. 2010, 107: 22623-22628.
Scher MB, Vaquero A, Reinberg D: SirT3 is a nuclear NAD+-dependent histone deacetylase that translocates to the mitochondria upon cellular stress. Genes Dev. 2007, 21: 920-928.
Someya S, Yu W, Hallows WC, Xu J, Vann JM, Leeuwenburgh C, Tanokura M, Denu JM, Prolla TA: Sirt3 mediates reduction of oxidative damage and prevention of age-related hearing loss under caloric restriction. Cell. 2010, 143: 802-812.
Schwer B, Schumacher B, Lombard DB, Xiao C, Kurtev MV, Gao J, Schneider JI, Chai H, Bronson RT, Tsai LH, Deng CX, Alt FW: Neural sirtuin 6 (Sirt6) ablation attenuates somatic growth and causes obesity. Proc Natl Acad Sci USA. 2010, 107: 21790-21794.
McCord RA, Michishita E, Hong T, Berber E, Boxer LD, Kusumoto R: SIRT6 stabilizes DNA-dependent protein kinase at chromatin for DNA double-strand break repair. Aging. 2009, 1: 109-121.
Kazantsev AG, Thompson LM: Therapeutic application of histone deacetylase inhibitors for central nervous system disorders. Nat Rev Drug Discov. 2008, 7: 854-868.
Sekhavat A, Sun JM, Davie JR: Competitive inhibition of histone deacetylase activity by trichostatin A and butyrate. Biochem Cell Biol. 2007, 85: 751-758.
Krämer OH, Zhu P, Ostendorff HP, Golebiewski M, Tiefenbach J, Peters MA, Brill B, Groner B, Bach I, Heinzel T, Göttlicher M: The histone deacetylase inhibitor valproic acid selectively induces proteasomal degradation of HDAC2. EMBO J. 2003, 22: 3411-3420.
Doyle K, Fitzpatrick FA: Redox signaling, alkylation (carbonylation) of conserved cysteines inactivates class I histone deacetylases 1, 2, and 3 and antagonizes their transcriptional repressor function. J Biol Chem. 2010, 285: 17417-17424.
Caito S, Rajendrasozhan S, Cook S, Chung S, Yao H, Friedman AE, Brookes PS, Rahman I: SIRT1 is a redox-sensitive deacetylase that is post-translationally modified by oxidants and carbonyl stress. FASEB J. 2010, 24: 3145-3159.
Chen CS, Wang YC, Yang HC, Huang PH, Kulp SK, Yang CC, Lu YS, Matsuyama S, Chen CY, Chen CS: Histone deacetylase inhibitors sensitize prostate cancer cells to agents that produce DNA double-strand breaks by targeting Ku70 acetylation. Cancer Res. 2007, 67: 5318-5327.
Zhang Y, Carr T, Dimtchev A, Zaer N, Dritschilo A, Jung M: Attenuated DNA damage repair by trichostatin A through BRCA1 suppression. Radiat Res. 2007, 168: 115-124.
Adimoolam S, Sirisawad M, Chen J, Thiemann P, Ford JM, Buggy JJ: HDAC inhibitor PCI-24781 decreases RAD51 expression and inhibits homologous recombination. Proc Natl Acad Sci USA. 2007, 104: 19482-19487.
Xu WS, Parmigiani RB, Marks PA: Histone deacetylase inhibitors: molecular mechanisms of action. Oncogene. 2007, 26: 5541-5552.
Johnstone RW, Licht JD: Histone deacetylase inhibitors in cancer therapy: is transcription the primary target?. Cancer Cell. 2003, 4: 13-18.
Taddei A, Roche D, Bickmore WA, Almouzni G: The effects of histone deacetylase inhibitors on heterochromatin: implications for anticancer therapy?. EMBO Rep. 2005, 6: 520-524.
Zain J, Kaminetzky D, O'Connor OA: Emerging role of epigenetic therapies in cutaneous T-cell lymphomas. Expert Rev Hematol. 2010, 3: 187-203.
Palmieri D, Lockman PR, Thomas FC, Hua E, Herring J, Hargrave E, Johnson M: Vorinostat inhibits brain metastatic colonization in a model of triple-negative breast cancer and induces DNA double-strand breaks. Clin Cancer Res. 2009, 15: 6148-6157.
Chen G, Li A, Zhao M, Gao Y, Zhou T, Xu Y, Du Z, Zhang X, Yu X: Proteomic analysis identifies protein targets responsible for depsipeptide sensitivity in tumor cells. J Proteome Res. 2008, 7: 2733-2742.
Rosato RR, Almenara JA, Maggio SC, Coe S, Atadja P, Dent P, Grant S: Role of histone deacetylase inhibitor-induced reactive oxygen species and DNA damage in LAQ-824/fludarabine antileukemic interactions. Mol Cancer Ther. 2008, 7: 3285-3297.
Dashwood RH, Myzak MC, Ho E: Dietary HDAC inhibitors: time to rethink weak ligands in cancer chemoprevention?. Carcinogenesis. 2006, 27: 344-349.
Dashwood RH, Ho E: Dietary histone deacetylase inhibitors: from cells to mice to man. Semin Cancer Biol. 2007, 17: 363-369.
Higdon JV, Delage B, Williams DE, Dashwood RH: Cruciferous vegetables and human cancer risk: epidemiologic evidence and mechanistic basis. Pharmacol Res. 2007, 55: 224-236.
Hayes JD, Kelleher MO, Eggleston IM: The cancer chemopreventive actions of phytochemicals derived from glucosinolates. Eur J Nutr. 2008, 47: 73-88.
Myzak MC, Karplus PA, Chung FL, Dashwood RH: A novel mechanism of chemoprotection by sulforaphane: inhibition of histone deacetylase. Cancer Res. 2004, 64: 5767-5774.
Lea MA, Randolph VM, Lee JE, desBordes C: Induction of histone acetylation in mouse erythroleukemia cells by some organosulfur compounds including allyl isothiocyanate. Int J Cancer. 2001, 92: 784-789.
Batra S, Sahu RP, Kandala PK, Srivastava SK: Benzyl isothiocyanate-mediated inhibition of histone deacetylase leads to NF-kappaB turnoff in human pancreatic carcinoma cells. Mol Cancer Ther. 2010, 9: 1596-1608.
Ma X, Fang Y, Beklemisheva A, Dai W, Feng J, Ahmed T, Liu D, Chiao JW: Phenylhexyl isothiocyanate inhibits histone deacetylases and remodels chromatins to induce growth arrest in human leukemia cells. Int J Oncol. 2006, 28: 1287-1293.
Wang LG, Liu XM, Fang Y, Dai W, Chiao FB, Puccio GM, Feng J, Liu D, Chiao JW: De-repression of the p21 promoter in prostate cancer cells by an isothiocyanate via inhibition of HDACs and c-Myc. Int J Oncol. 2008, 33: 375-380.
Meeran SM, Patel SN, Tollefsbol TO: Sulforaphane causes epigenetic repression of hTERT expression in human breast cancer cell lines. PLoS One. 2010, 5: e11457-
Rajendran P, Delage B, Dashwood WM, Yu TW, Wuth B, Williams DE, Ho E, Dashwood RH: Histone deacetylase turnover and recovery in sulforaphane-treated colon cancer cells: competing actions of 14-3-3 and Pin1 in HDAC3/SMRT corepressor complex dissociation/reassembly. Mol Cancer. 2011, 10: 68-
Clarke JD, Hsu A, Yu Z, Dashwood RH, Ho E: Differential effects of sulforaphane on histone deacetylases, cell cycle arrest and apoptosis in normal prostate cells versus hyperplastic and cancerous prostate cells. Mol Nutr Food Res. 2011
Kassie F, Laky B, Nobis E, Kundi M, Knasmüller S: Genotoxic effects of methyl isothiocyanate. Mutat Res. 2001, 490: 1-9.
Kassie F, Pool-Zobel B, Parzefall W, Knasmüller S: Genotoxic effects of benzyl isothiocyanate, a natural chemopreventive agent. Mutagenesis. 1999, 14: 595-604.
Kassie F, Knasmüller S: Genotoxic effects of allyl isothiocyanate (AITC) and phenethyl isothiocyanate (PEITC). Chem-Biol Interact. 2000, 127: 163-180.
Zhang R, Loganathan S, Humphreys I, Srivastava SK: Benzyl isothiocyanate-induced DNA damage causes G2/M cell cycle arrest and apoptosis in human pancreatic cancer cells. J Nutr. 2006, 136: 2728-2734.
Sekine-Suzuki E, Yu D, Kubota N, Okayasu R, Anzai K: Sulforaphane induces DNA double strand breaks predominantly repaired by homologous recombination pathway in human cancer cells. Biochem Biophys Res Commun. 2008, 377: 341-345.
Sestili P, Paolillo M, Lenzi M, Colombo E, Vallorani L, Casadei L, Martinelli C, Fimognari C: Sulforaphane induces DNA single strand breaks in cultured human cells. Mutat Res. 2010, 689: 65-73.
Andelová H, Rudolf E, Cervinka M: In vitro antiproliferative effects of sulforaphane on human colon cancer cell line SW620. Acta Medica (Hradec Kralove). 2007, 50: 171-176.
Singh SV, Herman-Antosiewicz A, Singh AV, Lew KL, Srivastava SK, Kamath R, Brown KD, Zhang L, Baskaran R: Sulforaphane-induced G2/M phase cell cycle arrest involves checkpoint kinase 2-mediated phosphorylation of cell division cycle 25C. J Biol Chem. 2004, 279: 25813-25822.
Zeng H, Trujillo ON, Moyer MP, Botnen JH: Prolonged sulforaphane treatment activates survival signaling in nontumorigenic NCM460 colon cells but apoptotic signaling in tumorigenic HCT116 colon cells. Nutr Cancer. 2011, 63: 248-255.
Xiao D, Powolny AA, Antosiewicz J, Hahm ER, Bommareddy A, Zeng Y, Desai D, Amin S, Herman-Antosiewicz A, Singh SV: Cellular responses to cancer chemopreventive agent D, L-sulforaphane in human prostate cancer cells are initiated by mitochondrial reactive oxygen species. Pharm Res. 2009, 26: 1729-1738.
Xiao D, Powolny AA, Moura MB, Kelley EE, Bommareddy A, Kim SH, Hahm ER, Normolle D, Van Houten B, Singh SV: Phenethyl isothiocyanate inhibits oxidative phosphorylation to trigger reactive oxygen species-mediated death of human prostate cancer cells. J Biol Chem. 2010, 285: 26558-26569.
Powolny AA, Singh SV: Differential response of normal (PrEC) and cancerous human prostate cells (PC-3) to phenethyl isothiocyanate-mediated changes in expression of antioxidant defense genes. Pharm Res. 2010, 27: 2766-2775.
Liu KC, Huang YT, Wu PP, Ji BC, Yang JS: The roles of AIF and Endo G in the apoptotic effects of benzyl isothiocyanate on DU 145 human prostate cancer cells via the mitochondrial signaling pathway. Int J Oncol. 2011, 38: 787-796.
Moon DO, Kang SH, Kim KC, Kim MO, Choi YH, Kim GY: Sulforaphane decreases viability and telomerase activity in hepatocellular carcinoma Hep3B cells through the reactive oxygen species-dependent pathway. Cancer Lett. 2010, 295: 260-266.
Sharma R, Sharma A, Chaudhary P, Pearce V, Vatsyayan R, Singh SV, Awasthi S, Awasthi YC: Role of lipid peroxidation in cellular responses to D, L-sulforaphane, a promising cancer chemopreventive agent. Biochemistry. 2010, 49: 3191-3202.
Mi L, Chung FL: Binding to protein by isothiocyanates: a potential mechanism for apoptosis induction in human non small lung cancer cells. Nutr Cancer. 2008, 60: 12-20.
Ahmad A, Sakr WA, Rahman KM: Anticancer properties of indole compounds: mechanism of apoptosis induction and role in chemotherapy. Curr Drug Targets. 2010, 11: 652-666.
Brew CT, Aronchik I, Hsu JC, Sheen JH, Dickson RB, Bjeldanes LF, Firestone GL: Indole-3-carbinol activates the ATM signaling pathway independent of DNA damage to stabilize p53 and induce G1 arrest of human mammary epithelial cells. Int J Cancer. 2006, 118: 857-868.
Wu Y, Feng X, Jin Y, Wu Z, Hankey W, Paisie C, Li L, Liu F, Barsky SH, Zhang W, Ganju R, Zou X: A novel mechanism of indole-3-carbinol effects on breast carcinogenesis involves induction of Cdc25A degradation. Cancer Prev Res. 2010, 3: 818-828.
Bhatnagar N, Li X, Chen Y, Zhou X, Garrett SH, Guo B: 3,3'-diindolylmethane enhances the efficacy of butyrate in colon cancer prevention through down-regulation of survivin. Cancer Prev Res. 2009, 2: 581-589.
Li Y, Li X, Guo B: Chemopreventive agent 3,3'-diindolylmethane selectively induces proteasomal degradation of class I histone deacetylases. Cancer Res. 2010, 70: 646-654.
Fan S, Meng Q, Auborn K, Carter T, Rosen EM: BRCA1 and BRCA2 as molecular targets for phytochemicals indole-3-carbinol and genistein in breast and prostate cancer cells. Br J Cancer. 2006, 94: 407-426.
Auborn KJ, Fan S, Rosen EM, Goodwin L, Chandraskaren A, Williams DE, Chen D, Carter TH: Indole-3-carbinol is a negative regulator of estrogen. J Nutr. 2003, 133: 2470S-2475S.
Pajak B, Gajkowska B, Orzechowski A: Molecular basis of parthenolide-dependent proapoptotic activity in cancer cells. Folia Histochem Cytobiol. 2008, 46: 129-135.
Gopal YN, Arora TS, Van Dyke MW: Parthenolide specifically depletes histone deacetylase 1 protein and induces cell death through ataxia telangiectasia mutated. Chem Biol. 2007, 14: 813-823.
Gopal YN, Chanchorn E, Van Dyke MW: Parthenolide promotes the ubiquitination of MDM2 and activates p53 cellular functions. Mol Cancer Ther. 2009, 8: 552-562.
Balasubramanyam K, Swaminathan V, Ranganathan A, Kundu TK: Small molecule modulators of histone acetyltransferase p300. J Biol Chem. 2003, 278: 19134-19140.
Sun Y, Jiang X, Chen S, Price BD: Inhibition of histone acetyltransferase activity by anacardic acid sensitizes tumor cells to ionizing radiation. FEBS Lett. 2006, 580: 4353-4356.
Ruotolo R, Tosi F, Vernarecci S, Ballario P, Mai A, Filetici P, Ottonello S: Chemogenomic profiling of the cellular effects associated with histone H3 acetylation impairment by a quinoline-derived compound. Genomics. 2010, 96: 272-280.
Kim MK, Shin JM, Eun HC, Chung JH: The role of p300 histone acetyltransferase in UV-induced histone modifications and MMP-1 gene transcription. PLoS One. 2009, 4: e4864-
Iciek M, Kwiecień I, Włodek L: Biological properties of garlic and garlic-derived organosulfur compounds. Environ Mol Mutagen. 2009, 50: 247-65.
Nian H, Delage B, Ho E, Dashwood RH: Modulation of histone deacetylase activity by dietary isothiocyanates and allyl sulfides: studies with sulforaphane and garlic organosulfur compounds. Environ Mol Mutagen. 2009, 50: 213-221.
Lea MA, Randolph VM, Patel M: Increased acetylation of histones induced by diallyl disulfide and structurally related molecules. Int J Oncol. 1999, 15: 347-352.
Lea MA, Randolph VM: Induction of histone acetylation in rat liver and hepatoma by organosulfur compounds including diallyl disulfide. Anticancer Res. 2001, 21: 2841-2845.
Lea MA, Rasheed M, Randolph VM, Khan F, Shareef A, desBordes C: Induction of histone acetylation and inhibition of growth of mouse erythroleukemia cells by S-allylmercaptocysteine. Nutr Cancer. 2002, 43: 90-102.
Nian H, Delage B, Pinto JT, Dashwood RH: Allyl mercaptan, a garlic-derived organosulfur compound, inhibits histone deacetylase and enhances Sp3 binding on the P21WAF1 promoter. Carcinogenesis. 2008, 29: 1816-1824.
Wang HC, Yang JH, Hsieh SC, Sheen LY: Allyl sulfides inhibit cell growth of skin cancer cells through induction of DNA damage mediated G2/M arrest and apoptosis. J Agric Food Chem. 2010, 58: 7096-7103.
Ling H, Wen L, Ji XX, Tang YL, He J, Tan H, Xia H, Zhou JG, Su Q: Growth inhibitory effect and Chk1-dependent signaling involved in G2/M arrest on human gastric cancer cells induced by diallyl disulfide. Braz J Med Biol Res. 2010, 43: 271-278.
Herman-Antosiewicz A, Singh SV: Checkpoint kinase 1 regulates diallyl trisulfide-induced mitotic arrest in human prostate cancer cells. J Biol Chem. 2005, 280: 28519-28528.
Wang H, Zhao Y, Li L, McNutt MA, Wu L, Lu S, Yu Y, Zhou W, Feng J, Chai G, Yang Y, Zhu WG: An ATM- and Rad3-related (ATR) signaling pathway and a phosphorylation-acetylation cascade are involved in activation of p53/p21Waf1/Cip1 in response to 5-aza-2'-deoxycytidine treatment. J Biol Chem. 2008, 283: 2564-2574.
Konkimalla VS, Wang G, Kaina B, Efferth T: Microarray-based expression of DNA repair genes does not correlate with growth inhibition of cancer cells by natural products derived from traditional Chinese medicine. Cancer Genomics Proteomics. 2008, 5: 79-84.
Yi L, Ji XX, Lin M, Tan H, Tang Y, Wen L, Ma YH, Su Q: Diallyl disulfide induces apoptosis in human leukemia HL-60 cells through activation of JNK mediated by reactive oxygen. Pharmazie. 2010, 65: 693-698.
Aquilano K, Filomeni G, Baldelli S, Piccirillo S, De Martino A, Rotilio G, Ciriolo MR: Neuronal nitric oxide synthase protects neuroblastoma cells from oxidative stress mediated by garlic derivatives. J Neurochem. 2007, 101: 1327-1337.
Lu HF, Sue CC, Yu CS, Chen SC, Chen GW, Chung JG: Diallyl disulfide (DADS) induced apoptosis undergo caspase-3 activity in human bladder cancer T24 cells. Food Chem Toxicol. 2004, 42: 1543-1552.
Sriram N, Kalayarasan S, Ashokkumar P, Sureshkumar A, Sudhandiran G: Diallyl sulfide induces apoptosis in Colo 320 DM human colon cancer cells: involvement of caspase-3, NF-kappaB, and ERK-2. Mol Cell Biochem. 2008, 311: 157-165.
Hatfield DL, Yoo MH, Carlson BA, Gladyshev VN: Selenoproteins that function in cancer prevention and promotion. Biochim Biophys Acta. 2009, 1790: 1541-1545.
Zeng H, Combs GF: Selenium as an anticancer nutrient: roles in cell proliferation and tumor cell invasion. J Nutr Biochem. 2008, 19: 1-7.
Clark LC, Combs GF, Turnbull BW, Slate EH, Chalker DK, Chow J, Davis LS: Effects of selenium supplementation for cancer prevention in patients with carcinoma of the skin. A randomized controlled trial. Nutritional Prevention of Cancer Study Group. JAMA. 1996, 276: 1957-1963.
Sanmartín C, Plano D, Palop JA: Selenium compounds and apoptotic modulation: a new perspective in cancer therapy. Mini Rev Med Chem. 2008, 8: 1020-1031.
Xiang N, Zhao R, Song G, Zhong W: Selenite reactivates silenced genes by modifying DNA methylation and histones in prostate cancer cells. Carcinogenesis. 2008, 29: 2175-2181.
Lee JI, Nian H, Cooper AJ, Sinha R, Dai J, Bisson WH, Dashwood RH, Pinto JT: Alpha-keto acid metabolites of naturally occurring organoselenium compounds as inhibitors of histone deacetylase in human prostate cancer cells. Cancer Prev Res. 2009, 2: 683-693.
Nian H, Bisson WH, Dashwood WM, Pinto JT, Dashwood RH: Alpha-keto acid metabolites of organoselenium compounds inhibit histone deacetylase activity in human colon cancer cells. Carcinogenesis. 2009, 30: 1416-1423.
Goulet AC, Watts G, Lord JL, Nelson MA: Profiling of selenomethionine responsive genes in colon cancer by microarray analysis. Cancer Biol Ther. 2007, 6: 494-503.
Zhou N, Xiao H, Li TK, Nur-E-Kamal A, Liu LF: DNA damage-mediated apoptosis induced by selenium compounds. J Biol Chem. 2003, 278: 29532-29537.
Qi Y, Schoene NW, Lartey FM, Cheng WH: Selenium compounds activate ATM-dependent DNA damage response via the mismatch repair protein hMLH1 in colorectal cancer cells. J Biol Chem. 2010, 285: 33010-33017.
Wu M, Kang MM, Schoene NW, Cheng WH: Selenium compounds activate early barriers of tumorigenesis. J Biol Chem. 2010, 285: 12055-12062.
Lee JS: Functional link between DNA damage responses and transcriptional regulation by ATM in response to a histone deacetylase inhibitor TSA. Cancer Res Treat. 2007, 39: 116-24.
Kato MA, Finley DJ, Lubitz CC, Zhu B, Moo TA, Loeven MR, Ricci JA, Zarnegar R, Katdare M, Fahey TJ: Selenium decreases thyroid cancer cell growth by increasing expression of GADD153 and GADD34. Nutr Cancer. 2010, 62: 66-73.
Jiang W, Jiang C, Pei H, Wang L, Zhang J, Hu H, Lü J: In vivo molecular mediators of cancer growth suppression and apoptosis by selenium in mammary and prostate models: lack of involvement of gadd genes. Mol Cancer Ther. 2009, 8: 682-691.
Chen T, Wong YS: Selenocystine induces reactive oxygen species-mediated apoptosis in human cancer cells. Biomed Pharmacother. 2009, 63: 105-113.
Kotsopoulos J, Chen Z, Vallis KA, Poll A, Ghadirian P, Kennedy G, Ainsworth P, Narod SA: Toenail selenium status and DNA repair capacity among female BRCA1 mutation carriers. Cancer Causes Control. 2010, 21: 679-687.
Katiyar S, Elmets CA, Katiyar SK: Green tea and skin cancer: photoimmunology, angiogenesis and DNA repair. J Nutr Biochem. 2007, 18: 287-296.
Pandey M, Shukla S, Gupta S: Promoter demethylation and chromatin remodeling by green tea polyphenols leads to re-expression of GSTP1 in human prostate cancer cells. Int J Cancer. 2010, 126: 2520-2533.
Nandakumar V, Vaid M, Katiyar SK: (-)-Epigallocatechin-3-gallate reactivates silenced tumor suppressor genes, Cip1/p21 and p16INK4a, by reducing DNA methylation and increasing histones acetylation in human skin cancer cells. Carcinogenesis. 2011, 32: 537-544.
Li Y, Yuan YY, Meeran SM, Tollefsbol TO: Synergistic epigenetic reactivation of estrogen receptor-α (ERα) by combined green tea polyphenol and histone deacetylase inhibitor in ERα-negative breast cancer cells. Mol Cancer. 2010, 9: 274-
Choi KC, Jung MG, Lee YH, Yoon JC, Kwon SH, Kang HB: Epigallocatechin-3-gallate, a histone acetyltransferase inhibitor, inhibits EBV-induced B lymphocyte transformation via suppression of RelA acetylation. Cancer Res. 2009, 69: 583-592.
Balasubramanian S, Adhikary G, Eckert RL: The Bmi-1 polycomb protein antagonizes the (-)-Epigallocatechin-3-Gallate dependent suppression of skin cancer cell survival. Carcinogenesis. 2010, 31: 496-503.
Delage B, Dashwood RH: Dietary manipulation of histone structure and function. Annu Rev Nutr. 2008, 28: 347-366.
Li GX, Chen YK, Hou Z, Xiao H, Jin H, Lu G, Lee MJ, Liu B, Guan F, Yang Z, Yu A, Yang CS: Pro-oxidative activities and dose-response relationship of (-)-epigallocatechin-3-gallate in the inhibition of lung cancer cell growth: a comparative study in vivo and in vitro. Carcinogenesis. 2010, 31: 902-910.
Suganuma M, Saha A, Fujiki H: New cancer treatment strategy using combination of green tea catechins and anticancer drugs. Cancer Sci. 2011, 102: 317-323.
Kurland JF, Tansey WP: Myc-mediated transcriptional repression by recruitment of histone deacetylase. Cancer Res. 2008, 68: 3624-3629.
Scuto A, Kirschbaum M, Kowolik C, Kretzner L, Juhasz A, Atadja P, Pullarkat V, Bhatia R, Forman S, Yen Y, Jove R: The novel histone deacetylase inhibitor, LBH589, induces expression of DNA damage response genes and apoptosis in Ph- acute lymphoblastic leukemia cells. Blood. 2008, 111: 5093-5100.
Ahmad KA, Harris NH, Johnson AD, Lindvall HC, Wang G, Ahmed K: Protein kinase CK2 modulates apoptosis induced by resveratrol and epigallocatechin-3-gallate in prostate cancer cells. Mol Cancer Ther. 2007, 6: 1006-1012.
Fang MZ, Wang Y, Ai N, Hou Z, Sun Y, Lu H: Tea polyphenol (-)-epigallocatechin-3-gallate inhibits DNA methyltransferase and reactivates methylation-silenced genes in cancer cell lines. Cancer Res. 2003, 63: 7563-7570.
Lee WJ, Shim JY, Zhu BT: Mechanisms for the inhibition of DNA methyltransferases by tea catechins and bioflavonoids. Mol Pharmacol. 2005, 68: 1018-1030.
Wang HF, Chuang SM, Hsiao CC, Cherng SH: A synergistic effect of GABA tea and copper(II) on DNA breakage in human peripheral lymphocytes. Food Chem Toxicol. 2011, 49: 955-962.
Aggarwal BB: Targeting inflammation-induced obesity and metabolic diseases by curcumin and other nutraceuticals. Annu Rev Nutr. 2010, 30: 173-199.
Padhye S, Chavan D, Pandey S, Deshpande J, Swamy KV, Sarkar FH: Perspectives on chemopreventive and therapeutic potential of curcumin analogs in medicinal chemistry. Mini Rev Med Chem. 2010, 10: 372-387.
Bar-Sela G, Epelbaum R, Schaffer M: Curcumin as an anti-cancer agent: review of the gap between basic and clinical applications. Curr Med Chem. 2010, 17: 190-197.
Epstein J, Sanderson IR, Macdonald TT: Curcumin as a therapeutic agent: the evidence from in vitro, animal and human studies. Br J Nutr. 2010, 103: 1545-1557.
Fu S, Kurzrock R: Development of curcumin as an epigenetic agent. Cancer. 2010, 116: 4670-4676.
Chung S, Yao H, Caito S, Hwang JW, Arunachalam G, Rahman I: Regulation of SIRT1 in cellular functions: role of polyphenols. Arch Biochem Biophys. 2010, 501: 79-90.
Li Y, Kong D, Wang Z, Sarkar FH: Regulation of microRNAs by natural agents: an emerging field in chemoprevention and chemotherapy research. Pharm Res. 2010, 27: 1027-1041.
Marcu MG, Jung YJ, Lee S, Chung EJ, Lee MJ, Trepel J, Neckers L: Curcumin is an inhibitor of p300 histone acetylatransferase. Med Chem. 2006, 2: 169-174.
Balasubramanyam K, Varier RA, Altaf M, Swaminathan V, Siddappa NB, Ranga U: Curcumin, a novel p300/CREB-binding protein-specific inhibitor of acetyltransferase, represses the acetylation of histone/nonhistone proteins and histone acetyltransferase-dependent chromatin transcription. J Biol Chem. 2004, 279: 51163-51171.
Kang J, Chen J, Shi Y, Jia J, Zhang Y: Curcumin-induced histone hypoacetylation: the role of reactive oxygen species. Biochem Pharmacol. 2005, 69: 1205-1213.
Mai A, Rotili D, Tarantino D, Ornaghi P, Tosi F, Vicidomini C: Small molecule inhibitors of histone acetyltransferase activity: identification and biological properties. J Med Chem. 2006, 49: 6897-6907.
Lv BH, Zhang L, Zhu CC, Liu J: Inhibition of curcumin on histone deacetylase and expression promotion of P21 (WAF1/CIP1) in HepG2 cells. Zhongguo Zhong Yao Za Zhi. 2007, 32: 2051-2055.
Chen Y, Shu W, Chen W, Wu Q, Liu H, Cui G: Curcumin, both histone deacetylase and p300/CBP-specific inhibitor, represses the activity of nuclear factor kappa B and Notch 1 in Raji cells. Basic Clin Pharmacol Toxicol. 2007, 101: 427-433.
Bora-Tatar G, Dayangac-Erden D, Demir AS, Dalkara S, Yelekci K, Erdem-Yurter H: Molecular modifications on carboxylic acid derivatives as potent histone deacetylase inhibitors: Activity and docking studies. Bioorg Med Chem. 2009, 17: 5219-5228.
Meja KK, Rajendrasozhan S, Adenuga D, Biswas SK, Sundar IK: Curcumin restores corticosteroid function in monocytes exposed to oxidants by maintaining HDAC2. Am J Respir Cell Mol Biol. 2008, 39: 312-323.
Rowe DL, Ozbay T, O'Regan RM, Nahta R: Modulation of the BRCA1 protein and induction of apoptosis in triple negative breast cancer cell lines by the polyphenolic compound Curcumin. Breast Cancer (Auckl). 2009, 3: 61-75.
Sahu RP, Batra S, Srivastava SK: Activation of ATM/Chk1 by curcumin causes cell cycle arrest and apoptosis in human pancreatic cancer cells. Br J Cancer. 2009, 100: 425-433.
Saha A, Kuzuhara T, Echigo N, Fujii A, Suganuma M, Fujiki H: Apoptosis of human lung cancer cells by curcumin mediated through up-regulation of growth arrest and DNA damage inducible genes 45 and 153. Biol Pharm Bull. 2010, 33: 1291-1299.
Lin SS, Huang HP, Yang JS, Wu JY, Hsia TC: DNA damage and endoplasmic reticulum stress mediated curcumin-induced cell cycle arrest and apoptosis in human lung carcinoma A-549 cells through the activation caspases cascade- and mitochondrial-dependent pathway. Cancer Lett. 2008, 272: 77-90.
Sun Y, Jiang X, Chen S, Fernandes N, Price BD: A role for the Tip60 histone acetyltransferase in the acetylation and activation of ATM. Proc Natl Acad Sci USA. 2005, 102: 13182-13187.
Chirnomas D, Taniguchi T, de la Vega M, Vaidya AP, Vasserman M: Chemosensitization to cisplatin by inhibitors of the Fanconi anemia/BRCA pathway. Mol Cancer Ther. 2006, 5: 952-961.
Dhandapani KM, Mahesh VB, Brann DW: Curcumin suppresses growth and chemoresistance of human glioblastoma cells via AP-1 and NFkappaB transcription factors. J Neurochem. 2007, 102: 522-538.
Cao J, Liu Y, Jia L, Zhou HM, Kong Y, Yang G, Jiang LP, Li QJ, Zhong LF: Curcumin induces apoptosis through mitochondrial hyperpolarization and mtDNA damage in human hepatoma G2 cells. Free Radic Biol Med. 2007, 43: 968-975.
Hail N: Mitochondrial reactive oxygen species affect sensitivity to curcumin-induced apoptosis. Free Radic Biol Med. 2008, 44: 1382-1393.
Hilchie AL, Furlong SJ, Sutton K, Richardson A, Robichaud MR, Giacomantonio CA, Ridgway ND, Hoskin DW: Curcumin-induced apoptosis in PC3 prostate carcinoma cells is caspase-independent and involves cellular ceramide accumulation and damage to mitochondria. Nutr Cancer. 2010, 62: 379-389.
Athar M, Back JH, Kopelovich L, Bickers DR, Kim AL: Multiple molecular targets of resveratrol: Anti-carcinogenic mechanisms. Arch Biochem Biophys. 2009, 486: 95-102.
Howitz KT, Bitterman KJ, Cohen HY, Lamming DW, Lavu S, Wood JG: Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature. 2003, 425: 191-196.
Wood JG, Rogina B, Lavu S, Howitz K, Helfand SL, Tatar M: Sirtuin activators mimic caloric restriction and delay ageing in metazoans. Nature. 2004, 430: 686-689.
Beher D, Wu J, Cumine S, Kim KW, Lu SC, Atangan L: Resveratrol is not a direct activator of SIRT1 enzyme activity. Chem Biol Drug Des. 2009, 74: 619-624.
Malik R, Kashyap A, Bansal K, Sharma P, Rayasam GV, Davis JA, Bora RS, Ray A, Saini KS: Comparative deacetylase activity of wild type and mutants of SIRT1. Biochem Biophys Res Commun. 2010, 391: 739-743.
Boily G, He XH, Pearce B, Jardine K, McBurney MW: SirT1-null mice develop tumors at normal rates but are poorly protected by resveratrol. Oncogene. 2009, 28: 2882-2893.
Wang RH, Zheng Y, Kim HS, Xu X, Cao L, Luhasen T: Interplay among BRCA1, SIRT1, and Survivin during BRCA1-associated tumorigenesis. Mol Cell. 2008, 32: 11-20.
Kai L, Samuel SK, Levenson AS: Resveratrol enhances p53 acetylation and apoptosis in prostate cancer by inhibiting MTA1/NuRD complex. Int J Cancer. 2010, 126: 1538-1548.
Gatz SA, Keimling M, Baumann C, Dörk T, Debatin KM, Fulda S, Wiesmüller L: Resveratrol modulates DNA double-strand break repair pathways in an ATM/ATR-p53- and -Nbs1-dependent manner. Carcinogenesis. 2008, 29: 519-527.
Leone S, Cornetta T, Basso E, Cozzi R: Resveratrol induces DNA double-strand breaks through human topoisomerase II interaction. Cancer Lett. 2010, 295: 167-172.
Rusin M, Zajkowicz A, Butkiewicz D: Resveratrol induces senescence-like growth inhibition of U-2 OS cells associated with the instability of telomeric DNA and upregulation of BRCA1. Mech Ageing Dev. 2009, 130: 528-537.
Tyagi A, Singh RP, Agarwal C, Siriwardana S, Sclafani RA, Agarwal R: Resveratrol causes Cdc2-tyr15 phosphorylation via ATM/ATR-Chk1/2-Cdc25C pathway as a central mechanism for S phase arrest in human ovarian carcinoma Ovcar-3 cells. Carcinogenesis. 2005, 26: 1978-1987.
Azmi AS, Bhat SH, Hadi SM: Resveratrol-Cu(II) induced DNA breakage in human peripheral lymphocytes: implications for anticancer properties. FEBS Lett. 2005, 579: 3131-3135.
Hadi SM, Ullah MF, Azmi AS, Ahmad A, Shamim U, Zubair H, Khan HY: Resveratrol mobilizes endogenous copper in human peripheral lymphocytes leading to oxidative DNA breakage: a putative mechanism for chemoprevention of cancer. Pharm Res. 2010, 27: 979-988.
Kaur M, Agarwal R, Agarwal C: Grape seed extract induces anoikis and caspase-mediated apoptosis in human prostate carcinoma LNCaP cells: possible role of ataxia telangiectasia mutated-p53 activation. Mol Cancer Ther. 2006, 5: 1265-1274.
Guha P, Dey A, Sen R, Chatterjee M, Chattopadhyay S, Bandyopadhyay SK: Intracellular GSH depletion triggered mitochondrial Bax translocation to accomplish resveratrol-induced apoptosis in the U937 cell line. J Pharmacol Exp Ther. 2011, 336: 206-214.
Banerjee S, Li Y, Wang Z, Sarkar FH: Multi-targeted therapy of cancer by genistein. Cancer Lett. 2008, 269: 226-242.
Li Y, Tollefsbol TO: Impact on DNA methylation in cancer prevention and therapy by bioactive dietary components. Curr Med Chem. 2010, 17: 2141-2151.
Kikuno N, Shiina H, Urakami S, Kawamoto K, Hirata H, Tanaka Y: Genistein mediated histone acetylation and demethylation activates tumor suppressor genes in prostate cancer cells. Int J Cancer. 2008, 123: 552-560.
Basak S, Pookot D, Noonan EJ, Dahiya R: Genistein down-regulates androgen receptor by modulating HDAC6-Hsp90 chaperone function. Mol Cancer Ther. 2008, 7: 3195-3202.
Ye R, Goodarzi AA, Kurz EU, Saito S, Higashimoto Y, Lavin MF, Appella E, Anderson CW, Lees-Miller SP: The isoflavonoids genistein and quercetin activate different stress signaling pathways as shown by analysis of site-specific phosphorylation of ATM, p53 and histone H2AX. DNA Repair. 2004, 3: 235-244.
Ding H, Duan W, Zhu WG, Ju R, Srinivasan K, Otterson GA, Villalona-Calero MA: P21 response to DNA damage induced by genistein and etoposide in human lung cancer cells. Biochem Biophys Res Commun. 2003, 305: 950-956.
Chang KL, Kung ML, Chow NH, Su SJ: Genistein arrests hepatoma cells at G2/M phase: involvement of ATM activation and upregulation of p21waf1/cip1 and Wee1. Biochem Pharmacol. 2004, 67: 717-726.
Ouyang G, Yao L, Ruan K, Song G, Mao Y, Bao S: Genistein induces G2/M cell cycle arrest and apoptosis of human ovarian cancer cells via activation of DNA damage checkpoint pathways. Cell Biol Int. 2009, 33: 1237-1244.
Shiau RJ, Chen KY, Wen YD, Chuang CH, Yeh SL: Genistein and beta-carotene enhance the growth-inhibitory effect of trichostatin A in A549 cells. Eur J Nutr. 2010, 49: 19-25.
Ide H, Jingsong Y, Yan L, China T, Kumamoto T, Koseki T, Muto S, Horie S: Testosterone augments polyphenol-induced DNA damage response in prostate cancer cell line, LNCaP. Cancer Sci. 2011, 102: 468-471.
Vauzour D, Vafeiadou K, Rice-Evans C, Cadenas E, Spencer JP: Inhibition of cellular proliferation by the genistein metabolite 5,7,3',4'-tetrahydroxyisoflavone is mediated by DNA damage and activation of the ATR signalling pathway. Arch Biochem Biophys. 2007, 468: 159-166.
Regenbrecht CR, Jung M, Lehrach H, Adjaye J: The molecular basis of genistein-induced mitotic arrest and exit of self-renewal in embryonal carcinoma and primary cancer cell lines. BMC Med Genomics. 2008, 1: 49-
Tominaga Y, Wang A, Wang RH, Wang X, Cao L, Deng CX: Genistein inhibits Brca1 mutant tumor growth through activation of DNA damage checkpoints, cell cycle arrest, and mitotic catastrophe. Cell Death Differ. 2007, 14: 472-479.
Ismail IA, Kang KS, Lee HA, Kim JW, Sohn YK: Genistein-induced neuronal apoptosis and G2/M cell cycle arrest is associated with MDC1 up-regulation and PLK1 down-regulation. Eur J Pharmacol. 2007, 575: 12-20.
Ullah MF, Shamim U, Hanif S, Azmi AS, Hadi SM: Cellular DNA breakage by soy isoflavone genistein and its methylated structural analogue biochanin A. Mol Nutr Food Res. 2009, 53: 1376-1385.
Ye R, Bodero A, Zhou BB, Khanna KK, Lavin MF, Lees-Miller SP: The plant isoflavanoid genistein activates p53 and Chk2 in an ATM-dependent manner. J Biol Chem. 2001, 276: 4828-4833.
Leung HY, Yung LH, Poon CH, Shi G, Lu AL, Leung LK: Genistein protects against polycyclic aromatic hydrocarbon-induced oxidative DNA damage in non-cancerous breast cells MCF-10A. Br J Nutr. 2009, 101: 257-262.
Lee WJ, Chen YR, Tseng TH: Quercetin induces FasL-related apoptosis, in part, through promotion of histone H3 acetylation in human leukemia HL-60 cells. Oncol Rep. 2011, 25 (2): 583-591.
Yamashita N, Tanemura H, Kawanishi S: Mechanism of oxidative DNA damage induced by quercetin in the presence of Cu(II). Mutat Res. 1999, 425: 107-115.
Tan J, Wang B, Zhu L: DNA binding and oxidative DNA damage induced by a quercetin copper(II) complex: potential mechanism of its antitumor properties. J Biol Inorg Chem. 2009, 14: 727-739.
Izzard RA, Jackson SP, Smith GC: Competitive and noncompetitive inhibition of the DNA-dependent protein kinase. Cancer Res. 1999, 59: 2581-2586.
Sebova K, Fridrichova I: Epigenetic tools in potential anticancer therapy. Anticancer Drugs. 2010, 21: 565-577.
Kim MS, Blake M, Baek JH, Kohlhagen G, Pommier Y, Carrier F: Inhibition of histone deacetylase increases cytotoxicity to anticancer drugs targeting DNA. Cancer Res. 2003, 63: 7291-7300.
Ozaki K, Kishikawa F, Tanaka M, Sakamoto T, Tanimura S, Kohno M: Histone deacetylase inhibitors enhance the chemosensitivity of tumor cells with cross-resistance to a wide range of DNA damaging drugs. Cancer Sci. 2008, 99: 376-384.
Lee JH, Choy ML, Ngo L, Foster SS, Marks PA: Histone deacetylase inhibitor induces DNA damage, which normal but not transformed cells can repair. Proc Natl Acad Sci USA. 2010, 107: 14639-14644.
Yu D, Sekine-Suzuki E, Xue L, Fujimori A, Kubota N, Okayasu R: Chemopreventive agent sulforaphane enhances radiosensitivity in human tumor cells. Int J Cancer. 2009, 125: 1205-1211.
Pettazzoni P, Pizzimenti S, Toaldo C, Sotomayor P, Tagliavacca L: Induction of cell cycle arrest and DNA damage by the HDAC inhibitor panobinostat (LBH589) and the lipid peroxidation end product 4-hydroxynonenal in prostate cancer cells. Free Radic Biol Med. 2011, 50: 313-322.
Sahu RP, Epperly MW, Srivastava SK: Benzyl isothiocyanate sensitizes human pancreatic cancer cells to radiation therapy. Front Biosci (Elite Ed). 2009, 1: 568-576.
Hu Y, Lu W, Chen G, Zhang H, Jia Y, Wei Y, Yang H, Zhang W: Overcoming resistance to histone deacetylase inhibitors in human leukemia with the redox modulating compound β-phenylethyl isothiocyanate. Blood. 2010, 116: 2732-2741.
Sukumari-Ramesh S, Singh N, Jensen MA, Dhandapani KM, Vender JR: Anacardic acid induces caspase-independent apoptosis and radiosensitizes pituitary adenoma cells. J Neurosurg. 2011
Xiao H, Zhang KJ: Antiproliferative effect of curcumin combined with cyclophosmide on the growth of human lymphoma cell line HT/CTX with drug resistance and its relation with FA/BRCA pathway. Zhongguo Shi Yan Xue Ye Xue Za Zhi. 2008, 16: 804-808.
Xiao H, Xiao Q, Zhang K, Zuo X, Shrestha UK: Reversal of multidrug resistance by curcumin through FA/BRCA pathway in multiple myeloma cell line MOLP-2/R. Ann Hematol. 2010, 89: 399-404.
Du B, Jiang L, Xia Q, Zhong L: Synergistic inhibitory effects of curcumin and 5-fluorouracil on the growth of the human colon cancer cell line HT-29. Chemotherapy. 2006, 52: 23-28.
Lev-Ari S, Strier L, Kazanov D, Madar-Shapiro L, Dvory-Sobol H: Celecoxib and curcumin synergistically inhibit the growth of colorectal cancer cells. Clin Cancer Res. 2005, 11: 6738-6744.
Ji Z: Targeting DNA damage and repair by curcumin. Breast Cancer. 2010, 4: 1-3.
Tsai MS, Weng SH, Kuo YH, Chiu YF, Lin YW: Synergistic effect of curcumin and cisplatin via downregulation of thymidine phosphorylase and ERCC1. Mol Pharmacol. 2011, 80: 136-46.
Giommarelli C, Zuco V, Favini E, Pisano C, Dal Piaz F, De Tommasi N, Zunino F: The enhancement of antiproliferative and proapoptotic activity of HDAC inhibitors by curcumin is mediated by Hsp90 inhibition. Cell Mol Life Sci. 2010, 67: 995-1004.
Podhorecka M, Halicka D, Klimek P, Kowal M, Chocholska S, Dmoszynska A: Resveratrol increases rate of apoptosis caused by purine analogues in malignant lymphocytes of chronic lymphocytic leukemia. Ann Hematol. 2011, 90: 173-183.
Wolff GL, Kodell RL, Moore SR, Cooney CA: Maternal epigenetics and methyl supplements affect agouti gene expression in Avy/a mice. FASEB J. 1998, 12: 949-957.
Poirier LA: Methyl group deficiency in hepatocarcinogenesis. Drug Metab Rev. 1994, 26: 185-99.
Davis CD, Uthus EO, Finley JW: Dietary selenium and arsenic affect DNA methylation in vitro in Caco-2 cells and in vivo in rat liver and colon. J Nutr. 2000, 130: 2903-2909.
Weaver IC, Cervoni N, Champagne FA, D'Alessio AC, Sharma S, Seckl JR: Epigenetic programming by maternal behavior. Nat Neurosci. 2004, 7: 847-854.
Weaver IC, Meaney MJ, Szyf M: Maternal care effects on the hippocampal transcriptome and anxiety-mediated behaviors in the offspring that are reversible in adulthood. Proc Natl Acad Sci USA. 2006, 103: 3480-3485.
Murugan RS, Vinothini G, Hara Y, Nagini S: Black tea polyphenols target matrix metalloproteinases, RECK, proangiogenic molecules and histone deacetylase in a rat hepatocarcinogenesis model. Anticancer Res. 2009, 29: 2301-2305.
Cosio BG, Mann B, Ito K, Jazrawi E, Barnes PJ, Chung KF: Histone acetylase and deacetylase activity in alveolar macrophages and blood mononocytes in asthma. Am J Respir Crit Care Med. 2004, 170: 141-147.
Cosio BG, Tsaprouni L, Ito K, Jazrawi E, Adcock IM, Barnes PJ: Theophylline restores histone deacetylase activity and steroid responses in COPD macrophages. J Exp Med. 2004, 200: 689-695.
Cosio BG, Iglesias A, Rios A, Noguera A, Sala E, Ito K: Low-dose theophylline enhances the anti-inflammatory effects of steroids during exacerbations of COPD. Thorax. 2009, 64: 424-429.
Ito K, Lim S, Caramori G, Cosio B, Chung KF, Adcock IM: A molecular mechanism of action of theophylline: Induction of histone deacetylase activity to decrease inflammatory gene expression. Proc Natl Acad Sci USA. 2002, 99: 8921-8926.
Priyadarsini RV, Vinothini G, Murugan RS, Manikandan P, Nagini S: The flavonoid Quercetin modulates the hallmark capabilities of hamster buccal pouch tumors. Nutr Cancer. 2011, 63: 218-226.
Hu R, Khor TO, Shen G, Jeong WS, Hebbar V, Chen C: Cancer chemoprevention of intestinal polyposis in ApcMin/+ mice by sulforaphane, a natural product derived from cruciferous vegetable. Carcinogenesis. 2006, 27: 2038-2046.
Myzak MC, Dashwood WM, Orner GA, Ho E, Dashwood RH: Sulforaphane inhibits histone deacetylase in vivo and suppresses tumorigenesis in Apc-minus mice. FASEB J. 2006, 20: 506-508.
Myzak MC, Tong P, Dashwood WM, Dashwood RH, Ho E: Sulforaphane retards the growth of human PC-3 xenografts and inhibits HDAC activity in human subjects. Exp Biol Med. 2007, 232: 227-234.
Clarke JD, Hsu A, Williams DE, Dashwood RH, Stevens JF, Yamamoto M, Ho E: Metabolism and tissue distribution of sulforaphane in Nrf2 knockout and wild-type mice. Pharm Res. 2011,
Ji Y, Kuo Y, Morris ME: Pharmacokinetics of dietary phenethylisothiocyanate in rats. Pharm Res. 2005, 22: 1658-1666.
Ye L, Dinkova-Kostova AT, Wade KL, Zhang Y, Shapiro TA, Talalay P: Quantitative determination of dithiocarbamates in human plasma, serum, erythrocytes and urine: pharmacokinetics of broccoli sprout isothiocyanates in humans. Clin Chim Acta. 2002, 316: 43-53.
Druesne-Pecollo N, Chaumontet C, Pagniez A, Vaugelade P, Bruneau A, Thomas M: In vivo treatment by diallyl disulfide increases histone acetylation in rat colonocytes. Biochem Biophys Res Commun. 2007, 354: 140-147.
Zhao J, Huang WG, He J, Tan H, Liao QJ, Su Q: Diallyl disulfide suppresses growth of HL-60 cell through increasing histone acetylation and p21WAF1 expression in vivo and in vitro. Acta Pharmacol Sin. 2006, 27: 1459-1466.
Tyagi A, Gu M, Takahata T, Frederick BA, Agarwal C, Siriwardana S, Agarwal R, Sclafani RA: Resveratrol selectively induces DNA damage, independent of Smad4 expression, in its efficacy against human head and neck squamous cell carcinoma. Clin Cancer Res. 2011,
Vanhees K, de Bock L, Godschalk RW, van Schooten FJ, van Waalwijk van Doorn-Khosrovani SB: Prenatal exposure to flavonoids: implication for cancer risk. Toxicol Sci. 2011, 120: 59-67.
Toyoizumi T, Sekiguchi H, Takabayashi F, Deguchi Y, Masuda S, Kinae N: Induction effect of coadministration of soybean isoflavones and sodium nitrite on DNA damage in mouse stomach. Food Chem Toxicol. 2010, 48: 2585-2591.
Amin AR, Wang D, Zhang H, Peng S, Shin HJ, Brandes JC, Tighiouart M, Khuri FR, Chen ZG, Shin DM: Enhanced anti-tumor activity by the combination of the natural compounds (-)-epigallocatechin-3-gallate and luteolin: potential role of p53. J Biol Chem. 2010, 285: 34557-34565.
Mueller S, Yang X, Sottero TL, Gragg A, Prasad G, Polley MY, Weiss WA, Matthay KK, Davidoff AM, DuBois SG, Haas-Kogan DA: Cooperation of the HDAC inhibitor vorinostat and radiation in metastatic neuroblastoma: efficacy and underlying mechanisms. Cancer Lett. 2011, 306: 223-229.
Di Micco R, Sulli G, Dobreva M, Liontos M, Botrugno OA, Gargiulo G, dal Zuffo R, Matti V: Interplay between oncogene-induced DNA damage response and heterochromatin in senescence and cancer. Nat Cell Biol. 2011, 13: 292-302.
Baschnagel A, Russo A, Burgan WE, Carter D, Beam K, Palmieri D, Steeg PS, Tofilon P, Camphausen K: Vorinostat enhances the radiosensitivity of a breast cancer brain metastatic cell line grown in vitro and as intracranial xenografts. Mol Cancer Ther. 2009, 8: 1589-1595.
Camphausen K, Scott T, Sproull M, Tofilon PJ: Enhancement of xenograft tumor radiosensitivity by the histone deacetylase inhibitor MS-275 and correlation with histone hyperacetylation. Clin Cancer Res. 2004, 10: 6066-6071.
Camphausen K, Cerna D, Scott T, Sproull M, Burgan WE, Cerra MA, Fine H, Tofilon PJ: Enhancement of in vitro and in vivo tumor cell radiosensitivity by valproic acid. Int J Cancer. 2005, 114: 380-386.
Geng L, Cuneo KC, Fu A, Tu T, Atadja PW, Hallahan DE: Histone deacetylase (HDAC) inhibitor LBH589 increases duration of gamma-H2AX foci and confines HDAC4 to the cytoplasm in irradiated nonsmall cell lung cancer. Cancer Res. 2006, 66: 11298-11304.
Cuneo KC, Fu A, Osusky K, Huamani J, Hallahan DE, Geng L: Histone deacetylase inhibitor NVP-LAQ824 sensitizes human nonsmall cell lung cancer to the cytotoxic effects of ionizing radiation. Anticancer Drugs. 2007, 18: 793-800.
Entin-Meer M, Yang X, VandenBerg SR, Lamborn KR, Nudelman A, Rephaeli A, Haas-Kogan DA: In vivo efficacy of a novel histone deacetylase inhibitor in combination with radiation for the treatment of gliomas. Neuro Oncol. 2007, 9: 82-88.
Lopez G, Liu J, Ren W, Wei W, Wang S, Lahat G, Zhu QS, Bornmann WG, McConkey DJ, Pollock RE, Lev DC: Combining PCI-24781, a novel histone deacetylase inhibitor, with chemotherapy for the treatment of soft tissue sarcoma. Clin Cancer Res. 2009, 15: 3472-3483.
Purrucker JC, Fricke A, Ong MF, Rübe C, Rübe CE, Mahlknecht U: HDAC inhibition radiosensitizes human normal tissue cells and reduces DNA Double-Strand Break repair capacity. Oncol Rep. 2010, 23: 263-269.
Wang LH, Pfister TD, Parchment RE, Kummar S, Rubinstein L, Evrard YA, Gutierrez ME, Murgo AJ, Tomaszewski JE, Doroshow JH, Kinders RJ: Monitoring drug induced gammaH2AX as a pharmacodynamic biomarker in individual circulating tumor cells. Clin Cancer Res. 2010, 16: 1073-1084.
Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, Mortimer P: Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009, 361: 123-134.
Redon CE, Nakamura AJ, Zhang YW, Ji JJ, Bonner WM, Kinders RJ, Parchment RE, Doroshow JH, Pommier Y: Histone gammaH2AX and poly(ADP-ribose) as clinical pharmacodynamic biomarkers. Clin Cancer Res. 2010, 16: 4532-4542.
Zupkovitz G, Grausenburger R, Brunmeir R, Senese S, Tischler J: The cyclin-dependent kinase inhibitor p21 is a crucial target for histone deacetylase 1 as a regulator of cellular proliferation. Mol Cell Biol. 2010, 30: 1171-1181.
Acknowledgements
This work was supported by grants CA90890, CA65525, CA122906, CA122959, CA80176 from National Institutes of Health, and Center grant P30 ES00210 from National Institute of Environmental Health Sciences.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
PR wrote the first draft of the article. EH, DEW and RHD edited and finalized the manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Rajendran, P., Ho, E., Williams, D.E. et al. Dietary phytochemicals, HDAC inhibition, and DNA damage/repair defects in cancer cells. Clin Epigenet 3, 4 (2011). https://doi.org/10.1186/1868-7083-3-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1868-7083-3-4