
Abstract
Recent studies have implicated potentially significant roles for astrocytes in the pathogenesis of neurodevelopmental disorders. Astrocytes undergo a dramatic maturation process following early differentiation from which typical morphology and important functions are acquired. Despite significant progress in understanding their early differentiation, very little is known about how astrocytes become functionally mature. In addition, whether functional maturation of astrocytes is disrupted in neurodevelopmental disorders and the consequences of this disruption remains essentially unknown. In this review, we discuss our perspectives about how astrocyte developmental maturation is regulated, and how disruption of the astrocyte functional maturation process, especially alterations in their ability to regulate glutamate homeostasis, may alter synaptic physiology and contribute to the pathogenesis of neurodevelopmental disorders.
Similar content being viewed by others

Review
Neurodevelopmental disorders are a wide range of clinical conditions that impair growth and development of the central nervous system (CNS) during early postnatal stages. In particular, autism spectrum disorder (ASD), a class of conditions defined by deficits in social behavior and communication [1], represents a major category of neurodevelopmental disorders. Although the exact causes for these disorders remain largely unknown, earlier genome-wide association studies (GWAS) [2–4] to recent exosome sequencing [5–8] have associated over 1,000 genes with various forms of neurodevelopmental disorders [9], including ASD, implicating a strong genetic component in their etiology. In particular, genetic mutations of methyl CpG binding protein 2 (MeCP2) [10] and fragile X mental retardation 1 (fmr1) [11] genes have been identified to cause the monogenic neurodevelopmental disorders Rett syndrome and Fragile X syndrome (FXS), respectively. Interestingly, both Rett syndrome and FXS significantly resemble typical autistic phenotypes [12, 13]. The studies on MeCP2/fragile X mental retardation protein (FMRP, protein product of fmr1 gene) function and how their mutations cause Rett syndrome and FXS therefore provide valuable clues to the pathogenesis of the sporadic forms of neurodevelopmental disorders. In addition to the identification of genetic risk factors, direct morphometric, neuropathological, and functional neuroimaging studies have characterized important pathological features in autistic brains, such as enlarged brain size (head circumference), decreased numbers of cerebellar purkinje neurons, increased inflammatory responses, and altered brain connectivity and activity (increased susceptibility to seizures) [14, 15].
Although pathological features of autistic brains are mostly neurocentric, it has gradually become evident from recent studies that (astro)glial cells are likely to be active components in the pathogenesis of ASD and other neurodevelopmental disorders [16, 17]. Astrocytes are known to significantly modulate synaptogenesis during development [18–20] and play diverse and active roles in synaptic physiology in the adult brain [21, 22]. Despite the essential roles of astrocytes at functional synapses, how astrocytes are generated and become morphologically and functionally mature during development remains largely uncharacterized. It also remains unknown whether astrocyte development becomes impaired in neurodevelopmental disorders and how impairment in astrocyte development may contribute to pathological features observed in various neurodevelopmental disorders. In this review, we will summarize recent progress in understanding astrocyte development and their involvement in neurodevelopmental disorders. Although several recent excellent reviews have described the involvement of astrocytes in neurodevelopmental disorders [16, 17], we will give our own perspectives about the morphological and functional maturation of astrocytes during development, how astrocyte developmental maturation is regulated, and how the disruption of the functional maturation process of astrocytes, especially alterations in their ability to regulate glutamate homeostasis, may alter synaptic physiology and contribute to the pathogenesis of neurodevelopmental disorders.
Developmental maturation of astrocytes in the CNS
All of the CNS cell types except the microglia are differentiated from neural stem cells (NSC) at different stages of embryogenesis. It is well characterized that glial cells, including astrocytes are generated following neuronal differentiation from NSC during early embryogenesis. The peak of astrogliogenesis occurs late prenatal to early postnatal stages, that is, E18-P7 in various rodent CNS regions [23, 24]. The developmental stages referred in this review are based on the mouse CNS development unless otherwise stated. Recent studies have found several pathways/mechanisms that are involved in astrocyte fate specification from NSC, including Janus kinase (JAK)/Signal Transducer and Activator of Transcription (STAT) [25], Bone morphogenetic protein (BMP)-SMAD [26, 27], Notch [28], and Nuclear factor IA (NFIA) [29]. These pathways are activated by the extrinsic gliogenesis signals, including Ciliary neurotrophic factor (CNTF), Cardiotrophin 1 (CT- 1) [30], and Leukemia inhibitory factor (LIF) [31], which are secreted from early differentiated neurons or late-stage NSCs. These signals also induce intrinsic changes, especially the epigenetic modifications to open the appropriate chromatin regions [31], which allows binding of transcriptional factors (STAT, SMAD, or NFIA) [32] to astroglial gene promoters (such as GFAP, S-100β, and GLAST) to induce their expression [27, 29, 33]. Although early in vitro studies suggest the presence of bipotent glial restricted cells (GRP) that can generate both oligodendrocytes and astrocytes [34], recent in vivo studies have shown that GRP is likely to be a minor pathway in gliogenesis [35]. The identity of astrocyte progenitor cells in vivo remains elusive. Interestingly, a recent study has shown that local proliferation of differentiated astrocytes is the major source of astroglia in the postnatal cortex [36]. Early produced astrocytes continue cell divisions while also undergo differentiation, implicating a progenitor status of early produced astrocytes [36].
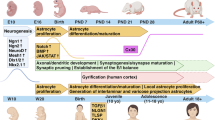
Adult astrocytes, regardless of their anatomical location (gray or white matter), typically occupy a large and non-overlapping domain, which is composed of a large number of branches and fine processes [37]. In particular, these fine processes (typically <50 nm in diameter) are typically distal from the soma and are generally GFAP immunostaining negative. These fine processes, also termed ‘peripheral astrocyte processes (PAPs)’ [38], constitute roughly 50% of the mature astrocyte volume and 80% of the surface area [39, 40], permitting sufficient insertion of various membrane proteins such as ion channels, ligand receptors, and transporters. PAPs extensively contact synapses and are considered primary sites for active astrocyte and neuron signaling [41]. Although a significant number of astrocytes are largely generated during the first week postnatally [36, 42], these fine processes (PAPs) are not induced in these astrocytes until several weeks later (Figure 1) [31, 43]. The astroglial network, indicated by the interacting PAPs, is also formed at later developmental stages (P14 to P26, Figure 1). In addition to morphological maturation, several important astroglial genes, including glutamate transporter GLT1 (rodent analog of human excitatory amino acid transporter 2) [44, 45], connexin 43 and 30 [46], and inwardly rectifying potassium channel Kir4.1 [47, 48], are also induced in astrocytes within 3 to 4 weeks after birth, suggesting that astrocytes also undergo dramatic molecular changes during developmental maturation. Interestingly, these astroglial genes represent some of most characteristic and important functions of astrocytes in the CNS. For example, GLT1 is the physiologically dominant glutamate transporter in the CNS to clear extracellular glutamate; connexin 43 and 30 are the major components of the astrocyte gap-junction which forms the astrocyte network; Kir4.1 is an important potassium channel in astrocytes that is critical for maintaining the K+ gradient for proper glutamate uptake and also significantly buffers activity-induced K+ release. Induction of these genes during development, together with the growth of PAPs in astrocytes, suggests that astrocytes undergo a developmental maturation phase from the first week to the following 2 to 3 weeks postnatally to acquire their unique morphology and molecular function. Notably, this is a developmental stage that follows the early astrocyte fate specification and differentiation. Moreover, the molecular maturation is tightly associated with the morphological maturation of astrocytes, as these important proteins mentioned above are all membrane proteins that are primarily localized on the surface of the fine processes for their proper functions in astrocytes.

Morphological maturation of astrocytes during postnatal development. (A) Growth of cortical astrocyte processes illustrated with tdTomato reporter on EAAT2 tdTomato transgenic mice during postnatal development; Scale bar: 100 μm; (B) A magnified view of typical astrocyte morphology at P4 and P30 from cortex of EAAT2 tdTomato mice. Scale bar: 50 μm.
Despite the dramatic changes astrocytes undergo during the maturation phase, the mechanisms for astrocyte developmental maturation remain largely unknown. Neuronal signaling has been implicated in early astrocyte fate specification by secreting the extrinsic signals like CT-1 [30] from early-differentiated neurons to induce astrocyte differentiation. Interestingly, although GLT1 is the predominant glutamate transporter in adult brain, cultured astrocytes express minimal levels of GLT1 [49]. Instead, GLAST (rodent analog of human excitatory amino acid transporter 1, EAAT1) is the dominant glutamate transporter in cultured astrocytes [50]. However, when astrocytes are co-cultured with neurons, GLT1 expression in astrocytes is highly induced in a dose-dependent manner (Figure 2) [51, 52]. Similarly, expression of connexin 43/30 is also induced in astrocytes when co-cultured with neurons [53, 54]. As cultured astrocytes share a more similar gene profile with immature astrocytes in vivo (P1-P7) [55], neuron-dependent induction of these astroglial genes in cultured astrocytes indicates that neuronal signaling is likely to play important roles in induction of astrocyte gene expression during development in vivo. Recent gene expression profile studies in astrocytes that are acutely isolated from the brain have revealed that developing astrocytes express many neurotransmitter receptors [55, 56], allowing them to receive neuronal signals. Indeed, a substantial body of literature has reported that neuronal activity triggers Ca2+ changes in astrocytes in vitro and in vivo[57, 58], though the specific metabotropic glutamate receptor that mediates this response varies significantly between immature (GRM5) and mature astrocytes (GRM3) [59]. In addition, although it is unclear whether neuronal activity modulates PAPs formation in astrocytes during developmental maturation, direct application of glutamate induces rapid filopodia motility in cultured astrocytes [60]. Neuronal activity has also been closely associated with structural plasticity of adult astrocytes in vivo[61]. Both secreted neuronal signals, such as activity-dependent neurotransmitters and small protein CT-1 and contact-dependent signaling, such as Notch-JAG1 interaction [62], can play important roles in astrocyte developmental maturation. Molecular mechanisms for astrocyte maturation, especially downstream pathways that mediate potential neuronal signaling are essentially unknown. As various pathways/mechanisms involved in early astrocyte specification are characterized (described above), it would be interesting to test whether any of these pathways also regulate developmental maturation of astrocytes at a later stage.

Expression of GLT1 in cultured astrocytes is induced by co-cultured neurons in a dose-dependent manner.
Roles of astrocytes in neurodevelopmental disorders
The first evidence of potential astrocyte abnormalities in neurodevelopmental disorders was from biochemical analysis of patient brain samples and screening of genetic risk factors for various forms of neurodevelopmental disorders. Astrogliosis, indicated by increased GFAP expression, was found in the cerebellar cortex of autistic brains [63], though neuronal degeneration was not generally observed in brains of neurodevelopmental disorder patients. Expression changes of a few other astroglial proteins were also observed in brain samples of ASD patients. For example, increased EAAT2 and EAAT1 expression was found in cerebellum of autistic patients [64]. Significantly increased connexin 43 was found in the superior frontal cortex, while decreased aquaporin 4 was found in the cerebellum of autistic brains [65]. These astroglial changes in autistic brains imply that astrocytes are likely to be involved in neurodevelopmental disorders. In addition, genetic studies have found associations between specific nucleotide polymorphisms in the EAAT1 sequence with severity of repetitive behaviors and anxiety in ASD children [66]. Mutations of Kir4.1 were also found in a subset of autism patients [67], which likely affects the K+ homeostasis in the brain and increases the seizure susceptibility in autism patients. Despite these clinical studies, it is important to note that specific astrocyte-related mechanisms involved in the pathogenesis of neurodevelopmental disorders remain to be characterized.
Significant progress in understanding the roles of astrocytes in neurodevelopmental disorders has recently been made from animal (mouse) models of two typical monogenic neurodevelopmental disorders: Rett syndrome and FXS. Loss-of-function mutations in MeCP2 are the primary cause for the vast majority of Rett syndrome patients [10]. The fragile X syndrome is caused by the transcriptional silencing of FMRP expression, as a result of hypermethylation on the abnormally high number (>200) of trinucleotide CCG insertions at the fmr1 gene locus [11]. Both MeCP2−/−[68, 69] and fmr1−/−[70] mice were later developed that recapitulate typical clinical symptoms of Rett syndrome or FXS, respectively. Extensive studies have been carried out in these mouse models to understand the functions of MeCP2 and FMRP in brain and how their loss-of-function mutations alter dendrite morphology and synaptic physiology, which underlies the clinical phenotypes of Rett syndrome and FXS [71, 72]. Using these mouse models, recent studies have started to unveil the pathogenic roles of astrocytes in neurodevelopmental disorders. Astrocytes derived from MeCP2−/− mice can significantly affect normal neuronal development [73]. Wild-type hippocampal neurons co-cultured with MeCP2-deficient astrocytes or treated with astrocyte conditioned medium (ACM) collected from MeCP2-deficient astrocytes exhibit abnormally stunted dendrites [73]. Similarly, astrocytes derived from fmr1−/−− mice also induce developmental delays in dendrite maturation and synaptic protein expression of hippocampal neuronal dendrites in co-cultures [74, 75]. Excessive neurotrophin-3 (NT-3), but not other growth factors secreted from fmr1−/− astrocytes, was later suggested to reduce the dendrites of neurons and synaptic protein levels in FXS condition [76]. In addition, loss of MeCP2 in astrocytes appears to be dependent upon the astroglial gap-junction [77] and it induces expression changes of several important astroglial genes [78]. Most interestingly, selective restoration of MeCP2 in astrocytes in vivo using the Cre-loxP recombination system significantly improves locomotion and anxiety levels, and restores respiratory abnormalities to a normal pattern [79]. At the cellular level, re-expressed MeCP2 in astrocytes also restores normal dendritic morphology and increases levels of the vesicular glutamate transporter VGLUT1 [79]. These results clearly suggest that astrocytes play important pathogenic roles in Rett syndrome and they should also be considered as important therapeutic targets, in addition to neurons [16].
Glutamate homeostasis is essential for brain physiology. Proper glutamatergic signaling is important in regulating neurite outgrowth, synaptogenesis, neuronal migration, differentiation, and cell death in the developing brain [80–82]. Given its important and diverse functions in development, it is not surprising that altered glutamatergic signaling may significantly contribute to the pathogenesis of neurodevelopmental disorders. Indeed, several genetic studies have associated variations of glutamate receptor genes (GluR6, GRM8, and GRIN2A) with ASD [83–85]. In addition, studies using fmr1−/− mice have characterized the abnormally increased group I mGluR (especially mGluR5) activation-induced protein synthesis [72, 86], which underlies several typical abnormalities observed in FXS, such as enhanced mGluR-dependent long-term depression (LTD) [87], induction of elongated/immature dendrites [88], and increased susceptibility to audiogenic seizure [89], and so on. Although FMRP is generally considered a translation suppressor and increased mGluR1/5-dependent dendritic protein synthesis is largely due to the loss of FMRP in neurons in FXS [86], the observation that genetic or pharmacological inhibition of mGluR5 activation effectively and significantly reduces mGluR1/5-dependent dendritic protein synthesis in fmr1−/− mice [90, 91] also implicates that mGluR1/5 activation is abnormally enhanced in FXS. In addition, inhibition of mGluR5 signaling also significantly suppresses repetitive behaviors and social behavior deficits and reverses elevated stereotypical and anxiety-like behaviors in mouse models of idiopathic autism [92], suggesting that abnormally enhanced mGluR5 activation may also contribute to some forms of idiopathic autism. In the mammalian CNS, neuronal mGluR1/5 is preferentially localized on the peri-synaptic surface membrane, thus its activation is highly dependent upon the glutamate that is spilled out from the synaptic cleft [93]. Interestingly, the extracellular, especially the spilled glutamate levels are tightly regulated by astroglial glutamate transporters GLT1/GLAST (human EAAT2/EAAT1) [94]; therefore the activation of neuronal mGluR1/5 is actively modulated by astroglial GLT1/GLAST expression/activity. Indeed, pharmacological inhibition or genetic deletion of GLT1/GLAST activity potentiates postsynaptic neuronal mGluR activation [95], while upregulation of GLT1 expression severely impairs mGluR-dependent LTD at rat mossy fiber-CA3 synapses [96].
We have recently found a downregulation of GLT1/GLAST expression and reduced glutamate uptake in cortex of fmr1−/− mice during postnatal development and have shown that their downregulation contributes to the enhanced neuronal excitability observed in fmr1−/− mouse cortex [97]. These results suggest that the dysregulated GLT1/GLAST expression is likely to be an upstream, astrocyte-mediated mechanism leading to enhanced neuronal mGluR5 activation in FXS. As a result, enhanced neuronal mGluR5 activation may synergistically increase abnormal dendritic protein synthesis, together with the loss of FMRP-mediated suppression on protein synthesis in fmr1−/− neurons (Figure 3). It also explains the earlier observations that the basal levels of protein synthesis are elevated in fmr1−/− mice [86]. In addition, proper expression of GLT1 and GLAST is known to be essential for the normal development of the CNS [81] and GLT1−/− mice exhibit severe seizures as early as P14 [98]. Moreover, 30% of GLT1+/− GLAST+/− (double heterogeneous) mice with 50% of GLT1 and GLAST expression levels (and 30% increased extracellular glutamate levels [99]) exhibit behavioral and neuroanatomical abnormalities often observed in autism, including abnormal social interaction, seizures, and an enlarged amygdala and hippocampus [100]. As astroglial glutamate transporters, especially GLT1, are strongly induced during the functional maturation process, potential dysregulation of GLT1/GLAST during development exemplifies how alterations in the functional maturation of astrocytes could contribute to the pathogenesis of FXS and other neurodevelopmental disorders.

Dysregulation of astroglial glutamate transporters is likely to enhance neuronal mGluR1/5 activation and further increases downstream protein synthesis in FXS condition. Synaptic neuron to astrocyte communication in (A) fmr1+/+ and (B) fmr1−/− conditions. LTD: Long-term depression.
Astrocytes perform many other important functions in mammalian CNS, in addition to the clearance of extracellular glutamate. Developing but not mature astrocytes secrete thrombospondins 1 and 2 (TSP-1 and TSP-2), which significantly and specifically increases numbers of excitatory synapses [101]. Glypican 4 (Gpc4) and glypican 6 (Gpc6) were further identified from astrocyte conditioned medium to promote glutamate receptor clustering and receptivity, which facilitates the formation of postsynaptically functioning CNS synapses [18]. Additional synaptogenic molecules secreted from astrocytes are being characterized [102]. Although immature dendrite morphology and altered synaptic functions are hallmarks in many neurodevelopmental disorders, whether these developing astrocyte-dependent synaptogenic pathways are altered and their consequences in neurodevelopmental disorders remain to be examined. In addition, we have very limited knowledge about the molecular changes of astrocytes in neurodevelopmental disorders, especially from in vivo settings. Examination of molecular changes in astrocytes in vivo has been traditionally difficult; however, recent development of an array of astrocyte reporter mice (BAC GLT1 eGFP [103], BAC ALDH1L1 eGFP [55], EAAT2 tdTomato [104]) has allowed rapid isolation of in vivo astrocytes from mouse models of neurodevelopmental disorders through fluorescence activated cell sorting (FACS) and subsequent genome-wide transcriptional profiling. The development of translational ribosome affinity purification (TRAP) technique and ALDH1L1-TRAP mice also provides another convenient in vivo approach to isolate translating mRNAs from astrocytes in an unprecedented temporal and spatial manner [105, 106]. These tools will greatly facilitate the molecular characterization of potential developmental abnormalities of astrocytes in various neurodevelopmental disorders. Results from these studies will provide valuable clues about the pathways that regulate functional maturation of astrocytes during development and their molecular alterations in neurodevelopmental disorders.
Conclusions
In summary, we discuss the functional maturation of astrocyte during postnatal development, and the disruption of this maturation process, like the dysregulation of glutamate transporters may serve as an astrocyte-dependent mechanism for the pathogenesis of FXS and other neurodevelopmental disorders. Although the molecular mechanisms of astroglial maturation and how the disruption of this maturation process contributes to the pathogenesis of neurodevelopmental disorders remain to be investigated, we expect that the availability of novel in vivo tools for astrocyte study will greatly help answer these questions. Ultimately, a better understanding of the roles of (astro)glia in the pathogenesis of neurodevelopmental disorders will facilitate the search for cures for these disorders.
Abbreviations
- ASD:
-
Autism spectrum disorder
- BMP:
-
Bone morphogenetic protein
- CNS:
-
Central nervous system
- CNTF:
-
Ciliary neurotrophic factor
- CT-1:
-
Cardiotrophin 1
- EAAT:
-
Human excitatory amino acid transporter
- FACS:
-
Fluorescence activated cell sorting
- Fmr1:
-
Fragile X mental retardation 1
- FMRP:
-
Fragile X mental retardation protein
- FXS:
-
Fragile X syndrome
- GRM:
-
Metabotropic glutamate receptor
- GWAS:
-
Genome-wide association studies
- JAK:
-
Janus kinase
- Kir:
-
Inwardly rectifying potassium channel
- LIF:
-
Leukemia inhibitory factor
- LTD:
-
Long-term depression
- MeCP2:
-
Methyl CpG binding protein 2
- NFIA:
-
Nuclear factor IA
- NSC:
-
Neural stem cells
- NT-3:
-
Neurotrophin-3
- PAP:
-
Peripheral astrocyte processes
- STAT:
-
Signal Transducer and Activator of Transcription
- TRAP:
-
Translational ribosome affinity purification
- TSP:
-
Thrombospondin
- VGluT1:
-
Vesicular glutamate transporter 1.
References
Diagnostic and statistical manual of mental disorders. 1994, Washington, DC: American Psychiatric Association, 4
Wassink TH, Brzustowicz LM, Bartlett CW, Szatmari P: The search for autism disease genes. Ment Retard Dev Disabil Res Rev. 2004, 10: 272-283. 10.1002/mrdd.20041.
Li X, Zou H, Brown WT: Genes associated with autism spectrum disorder. Brain Res Bull. 2012, 88: 543-552. 10.1016/j.brainresbull.2012.05.017.
Weiss LA, Arking DE, Daly MJ, Chakravarti A: A genome-wide linkage and association scan reveals novel loci for autism. Nature. 2009, 461: 802-808. 10.1038/nature08490.
Luo R, Sanders SJ, Tian Y, Voineagu I, Huang N, Chu SH, Klei L, Cai C, Ou J, Lowe JK, Hurles ME, Devlin B, State MW, Geschwind DH: Genome-wide transcriptome profiling reveals the functional impact of rare de novo and recurrent CNVs in autism spectrum disorders. Am J Hum Genet. 2012, 91: 38-55. 10.1016/j.ajhg.2012.05.011.
Michaelson JJ, Shi Y, Gujral M, Zheng H, Malhotra D, Jin X, Jian M, Liu G, Greer D, Bhandari A, Wu W, Corominas R, Peoples A, Koren A, Gore A, Kang S, Lin GN, Estabillo J, Gadomski T, Singh B, Zhang K, Akshoomoff N, Corsello C, McCarroll S, Iakoucheva LM, Li Y, Wang J, Sebat J: Whole-genome sequencing in autism identifies hot spots for de novo germline mutation. Cell. 2012, 151: 1431-1442. 10.1016/j.cell.2012.11.019.
O'Roak BJ, Vives L, Fu W, Egertson JD, Stanaway IB, Phelps IG, Carvill G, Kumar A, Lee C, Ankenman K, Munson J, Hiatt JB, Turner EH, Levy R, O’Day DR, Krumm N, Coe BP, Martin BK, Borenstein E, Nickerson DA, Mefford HC, Doherty D, Akey JM, Bernier R, Eichler EE, Shendure J: Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science. 2012, 338: 1619-1622. 10.1126/science.1227764.
O’Roak BJ, Vives L, Girirajan S, Karakoc E, Krumm N, Coe BP, Levy R, Ko A, Lee C, Smith JD, Turner EH, Stanaway IB, Vernot B, Malig M, Baker C, Reilly B, Akey JM, Borenstein E, Rieder MJ, Nickerson DA, Bernier R, Shendure J, Eichler EE: Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature. 2012, 485: 246-250. 10.1038/nature10989.
Williams SC: Genetics: Searching for answers. Nature. 2012, 491: S4-S6. 10.1038/491S4a.
Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY: Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999, 23: 185-188. 10.1038/13810.
Verkerk AJ, Pieretti M, Sutcliffe JS, Fu YH, Kuhl DP, Pizzuti A, Reiner O, Richards S, Victoria MF, Zhang FP, Eussen BE, van Ommen G-JB, Blonden LAJ, Riggins GJ, Chastain JL, Kunst CB, Galjaard H, Caskey CT, Nelson DL, Oostra BA, Warren ST: Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell. 1991, 65: 905-914. 10.1016/0092-8674(91)90397-H.
Percy AK: Rett syndrome: exploring the autism link. Arch Neurol. 2011, 68: 985-989. 10.1001/archneurol.2011.149.
Oddi D, Crusio WE, D’Amato FR, Pietropaolo S: Monogenic mouse models of social dysfunction: Implications for autism. Behav Brain Res. 2013, 251: 75-84.
DiCicco-Bloom E, Lord C, Zwaigenbaum L, Courchesne E, Dager SR, Schmitz C, Schultz RT, Crawley J, Young LJ: The developmental neurobiology of autism spectrum disorder. J Neurosci. 2006, 26: 6897-6906. 10.1523/JNEUROSCI.1712-06.2006.
Silver WG, Rapin I: Neurobiological basis of autism. Pediatr Clin North Am. 2012, 59: 45-61. 10.1016/j.pcl.2011.10.010. x
McGann JC, Lioy DT, Mandel G: Astrocytes conspire with neurons during progression of neurological disease. Curr Opin Neurobiol. 2012, 22: 850-858. 10.1016/j.conb.2012.03.009.
Molofsky AV, Krencik R, Ullian EM, Tsai HH, Deneen B, Richardson WD, Barres BA, Rowitch DH: Astrocytes and disease: a neurodevelopmental perspective. Genes Dev. 2012, 26: 891-907. 10.1101/gad.188326.112.
Allen NJ, Bennett ML, Foo LC, Wang GX, Chakraborty C, Smith SJ, Barres BA: Astrocyte glypicans 4 and 6 promote formation of excitatory synapses via GluA1 AMPA receptors. Nature. 2012, 486: 410-414.
Liauw J, Hoang S, Choi M, Eroglu C, Sun GH, Percy M, Wildman-Tobriner B, Bliss T, Guzman RG, Barres BA, Steinberg GK: Thrombospondins 1 and 2 are necessary for synaptic plasticity and functional recovery after stroke. J Cereb Blood Flow Metab. 2008, 28: 1722-1732. 10.1038/jcbfm.2008.65.
Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, Micheva KD, Mehalow AK, Huberman AD, Stafford B, Sher A, Litke AM, Lambris JD, Smith SJ, John SW, Barres BA: The classical complement cascade mediates CNS synapse elimination. Cell. 2007, 131: 1164-1178. 10.1016/j.cell.2007.10.036.
Allen NJ, Barres BA: Neuroscience: Glia - more than just brain glue. Nature. 2009, 457: 675-677. 10.1038/457675a.
Barres BA: The mystery and magic of glia: a perspective on their roles in health and disease. Neuron. 2008, 60: 430-440. 10.1016/j.neuron.2008.10.013.
Tien AC, Tsai HH, Molofsky AV, McMahon M, Foo LC, Kaul A, Dougherty JD, Heintz N, Gutmann DH, Barres BA, Rowitch DH: Regulated temporal-spatial astrocyte precursor cell proliferation involves BRAF signalling in mammalian spinal cord. Development. 2012, 139: 2477-2487. 10.1242/dev.077214.
Sauvageot CM, Stiles CD: Molecular mechanisms controlling cortical gliogenesis. Curr Opin Neurobiol. 2002, 12: 244-249. 10.1016/S0959-4388(02)00322-7.
Bonni A, Sun Y, Nadal-Vicens M, Bhatt A, Frank DA, Rozovsky I, Stahl N, Yancopoulos GD, Greenberg ME: Regulation of gliogenesis in the central nervous system by the JAK-STAT signaling pathway. Science. 1997, 278: 477-483. 10.1126/science.278.5337.477.
Stipursky J, Francis D, Gomes FC: Activation of MAPK/PI3K/SMAD pathways by TGF-beta(1) controls differentiation of radial glia into astrocytes in vitro. Dev Neurosci. 2012, 34: 68-81. 10.1159/000338108.
Nakashima K, Takizawa T, Ochiai W, Yanagisawa M, Hisatsune T, Nakafuku M, Miyazono K, Kishimoto T, Kageyama R, Taga T: BMP2-mediated alteration in the developmental pathway of fetal mouse brain cells from neurogenesis to astrocytogenesis. Proc Natl Acad Sci U S A. 2001, 98: 5868-5873. 10.1073/pnas.101109698.
Nagao M, Sugimori M, Nakafuku M: Cross talk between notch and growth factor/cytokine signaling pathways in neural stem cells. Mol Cell Biol. 2007, 27: 3982-3994. 10.1128/MCB.00170-07.
Deneen B, Ho R, Lukaszewicz A, Hochstim CJ, Gronostajski RM, Anderson DJ: The transcription factor NFIA controls the onset of gliogenesis in the developing spinal cord. Neuron. 2006, 52: 953-968. 10.1016/j.neuron.2006.11.019.
Barnabe-Heider F, Wasylnka JA, Fernandes KJ, Porsche C, Sendtner M, Kaplan DR, Miller FD: Evidence that embryonic neurons regulate the onset of cortical gliogenesis via cardiotrophin-1. Neuron. 2005, 48: 253-265. 10.1016/j.neuron.2005.08.037.
Freeman MR: Specification and morphogenesis of astrocytes. Science. 2010, 330: 774-778. 10.1126/science.1190928.
Song MR, Ghosh A: FGF2-induced chromatin remodeling regulates CNTF-mediated gene expression and astrocyte differentiation. Nat Neurosci. 2004, 7: 229-235. 10.1038/nn1192.
Stipursky J, Gomes FC: TGF-beta1/SMAD signaling induces astrocyte fate commitment in vitro: implications for radial glia development. Glia. 2007, 55: 1023-1033. 10.1002/glia.20522.
Raff MC, Miller RH, Noble M: A glial progenitor cell that develops in vitro into an astrocyte or an oligodendrocyte depending on culture medium. Nature. 1983, 303: 390-396. 10.1038/303390a0.
Rowitch DH, Kriegstein AR: Developmental genetics of vertebrate glial-cell specification. Nature. 2010, 468: 214-222. 10.1038/nature09611.
Ge WP, Miyawaki A, Gage FH, Jan YN, Jan LY: Local generation of glia is a major astrocyte source in postnatal cortex. Nature. 2012, 484: 376-380. 10.1038/nature10959.
Bushong EA, Martone ME, Jones YZ, Ellisman MH: Protoplasmic astrocytes in CA1 stratum radiatum occupy separate anatomical domains. J Neurosci. 2002, 22: 183-192.
Derouiche A, Frotscher M: Peripheral astrocyte processes: monitoring by selective immunostaining for the actin-binding ERM proteins. Glia. 2001, 36: 330-341. 10.1002/glia.1120.
Wolff JR: The astrocyte as link between capillary and nerve cell. Triangle. 1970, 9: 153-164.
Chao TI RM, Wolff JR: The synapse-astrocyte boundary: anatomical basis for an integrative role of glia in synaptic transmission. Tripartite synapses: Synaptic transmission with glia. Edited by: Volterra A, Magistretti P, Haydon PG. 2002, Oxford, New York: Oxford University Press
Reichenbach AWH: Structural association of astrocytes with neurons and vasculature: defining territorial boundaries. Astrocytes in (patho)physiology of the nervous system. Edited by: Parpura VHP. 2009, New York: Springer, 251-286.
Bandeira F, Lent R, Herculano-Houzel S: Changing numbers of neuronal and non-neuronal cells underlie postnatal brain growth in the rat. Proc Natl Acad Sci U S A. 2009, 106: 14108-14113. 10.1073/pnas.0804650106.
Bushong EA, Martone ME, Ellisman MH: Maturation of astrocyte morphology and the establishment of astrocyte domains during postnatal hippocampal development. Int J Dev Neurosci. 2004, 22: 73-86. 10.1016/j.ijdevneu.2003.12.008.
Furuta A, Rothstein JD, Martin LJ: Glutamate transporter protein subtypes are expressed differentially during rat CNS development. J Neurosci. 1997, 17: 8363-8375.
Sutherland ML, Delaney TA, Noebels JL: Glutamate transporter mRNA expression in proliferative zones of the developing and adult murine CNS. J Neurosci. 1996, 16: 2191-2207.
Nagy JI, Patel D, Ochalski PA, Stelmack GL: Connexin30 in rodent, cat and human brain: selective expression in gray matter astrocytes, co-localization with connexin43 at gap junctions and late developmental appearance. Neuroscience. 1999, 88: 447-468. 10.1016/S0306-4522(98)00191-2.
Seifert G, Huttmann K, Binder DK, Hartmann C, Wyczynski A, Neusch C, Steinhauser C: Analysis of astroglial K+ channel expression in the developing hippocampus reveals a predominant role of the Kir4.1 subunit. J Neurosci. 2009, 29: 7474-7488. 10.1523/JNEUROSCI.3790-08.2009.
Higashimori H, Sontheimer H: Role of Kir4.1 channels in growth control of glia. Glia. 2007, 55: 1668-1679. 10.1002/glia.20574.
Schlag BD, Vondrasek JR, Munir M, Kalandadze A, Zelenaia OA, Rothstein JD, Robinson MB: Regulation of the glial Na+−dependent glutamate transporters by cyclic AMP analogs and neurons. Mol Pharmacol. 1998, 53: 355-369.
Duan S, Anderson CM, Stein BA, Swanson RA: Glutamate induces rapid upregulation of astrocyte glutamate transport and cell-surface expression of GLAST. J Neurosci. 1999, 19: 10193-10200.
Yang Y, Gozen O, Watkins A, Lorenzini I, Lepore A, Gao Y, Vidensky S, Brennan J, Poulsen D, Won Park J, Li Jeon N, Robinson MB, Rothstein JD: Presynaptic regulation of astroglial excitatory neurotransmitter transporter GLT1. Neuron. 2009, 61: 880-894. 10.1016/j.neuron.2009.02.010.
Swanson RA, Liu J, Miller JW, Rothstein JD, Farrell K, Stein BA, Longuemare MC: Neuronal regulation of glutamate transporter subtype expression in astrocytes. J Neurosci. 1997, 17: 932-940.
Koulakoff A, Ezan P, Giaume C: Neurons control the expression of connexin 30 and connexin 43 in mouse cortical astrocytes. Glia. 2008, 56: 1299-1311. 10.1002/glia.20698.
Rouach N, Glowinski J, Giaume C: Activity-dependent neuronal control of gap-junctional communication in astrocytes. J Cell Biol. 2000, 149: 1513-1526. 10.1083/jcb.149.7.1513.
Cahoy JD, Emery B, Kaushal A, Foo LC, Zamanian JL, Christopherson KS, Xing Y, Lubischer JL, Krieg PA, Krupenko SA, Thompson WJ, Barres BA: A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J Neurosci. 2008, 28: 264-278. 10.1523/JNEUROSCI.4178-07.2008.
Lovatt D, Sonnewald U, Waagepetersen HS, Schousboe A, He W, Lin JH, Han X, Takano T, Wang S, Sim FJ, Goldman SA, Nedergaard M: The transcriptome and metabolic gene signature of protoplasmic astrocytes in the adult murine cortex. J Neurosci. 2007, 27: 12255-12266. 10.1523/JNEUROSCI.3404-07.2007.
Agulhon C, Petravicz J, McMullen AB, Sweger EJ, Minton SK, Taves SR, Casper KB, Fiacco TA, McCarthy KD: What is the role of astrocyte calcium in neurophysiology?. Neuron. 2008, 59: 932-946. 10.1016/j.neuron.2008.09.004.
Wang X, Lou N, Xu Q, Tian GF, Peng WG, Han X, Kang J, Takano T, Nedergaard M: Astrocytic Ca2+ signaling evoked by sensory stimulation in vivo. Nat Neurosci. 2006, 9: 816-823. 10.1038/nn1703.
Sun W, McConnell E, Pare JF, Xu Q, Chen M, Peng W, Lovatt D, Han X, Smith Y, Nedergaard M: Glutamate-dependent neuroglial calcium signaling differs between young and adult brain. Science. 2013, 339: 197-200. 10.1126/science.1226740.
Lavialle M, Aumann G, Anlauf E, Prols F, Arpin M, Derouiche A: Structural plasticity of perisynaptic astrocyte processes involves ezrin and metabotropic glutamate receptors. Proc Natl Acad Sci U S A. 2011, 108: 12915-12919. 10.1073/pnas.1100957108.
Genoud C, Quairiaux C, Steiner P, Hirling H, Welker E, Knott GW: Plasticity of astrocytic coverage and glutamate transporter expression in adult mouse cortex. PLoS Biol. 2006, 4: e343-10.1371/journal.pbio.0040343.
Eiraku M, Tohgo A, Ono K, Kaneko M, Fujishima K, Hirano T, Kengaku M: DNER acts as a neuron-specific Notch ligand during Bergmann glial development. Nat Neurosci. 2005, 8: 873-880. 10.1038/nn1492.
Laurence JA, Fatemi SH: Glial fibrillary acidic protein is elevated in superior frontal, parietal and cerebellar cortices of autistic subjects. Cerebellum. 2005, 4: 206-210. 10.1080/14734220500208846.
Purcell AE, Jeon OH, Zimmerman AW, Blue ME, Pevsner J: Postmortem brain abnormalities of the glutamate neurotransmitter system in autism. Neurology. 2001, 57: 1618-1628. 10.1212/WNL.57.9.1618.
Fatemi SH, Folsom TD, Reutiman TJ, Lee S: Expression of astrocytic markers aquaporin 4 and connexin 43 is altered in brains of subjects with autism. Synapse. 2008, 62: 501-507. 10.1002/syn.20519.
Gadow KD, Roohi J, DeVincent CJ, Kirsch S, Hatchwell E: Glutamate transporter gene (SLC1A1) single nucleotide polymorphism (rs301430) and repetitive behaviors and anxiety in children with autism spectrum disorder. J Autism Dev Disord. 2010, 40: 1139-1145. 10.1007/s10803-010-0961-7.
Sicca F, Imbrici P, D’Adamo MC, Moro F, Bonatti F, Brovedani P, Grottesi A, Guerrini R, Masi G, Santorelli FM, Pessia M: Autism with seizures and intellectual disability: possible causative role of gain-of-function of the inwardly-rectifying K+ channel Kir4.1. Neurobiol Dis. 2011, 43: 239-247. 10.1016/j.nbd.2011.03.016.
Guy J, Hendrich B, Holmes M, Martin JE, Bird A: A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat Genet. 2001, 27: 322-326. 10.1038/85899.
Chen RZ, Akbarian S, Tudor M, Jaenisch R: Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat Genet. 2001, 27: 327-331. 10.1038/85906.
Fmr1 knockout mice: a model to study fragile X mental retardation. The Dutch-Belgian Fragile X Consortium. Cell. 1994, 78: 23-33.
Zoghbi HY: Postnatal neurodevelopmental disorders: meeting at the synapse?. Science. 2003, 302: 826-830. 10.1126/science.1089071.
Bear MF, Huber KM, Warren ST: The mGluR theory of fragile X mental retardation. Trends Neurosci. 2004, 27: 370-377. 10.1016/j.tins.2004.04.009.
Ballas N, Lioy DT, Grunseich C, Mandel G: Non-cell autonomous influence of MeCP2-deficient glia on neuronal dendritic morphology. Nat Neurosci. 2009, 12: 311-317. 10.1038/nn.2275.
Jacobs S, Nathwani M, Doering LC: Fragile X astrocytes induce developmental delays in dendrite maturation and synaptic protein expression. BMC Neurosci. 2010, 11: 132-10.1186/1471-2202-11-132.
Jacobs S, Doering LC: Astrocytes prevent abnormal neuronal development in the fragile x mouse. J Neurosci. 2010, 30: 4508-4514. 10.1523/JNEUROSCI.5027-09.2010.
Yang Q, Feng B, Zhang K, Guo YY, Liu SB, Wu YM, Li XQ, Zhao MG: Excessive astrocyte-derived neurotrophin-3 contributes to the abnormal neuronal dendritic development in a mouse model of fragile x syndrome. PLoS Genet. 2012, 8: e1003172-10.1371/journal.pgen.1003172.
Maezawa I, Swanberg S, Harvey D, LaSalle JM, Jin LW: Rett syndrome astrocytes are abnormal and spread MeCP2 deficiency through gap junctions. J Neurosci. 2009, 29: 5051-5061. 10.1523/JNEUROSCI.0324-09.2009.
Okabe Y, Takahashi T, Mitsumasu C, Kosai K, Tanaka E, Matsuishi T: Alterations of gene expression and glutamate clearance in astrocytes derived from an MeCP2-null mouse model of Rett syndrome. PLoS One. 2012, 7: e35354-10.1371/journal.pone.0035354.
Lioy DT, Garg SK, Monaghan CE, Raber J, Foust KD, Kaspar BK, Hirrlinger PG, Kirchhoff F, Bissonnette JM, Ballas N, Mandel G: A role for glia in the progression of Rett’s syndrome. Nature. 2011, 475: 497-500. 10.1038/nature10214.
Manent JB, Represa A: Neurotransmitters and brain maturation: early paracrine actions of GABA and glutamate modulate neuronal migration. Neuroscientist. 2007, 13: 268-279. 10.1177/1073858406298918.
Matsugami TR, Tanemura K, Mieda M, Nakatomi R, Yamada K, Kondo T, Ogawa M, Obata K, Watanabe M, Hashikawa T, Tanaka K: From the Cover: Indispensability of the glutamate transporters GLAST and GLT1 to brain development. Proc Natl Acad Sci U S A. 2006, 103: 12161-12166. 10.1073/pnas.0509144103.
Choudhury PR, Lahiri S, Rajamma U: Glutamate mediated signaling in the pathophysiology of autism spectrum disorders. Pharmacol Biochem Behav. 2012, 100: 841-849. 10.1016/j.pbb.2011.06.023.
Jamain S, Betancur C, Quach H, Philippe A, Fellous M, Giros B, Gillberg C, Leboyer M, Bourgeron T: Linkage and association of the glutamate receptor 6 gene with autism. Mol Psychiatry. 2002, 7: 302-310. 10.1038/sj.mp.4000979.
Serajee FJ, Zhong H, Nabi R, Huq AH: The metabotropic glutamate receptor 8 gene at 7q31: partial duplication and possible association with autism. J Med Genet. 2003, 40: e42-10.1136/jmg.40.4.e42.
Barnby G, Abbott A, Sykes N, Morris A, Weeks DE, Mott R, Lamb J, Bailey AJ, Monaco AP: Candidate-gene screening and association analysis at the autism-susceptibility locus on chromosome 16p: evidence of association at GRIN2A and ABAT. Am J Hum Genet. 2005, 76: 950-966. 10.1086/430454.
Bassell GJ, Warren ST: Fragile X syndrome: loss of local mRNA regulation alters synaptic development and function. Neuron. 2008, 60: 201-214. 10.1016/j.neuron.2008.10.004.
Huber KM, Gallagher SM, Warren ST, Bear MF: Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc Natl Acad Sci U S A. 2002, 99: 7746-7750. 10.1073/pnas.122205699.
Comery TA, Harris JB, Willems PJ, Oostra BA, Irwin SA, Weiler IJ, Greenough WT: Abnormal dendritic spines in fragile X knockout mice: maturation and pruning deficits. Proc Natl Acad Sci U S A. 1997, 94: 5401-5404. 10.1073/pnas.94.10.5401.
Hagerman PJ, Stafstrom CE: Origins of epilepsy in fragile X syndrome. Epilepsy Curr. 2009, 9: 108-112. 10.1111/j.1535-7511.2009.01309.x.
Michalon A, Sidorov M, Ballard TM, Ozmen L, Spooren W, Wettstein JG, Jaeschke G, Bear MF, Lindemann L: Chronic pharmacological mGlu5 inhibition corrects fragile X in adult mice. Neuron. 2012, 74: 49-56. 10.1016/j.neuron.2012.03.009.
Dolen G, Osterweil E, Rao BS, Smith GB, Auerbach BD, Chattarji S, Bear MF: Correction of fragile X syndrome in mice. Neuron. 2007, 56: 955-962. 10.1016/j.neuron.2007.12.001.
Silverman JL, Smith DG, Rizzo SJ, Karras MN, Turner SM, Tolu SS, Bryce DK, Smith DL, Fonseca K, Ring RH, Crawley JN: Negative allosteric modulation of the mGluR5 receptor reduces repetitive behaviors and rescues social deficits in mouse models of autism. Sci Transl Med. 2012, 4: 131ra151-
Huang YH, Bergles DE: Glutamate transporters bring competition to the synapse. Curr Opin Neurobiol. 2004, 14: 346-352. 10.1016/j.conb.2004.05.007.
Danbolt NC: Glutamate uptake. ProgNeurobiol. 2001, 65: 1-105.
Huang YH, Sinha SR, Tanaka K, Rothstein JD, Bergles DE: Astrocyte glutamate transporters regulate metabotropic glutamate receptor-mediated excitation of hippocampal interneurons. J Neurosci. 2004, 24: 4551-4559. 10.1523/JNEUROSCI.5217-03.2004.
Omrani A, Melone M, Bellesi M, Safiulina V, Aida T, Tanaka K, Cherubini E, Conti F: Up-regulation of GLT-1 severely impairs LTD at mossy fibre–CA3 synapses. J Physiol. 2009, 587: 4575-4588. 10.1113/jphysiol.2009.177881.
Higashimori H, Morel L, Huth J, Lindemann L, Dulla C, Taylor A, Freeman M, Yang Y: Astroglial FMRP-dependent translational down-regulation of mGluR5 underlies glutamate transporter GLT1 dysregulation in the fragile X Mouse. Hum Mol Genet. 2013, 22: 2041-2054. 10.1093/hmg/ddt055.
Tanaka K: Epilepsy and exacerbation of brain injury in mice lacking glutamate transporter GLT-1 (Vol 276, pg 1699, 1997). Science. 1997, 278: 21-21.
Takasaki C, Okada R, Mitani A, Fukaya M, Yamasaki M, Fujihara Y, Shirakawa T, Tanaka K, Watanabe M: Glutamate transporters regulate lesion-induced plasticity in the developing somatosensory cortex. J Neurosci. 2008, 28: 4995-5006. 10.1523/JNEUROSCI.0861-08.2008.
Tanaka K: Role of glutamate transporters in the pathophysiology of major mental illnesses. Nihon Shinkei Seishin Yakurigaku Zasshi. 2009, 29: 161-164.
Christopherson KS, Ullian EM, Stokes CC, Mullowney CE, Hell JW, Agah A, Lawler J, Mosher DF, Bornstein P, Barres BA: Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell. 2005, 120: 421-433. 10.1016/j.cell.2004.12.020.
Kucukdereli H, Allen NJ, Lee AT, Feng A, Ozlu MI, Conatser LM, Chakraborty C, Workman G, Weaver M, Sage EH, Barres BA, Eroglu C: Control of excitatory CNS synaptogenesis by astrocyte-secreted proteins Hevin and SPARC. Proc Natl Acad Sci U S A. 2011, 108: E440-E449. 10.1073/pnas.1104977108.
Regan MR, Huang YH, Kim YS, Dykes-Hoberg MI, Jin L, Watkins AM, Bergles DE, Rothstein JD: Variations in promoter activity reveal a differential expression and physiology of glutamate transporters by glia in the developing and mature CNS. J Neurosci. 2007, 27: 6607-6619. 10.1523/JNEUROSCI.0790-07.2007.
Yang Y, Vidensky S, Jin L, Jie C, Lorenzini I, Frankl M, Rothstein JD: Molecular comparison of GLT1+ and ALDH1L1+ astrocytes in vivo in astroglial reporter mice. Glia. 2011, 59: 200-207. 10.1002/glia.21089.
Doyle JP, Dougherty JD, Heiman M, Schmidt EF, Stevens TR, Ma G, Bupp S, Shrestha P, Shah RD, Doughty ML, Gong S, Greengard P, Heintz N: Application of a translational profiling approach for the comparative analysis of CNS cell types. Cell. 2008, 135: 749-762. 10.1016/j.cell.2008.10.029.
Heiman M, Schaefer A, Gong S, Peterson JD, Day M, Ramsey KE, Suarez-Farinas M, Schwarz C, Stephan DA, Surmeier DJ, Greenard P, Heintz N: A translational profiling approach for the molecular characterization of CNS cell types. Cell. 2008, 135: 738-748. 10.1016/j.cell.2008.10.028.
Acknowledgements
This work is supported by NIMH 099554 (to YY) and FRAXA Research Grant (to YY and HH).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
YY and HH contributed to the writing of the manuscript; HH and LM performed imaging and western experiments. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Yang, Y., Higashimori, H. & Morel, L. Developmental maturation of astrocytes and pathogenesis of neurodevelopmental disorders. J Neurodevelop Disord 5, 22 (2013). https://doi.org/10.1186/1866-1955-5-22
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1866-1955-5-22