
Abstract
Background
Miniature inverted-repeat transposable elements (MITEs) are short, nonautonomous DNA elements flanked by subterminal or terminal inverted repeats (TIRs) with no coding capacity. MITEs were originally recognized as important components of plant genomes, where they can attain extremely high copy numbers, and are also found in several animal genomes, including mosquitoes, fish and humans. So far, few MITEs have been described in Drosophila.
Results
Herein we describe the distribution and evolution of Mar, a MITE family of hAT transposons, in Drosophilidae species. In silico searches and PCR screening showed that Mar distribution is restricted to the willistoni subgroup of the Drosophila species, and a phylogenetic analysis of Mar indicates that this element may have originated prior to the diversification of these species. Most of the Mar copies in D. willistoni present conserved target site duplications and TIRs, indicating recent mobilization of these sequences. We also identified relic copies of potentially full-length Mar transposon in D. tropicalis and D. willistoni. The phylogenetic relationship among transposases from the putative full-length Mar and other hAT superfamily elements revealed that Mar is placed into the recently determined Buster group of hAT transposons.
Conclusion
On the basis of the obtained data, we can suggest that the origin of these Mar MITEs occurred before the subgroup willistoni speciation, which started about 5.7 Mya. The Mar relic transposase existence indicates that these MITEs originated by internal deletions and suggests that the full-length transposon was recently functional in D. willistoni, promoting Mar MITEs mobilization.
Similar content being viewed by others

Background
Transposable elements (TEs) are discrete segments of DNA distinguished by their ability to move and replicate within genomes[1]. TE-derived sequences are the most abundant components of several eukaryotic genomes. An increasing amount of evidences shows that TEs can play an important role in driving the evolution and genome complexity[2–6].
TEs can be divided into two classes based on their mechanism of transposition: class I comprises the retrotransposons that transpose through an RNA intermediate, and class II comprises the transposons that transpose through a DNA intermediate[7]. Class II transposons encode for the transposase enzyme, which specifically recognizes the element terminal inverted repeats (TIRs), excises the transposon and inserts it elsewhere in the host genome. Insertion in the genome results in target site duplications (TSDs). Depending on their ability to direct their own transposition, TEs from both classes can include both autonomous and nonautonomous copies. Autonomous TEs encode for the proteins required for their transposition, and nonautonomous TEs can be mobilized in trans using the enzymes produced by autonomous elements[7, 8].
Within the class II transposons, there is a special group of nonautonomous sequences, called miniature inverted-repeat transposable elements (MITEs), which can be present in high number of copies in some genomes. They are characterized by short sequences with no coding capacity, contain conserved TIRs, are flanked by TSDs produced by the insertion and probably originated from a subset of autonomous DNA transposons[9–12]. MITEs often include internal AT-rich sequences that are not homologous to their parental autonomous elements. They were first discovered in plants, but they have also been found in several animal genomes, including Caenorhabditis elegans, Drosophila, mosquitoes, fish and humans[13, 14].
The first MITE families described in Drosophila were Vege and Mar, both of which were discovered in D. willistoni[15]. These elements have 884 bp and 610 bp, respectively, and are AT-rich. Vege has 12-bp TIRs and Mar has 11 bp TIRs, and both elements are flanked by 8-bp TSDs. The initial tBLASTn and BLASTx analysis indicated that both elements have neither coding capacity nor significant sequence similarity to published sequences available at the time that the analysis was conducted. As MITEs have been grouped into TE superfamilies based on the length of their TIRs and TSDs, Vege and Mar were hypothesized to be members of the hAT superfamily[15]. Thus, Mar and Vege precursor elements are probably autonomous elements from the hAT superfamily; however, these precursors were not previously identified. The hAT superfamily is widely distributed in multicellular organisms, including plants, animals and fungi[16]. Members of this superfamily are flanked by 8-bp TSDs, have relatively short TIRs (5 to 27 bp) and are less than 4 kb in overall length[7]. Recently, the hAT superfamily was divided into two families, Ac and Buster, primarily due to differences in target site selection[17].
Little is known about MITEs in Drosophila.We investigated the presence and evolution of Mar in Drosophilidae species and characterized Mar copies from the D. willistoni genome. We show herein that Mar is restricted to the willistoni subgroup species and propose that Mar originated prior to the diversification of these species. In D. willistoni, we found evidence of recent mobilization and amplification. We also identified relic copies of a full-length Mar in D. tropicalis and D. willistoni, suggesting that the origin of the Mar MITEs occurred by internal deletion of an autonomous copy followed by amplification. In a phylogeny of hAT elements, full-length Mar forms a clade with Buster elements from bat, mosquito, sea urchin (Strongylocentrotus purpuratus), zebrafish (Danio rerio) and freshwater planarian (Schmidtea mediterranea), and not with other Drosophila hAT elements. The TSD consensus also indicates that Mar is a hAT element from the Buster family. As far as we know, this is the first Buster element described in Drosophila.
Results
Mar is restricted to the willistoni subgroup species
In silico searches for Mar homologous sequences were conducted in the following genomes: D. melanogaster, D. simulans, D. sechellia, D. yakuba, D. erecta, D. ficusphila, D. eugracilis, D. biarmipes, D. takahashii, D. elegans, D. rhopaloa, D. kikkawai, D. ananassae, D. bipectinata, D. pseudoobscura, D. persimilis, D. willistoni, D. mojavensis, D. virilis and D. grimshawi. As expected, sequences homologous to Mar were found in D. willistoni. In the other 19 available genomes, no sequences homologous to Mar were found. These available genomes comprise three species from the Drosophila subgenus and 16 from the Sophophora subgenus, including 14 species from the melanogaster group and 2 from the obscura group.
To expand the analysis of Mar distribution, we used PCR and Dot blot strategies in a large number of Drosophilidae species belonging to different Drosophila groups (Table1). A pair of primers, MarF and MarR, was used to amplify a 455-bp fragment of Mar (Figure1). PCR results showed amplification only in the species from the willistoni subgroup: D. willistoni, D. paulistorum, D. equinoxialis, D. insularis and D. tropicalis. The fragment lengths varied from roughly 270 bp to 450 bp for most species, but for D. tropicalis the amplified fragment was larger than expected (approximately 2,600 bp), suggesting the possibility of finding a full-length transposon. The Dot blot results (Additional file1 and Figure2) corroborated the PCR results, showing positive signals only in the willistoni subgroup species. Species from the bocainensis subgroup (also part of the willistoni group) presented a very weak signal, which may indicate the presence of highly divergent sequences related to Mar.

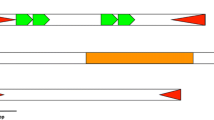
Schematic representation of the reconstructed full-length Mar compared to the canonical Mar element (MITE). Common regions are indicated, including terminal inverted repeats (black boxes). The transposase coding region with the Dimer_Tnp_hAT domain coding region is also shown. Below are the schematic representations of copies found in D. tropicalis and D. willistoni. Only indels of more than 12 nucleotides are represented. Arrows indicate the primer annealing regions. The primers MarF and MarR were used to amplify Mar from the willistoni group species, and Mar2F and Mar2R were used to sequence the D. tropicalis clones

Evolutionary relationships between the willistoni group species, based on[25], and the results obtained from the PCR and Dot blot screenings.
All cloned sequences (five from D. insularis, ten from D. paulistorum, five from D. equinoxialis, seven from D. willistoni and six from D. tropicalis) and those obtained by in silico searches (93 sequences from the D. willistoni genome) were used in the phylogenetic analysis to understand the evolutionary dynamics of Mar in the willistoni subgroup (GenBank accession number and scaffold coordinates of sequences are shown in Additional files2 and3). Figure3 shows the Neighbor-joining tree obtained for Mar, which can be compared with the host species phylogeny in Figure2. Two major groups, highlighted in the phylogeny, are composed of only very similar sequences from D. willistoni. Most of the sequences from D. equinoxialis, D. paulistorum and D. insularis are located in a group with very low branching support. The maximum likelihood (ML) and Bayesian trees show similar topologies (data not shown).

Mar Neighbor-joining tree. Bootstrap values are shown at the nodes; values smaller than 50 were omitted. Different species are highlighted with different colors, as shown in the legend. The two clades highlighted with rectangles represent two events of pronounced copy number amplification in D. willistoni. More information about these sequences is available in Additional files2 and3.
The Mar sequences present an overall mean divergence of 9.96%. Table2 shows the mean divergence of Mar sequences found within and between species. The intraspecies divergence ranged from 0.3% for D. tropicalis up to 8.6% for D. paulistorum. Concerning the interspecies divergence, the values varied from 8.5% between D. paulistorum and D. insularis to 16.3% between D. tropicalis and D. paulistorum. Lower levels of intraspecies divergence would be expected if the copies were recently transposed. The generally high divergence found within species and the interspersed distribution of species in the phylogeny can be explained by the presence of these sequences prior to the split of the species. On the other hand, in D. willistoni, we were able to evaluate a large number of copies, which enabled us to obtain a better view of Mar evolution. In spite of the presence of ancient Mar copies in D. willistoni, represented by their distinct positions in the phylogeny, there are two clear events of recent mobilization and pronounced amplification of Mar (highlighted clades in Figure3).
Mar copies from D. willistoni
We identified 93 Mar sequences in the D. willistoni genome (Additional file3). The exact number of copies is difficult to determine because the genome contains some small and fragmented copies that are not captured in the searches. Also, we cannot exclude the existence of duplicated scaffolds in the database, particularly the very short ones. Of the sequences identified, 74 (79%) contain 11-bp conserved TIRs (CAG(G/A)GGTAGGC), which are not perfect as they were described previously[15]. Only one sequence exhibited perfect TIRs. The majority of copies (79%) are flanked by 8-bp conserved TSDs, indicating recent mobilization of these sequences. The Mar element TSD consensus sequence (5′-nnnTAnnn-3′) matches that of the Buster element TSD consensus sequence. This strongly suggests that Mar belongs to the Buster family of hAT transposons. Analysis of Mar copies distribution throughout the genome reveals that 32 copies are found within a gene or less than 2 kb from a gene (Additional file4). Only a small region of Mar was found in a predicted coding sequence.
Putative full-length Mar
The amplified sequences from D. tropicalis were much longer than expected. We therefore used a second pair of primers, Mar2F and Mar2R, to sequence the entire fragment. We obtained six clones with good-quality sequences of approximately 2,480 bp with a 300-bp region homologous to Mar in the 3′ and few nucleotides in the 5′ region. These clones have 96% to 99% sequence identity and show a mean divergence of 10.5% from the corresponding region of the canonical Mar sequence. We cannot distinguish whether these clones are different copies or alleles from the same genome, or if there is polymorphism in the population. BLASTn analysis of these sequences showed significant sequence similarity to the Mar element and did not produce any other significant hit.
For all clones, the FGENESH program failed to identify a significant coding region. However, BLASTx searches revealed an intriguing similarity to proteins belonging to the TFII-I family in several distinct organisms, including Camponotus floridanus (insect), S. purpuratus (sea urchin), Anoliscaro linensis (lizard) and several fishes. The highest similarity corresponded to the general transcription factor II-I repeat domain-containing protein 2-like from Xenopus tropicalis (XP_002941054), and the BLAST alignments showed significant similarity (query coverage: 70%; E-value: equal to or less than 5e-92; mean similarity: 50%), except for the presence of stop codons in the D. tropicalis sequences. The D. tropicalis sequences also showed similarity to some transposase sequences, although they had lower similarity scores, confirming their TE origin. This X. tropicalis protein could be an element that was incorrectly annotated, since a CENSOR screening against a Repbase reference collection of repeats revealed 66% similarity with hAT-43_SM, an element from S. mediterranea. Alternatively, this protein could have resulted from the domestication of a hAT superfamily element that has not yet been described. There are several examples of elements from this superfamily being exapted to essential functions within the host genome[17, 18].
To better characterize the sequences found in D. tropicalis, we searched for similar sequences in the D. willistoni genome and found four sequences with significant similarity (mean of 88%). A schematic representation of these sequences can be observed in Figure1, and the scaffold coordinates are available in Additional file3. Three sequences (scaf94, scaf95 and scaf96) are shorter than those from D. tropicalis, but the other one (scaf72) drew our attention because its TIR sequences are identical to those found in the canonical Mar element and it is flanked by 8-bp TSDs with one mismatch (CTCTAC(C/T)C). Despite the fact that this appears to be a complete element, we were not able to find a significant coding region in this copy or in the shorter copies.
Next we aligned the canonical Mar, the D. tropicalis sequences and the Dwillistoni_scaf72 sequence from D. willistoni to obtain a consensus sequence by selecting the most common nucleotide in each position. Some slight modifications were made to the consensus sequence in an attempt to reconstruct a functional sequence with potential coding regions. An alignment of these sequences can be found in Additional file5. Using this approach and the FGENESH program, we were able to identify a well-defined exon predicted to encode a protein of 591 amino acids. As expected, a BLASTp search also showed significant similarity to the X. tropicalis protein (XP_002941054), and a hAT family dimerization domain was found in the carboxy terminal of the predicted protein (Additional file6). A schematic representation of this reconstructed Mar full-length element and the transposase coding region is shown in Figure1. The sequences of the entire reconstructed element, coding region and protein are available in Additional file7.
Although the D. willistoni complete copy (scaf72) has a large deletion in relation to the reconstructed copy and has no coding capacity, the presence of TIRs identical to those present in the canonical Mar element indicates that this is a relic of an autonomous full-length Mar. Not surprisingly, this sequence appears as a basal branch in the Mar phylogeny (Figure3). Moreover, the TSD sequences flanking this copy also match the Buster element TSD consensus sequence.
Mar position in the hAT elements phylogeny
To establish the relationship between the Mar consensus transposase and the hAT superfamily elements, we assembled the transposase sequences described in[17] along with other homologous sequences detected by a BLASTp search. In our analysis, the hAT transposase phylogenetic tree also revealed two major clusters of related sequences (Figure4), previously labeled Ac family and Buster family[17]. The Mar putative transposase fell within the Buster family. It forms a clade with Buster transposase sequences from bat (MlBuster1 and Myotis-hAT1), mosquito (AeBuster4), sea urchin Strongylocentrotus purpuratus (Sp-Buster-1,2,2b,c), zebrafish Danio rerio (hAT5_DR) and freshwater planarian Schmidtea mediterranea (sm_hAT3 and sm_hAT6). As expected, it is closed to the general transcription factor II-I repeat domain-containing protein 2-like from X. tropicalis (XP_002941054). All of the other Drosophila hAT elements belong to the Ac family. These data confirm that Mar belongs to the Buster family.

Neighbor-joining tree showing the relationships among transposase amino acid sequences from several hAT elements, including the putative Mar transposase . Bootstrap values are shown at the nodes. Values smaller than 50 were omitted. The identity of the sequences can be found in Additional file9.
Discussion
In Drosophila genomes, MITEs are not as abundant and diverse as in mosquitoes and plants. Herein we describe the evolution of Mar, a MITE family in Drosophila. It is important to note that the designation of MITE is not attributed to a common origin or a taxonomic level in TE classification. The designation of MITE is useful to describe this type of nonautonomous elements that share typical structural features: (1) short elements with no coding capacity, (2) can be present in a high number of copies, (3) contain TIRs, (4) are often located in or near genes and (5) are AT-rich mainly in the inner region[13, 19]. The D. willistoni Mar element shows these characteristics, but the number of copies is not as high as that of most of the MITE families. However, several MITE families exhibit more modest copy numbers[11, 20–24]. We were unable to analyze the number of copies or conservation of TIRs and TSDs in species other than D. willistoni from the willistoni group; hence we do not know if the Mar element spread successfully throughout other genomes. In D. tropicalis, no Mar MITE copies were found.
Mar sequences are present only in Drosophila species from the willistoni subgroup. In general, the Mar phylogeny showed very weak resolution, with a scattered distribution of sequences in different species. This could be indicative of horizontal transfer between species, a common process in TE evolution[22]. However, the species involved are very closely related, and some levels of incongruence were found between different phylogenies of the willistoni subgroup, which suggests that saturation, introgression and perhaps incompletely sorted ancestral polymorphisms due to rapid radiation may have occurred[25]. Considering that Mar is a multiple copy sequence, the Mar phylogeny supports the view that the origin of this MITE occurred after the separation of the willistoni and bocainensis subgroups, but before the subgroup willistoni speciation that began approximately 5.7 Mya[25]. At least in D. willistoni, recent transposition bursts have occurred. Some sequences distantly related to Mar may be present in species from the bocainensis subgroup, as suggested by the Dot blot screening.
In plants and mosquitoes, MITEs are frequently associated with host genes, indicating a potential role for these elements in gene regulation and genome organization[26–28]. We found several Mar copies in or near genes in the D. willistoni genome. Some of these insertions may be ancient copies present in the ancestor of the willistoni subgroup. However, most of the gene-associated copies of Mar contain conserved TIRs and TSDs, which suggests that these copies were recently inserted in the D. willistoni genome and had no time to accumulate mutations. Because of the recent mobilization of this element, Mar is a potentially powerful factor promoting intra- and interspecies variability in the willistoni group.
Our analysis revealed that the putative Mar transposase is related to the Buster family of hAT transposons, and the Mar element TSD consensus sequence (5′-nnnTAnnn-3′) also indicates that Mar is a Buster element. The Mar transposase similarity with TFII-I family proteins and transposons from several distinct organisms from divergent taxa raises questions regarding the Mar MITE origin. It is known that the Buster family consists of both active transposons and domesticated genes that have lost their TIRs but are highly conserved across species[17]. The full-length copy of Mar found in D. willistoni still retains the TIRs and probably represents an ancient copy of the autonomous Mar element rather than a domesticated gene. The intriguing discontinuous distribution of Buster family sequences across vertebrates and invertebrates is referred by some authors as a result of horizontal transfer between species[17, 29]. More studies are necessary to better understand the relationship between Mar and transposons from other species.
Considering the recent mobilization of Mar MITEs in D. willistoni, we suppose that there should be an active copy allowing the mobilization. Analysis of the coding capacity of the full-length copy of Mar suggests that it is no longer active. Thus, it remains uncertain whether this copy was responsible for the recent Mar mobility before it became inactive. We cannot exclude the possibility that there is, elsewhere in the D. willistoni genome or in other D. willistoni strains, a functional copy that could still provide a source of transposase for Mar MITE mobilization. Alternatively, another element may provide the transposase for Mar mobilization. Cross-mobilization is highly associated with the amplification of MITE families[23]. For instance, in rice, the MITE mPing (derived from the autonomous element Ping) can be mobilized by the related autonomous element Pong[24]. Additionally, another work recently showed cross-mobilization of MITEs from the Stowaway family by the Osmar transposase[23]. In insects, within the hAT superfamily of DNA transposons, cross-mobilization has been reported to the hobo element, which is able to mobilize the hermes transposon[30]. It would be expected that a hAT element would provide the transposase for Mar mobilization, since TIR similarity is an essential requirement for MITE transposition[31, 32]. de Freitas Ortiz and Loreto[33] characterized five different hAT elements in D. willistoni, of which three are potentially active. These elements were classified as Ac family members[17], and a comparison of their TSD consensus sequences and TIRs with those from the Mar element (Additional file8) does not support the hypothesis that some of these hAT elements could be responsible for Mar mobilization. The TSD consensus sequences of the Mar insertions indicate that they were mobilized by a Buster element. To our knowledge, Mar is the first Buster member described in Drosophila; however, more specific searches can identify new Buster elements in these species.
The origin of different MITE families is not completely clear, and distinct processes may be involved. One hypothesis is that the MITEs originated by the deletion of autonomous copies[34]. Our results suggest that Mar MITEs originated by deletion of a full-length copy and subsequent amplification.
Conclusions
Mar distribution is restricted to the willistoni subgroup species and probably originated prior to the diversification of these species. In D. willistoni, we found evidence of recent mobilization and amplification. We also identified nonautonomous copies of a full-length Mar element in D. tropicalis and D. willistoni, suggesting that the origin of the Mar MITEs may have occurred by internal deletion of an autonomous copy followed by amplification. These elements belong to the Buster family and represent the first element of this family identified in Drosophila.
Methods
In silico searches
Searches for Mar homologous sequences were conducted in the following genomes using BLASTn on FlyBase: D. melanogaster, D. simulans, D. sechellia, D. yakuba, D. erecta, D. ficusphila, D. eugracilis, D. biarmipes, D. takahashii, D. elegans, D. rhopaloa, D. kikkawai, D. ananassae, D. bipectinata, D. pseudoobscura, D. persimilis, D. willistoni, D. mojavensis, D. virilis and D. grimshawi[35]. The complete canonical Mar sequence (AF518731.1) was used as a query. The presence of conserved TIRs and TSDs in the Mar sequences from D. willistoni was analyzed by visual inspection of the sequence alignments. We analyzed all hits with an E-value lower than e-100. WebLogo was used for the TSD analysis[36]. Local BLASTn searches were performed against different sequence datasets of the D. willistoni genome (coding sequences, intron and gene extended 2,000-bp) to identify Mar insertions in gene regions.
PCR and Dot blot screening
We screened for the presence of Mar elements in 61 Drosophila species, as well as Zaprionus indianus, Z. tuberculatus, Scaptodrosophila latifasciaeformis and S. lebanonensis, using PCR and Dot blotting (Table1). DNA was extracted from 30 fresh adult flies using a phenol-chloroform protocol[37]. For the PCR reactions, two primers were designed to amplify a Mar element fragment of approximately 450 bp: MarF 5′-CGCGAATCGTATGTGAA-3′ and MarR 5′-CGATGTGAGCACGAAGTACA-3′ (Figure1). The PCR reactions (50 μl) were performed as follows: 50 ng of template DNA, 20 pM of each primer, 2.5 mM MgCl2 and 1 U Taq DNA polymerase. The amplification conditions were as follows: first denaturation at 92°C for 2 minutes, 30 cycles of denaturation at 92°C for 45 seconds, primer annealing at 55°C for 50 seconds and extension at 72°C for 1 minute, followed by extension at 72°C for 5 minutes.
For Dot blot hybridizations, samples of denatured DNA (1 μg) were transferred onto a nylon membrane (Hybond-N+; GE Healthcare Biosciences, Pittsburgh, PA, USA). The AlkPhos Direct Labelling and Detection System and the CDP-Star kit (GE Healthcare) were used to label and detect nucleic acids according to the manufacturer’s instructions. The PCR product of the Mar element from D. willistoni was used as the probe.
DNA cloning and sequencing
Amplified samples were visualized on a 0.8% agarose gel. The bands were purified using the GFX Purification Kit (GE Healthcare) and cloned using the TOPO-TA cloning vector (Invitrogen, Carlsbad, CA, USA). Cloned PCR products were sequenced using the universal primers M13 (forward and reverse) on a MegaBACE 500 sequencer. The dideoxy chain-termination reaction was performed using the DYEnamicET kit (GE Healthcare). Two additional primers were used for sequencing the D. tropicalis clones: Mar2F 5′-CGGACGAAAGGGTATTAACT-3′ and Mar2R 5′-GCCGTTACACTTGTTTCCTA-3′. Both DNA strands were sequenced at least twice or until a reliable sequence was obtained. The sequences from each clone were assembled using Gap4 software from the Stadenpackage[38]. The sequence accession numbers are available in Additional file2.
Sequence analysis
Nucleotide and amino acid sequences were aligned using the Muscle tool[39] with default parameters. Nucleotide sequences were used to construct phylogenies according to the following methods: Neighbor-joining and maximum likelihood using the Tamura three-parameter substitution model with a gamma parameter of 2.0 as indicated by model selection analysis. These analyses were implemented using MEGA 5 software[40]. Bayesian analysis was performed using MrBayes 3.1.2 with at least 2,000,000 generations and a burn-in region of 1,000 trees using the Hasegawa, Kishino and Yano (HKY) model with gamma distribution as suggested by the MrModel Test 2.3 program[41]. To calculate the average divergence within and between species, we used MEGA 5 software and the p- distance function[40].
To check whether the full-length Mar copies potentially encode a functional transposase, we used FGENESH[42] to predict the existence of coding regions and possible introns. CENSOR software[43] was used to screen query sequences against the reference collection of repeats in Repbase.
The transposase amino acid sequences from several hAT superfamily members were compared to the Mar consensus sequence from D. tropicalis. The protein sequences used were collected based on the work of Arensburger et al.[17] from several databases and one manuscript. These sequence identities are shown in Additional file9. The phylogenetic analysis was conducted using MEGA 5 software[40]. A Neighbor-joining method using the Jones-Taylor-Thornton (JTT) model (with a gamma parameter of 2.0) was used, as indicated by model selection analysis.
Abbreviations
- BLAST:
-
Basic Local Alignment Search Tool
- bp:
-
Base pairs
- MITE:
-
Miniature inverted-repeat transposable element
- PCR:
-
Polymerase chain reaction
- TIRs:
-
Terminal inverted repeats
- TSD:
-
Target site duplication.
References
Kidwell MG, Lisch DR: Perspective: transposable elements, parasitic DNA, and genome evolution. Evolution. 2001, 55: 1-24.
Deprá M, Valente VLS, Margis R, Loreto ELS: The hobo transposon and hobo-related elements are expressed as developmental genes in Drosophila. Gene. 2009, 448: 57-63. 10.1016/j.gene.2009.08.012.
Feschotte C, Pritham EJ: Mobile DNA: genomes under the influence. Genome Biol. 2006, 7: 320-10.1186/gb-2006-7-6-320.
Kazazian HH: Mobile elements: drivers of genome evolution. Science. 2004, 303: 1626-1632. 10.1126/science.1089670.
Loreto E, da Silva L, Zaha A, Valente V: Distribution of transposable elements in neotropical species of Drosophila. Genetica. 1998, 101: 153-165.
Thornburg BG, Gotea V, Makałowski W: Transposable elements as a significant source of transcription regulating signals. Gene. 2006, 365: 104-110.
Wicker T, Sabot F, Hua-Van A, Bennetzen JL, Capy P, Chalhoub B, Flavell A, Leroy P, Morgante M, Panaud O, Paux E, SanMiguel P, Schulman AH: A unified classification system for eukaryotic transposable elements. Nat Rev Genet. 2007, 8: 973-982. 10.1038/nrg2165.
Capy P, Bazin C, Langin T: Dynamics and Evolution of Transposable Elements. 1998, Landes Bioscience, Austin, TX, 197-
Bureau TE, Wessler SR: Tourist: a large family of small inverted repeat elements frequently associated with maize genes. Plant Cell. 1992, 4: 1283-1294.
Jiang N, Feschotte C, Zhang X, Wessler SR: Using rice to understand the origin and amplification of miniature inverted repeat transposable elements (MITEs). CurrOpinPlant Biol. 2004, 7: 115-119.
Quesneville H, Nouaud D, Anxolabéhère D: P elements and MITE relatives in the whole genome sequence of Anopheles gambiae. BMC Genomics. 2006, 7: 214-10.1186/1471-2164-7-214.
De FreitasOrtiz M, Loreto EL: The hobo-related elements in the melanogaster species group. GenetRes (Camb). 2008, 90: 243-252.
Feschotte C, Jiang N, Wessler SR: Plant transposable elements: where genetics meets genomics. Nat Rev Genet. 2002, 3: 329-341.
González J, Petrov D: MITEs––the ultimate parasites. Science. 2009, 325: 1352-1353. 10.1126/science.1179556.
Holyoake AJ, Kidwell MG: Vege and Mar: two novel hAT MITE families from Drosophila willistoni. Mol Biol Evol. 2003, 20: 163-167. 10.1093/molbev/msg023.
Rubin E, Lithwick G, Levy A: Structure and evolution of the hAT transposon superfamily. Genetics. 2001, 158: 949-957.
Arensburger P, Hice RH, Zhou L, Smith RC, Tom AC, Wright JA, Knapp J, O’Brochta DA, Craig NL, Atkinson PW: Phylogenetic and functional characterization of the hAT transposon superfamily. Genetics. 2011, 188: 45-57. 10.1534/genetics.111.126813.
Sinzelle L, Izsvák Z, Ivics Z: Molecular domestication of transposable elements: from detrimental parasites to useful host genes. Cell Mol Life Sc. 2009, 66: 1073-1093. 10.1007/s00018-009-8376-3.
Kuang H, Padmanabhan C, Li F, Kamei A, Bhaskar PB, Ouyang S, Jiang J, Buell CR, Baker B: Identification of miniature inverted-repeat transposable elements (MITEs) and biogenesis of their siRNAs in the Solanaceae: new functional implications for MITEs. Genome Res. 2009, 19: 42-56.
Grzebelus D, Gładysz M, Macko-Podgórni A, Gambin T, Golis B, Rakoczy R, Gambin A: Population dynamics of miniature inverted-repeat transposable elements (MITEs) in Medicago truncatula. Gene. 2009, 448: 214-220. 10.1016/j.gene.2009.06.004.
Xu J, Wang M, Zhang X, Tang F, Pan G, Zhou Z: Identification of NbME MITE families: potential molecular markers in the microsporidia Nosemabombycis. J Invertebr Pathol. 2010, 103: 48-52. 10.1016/j.jip.2009.10.011.
Loreto ELS, Carareto CMA, Capy P: Revisiting horizontal transfer of transposable elements in Drosophila. Heredity (Edinb). 2008, 100: 545-554. 10.1038/sj.hdy.6801094.
Yang G, Nagel DH, Feschotte C, Hancock CN, Wessler SR: Tuned for transposition: molecular determinants underlying the hyperactivity of a Stowaway MITE. Science. 2009, 325: 1391-1394. 10.1126/science.1175688.
Jiang N, Bao Z, Zhang X, Hirochika H, Eddy SR, McCouch SR, Wessler SR: An active DNA transposon family in rice. Nature. 2003, 421: 163-167. 10.1038/nature01214.
Robe LJ, Cordeiro J, Loreto ELS, Valente VLS: Taxonomic boundaries, phylogenetic relationships and biogeography of the Drosophila willistoni subgroup (Diptera: Drosophilidae). Genetica. 2010, 138: 601-617. 10.1007/s10709-009-9432-5.
Wessler SR, Bureau TE, White SE: LTR-retrotransposons and MITEs: important players in the evolution of plant genomes. Curr Opin Genet Dev. 1995, 5: 814-821. 10.1016/0959-437X(95)80016-X.
Mao L, Wood TC, Yu Y, Budiman MA, Tomkins J, Woo S, Sasinowski M, Presting G, Frisch D, Goff S, Dean RA, Wing RA: Rice transposable elements: a survey of 73,000 sequence-tagged-connectors. Genome Res. 2000, 10: 982-990. 10.1101/gr.10.7.982.
Tu Z: Three novel families of miniature inverted-repeat transposable elements are associated with genes of the yellow fever mosquito, Aedesaegypti. Proc Natl Acad Sci U S A. 1997, 94: 7475-7480. 10.1073/pnas.94.14.7475.
Gilbert C, Schaack S, Pace JK, Brindley PJ, Feschotte C: A role for host-parasite interactions in the horizontal transfer of transposons across phyla. Nature. 2010, 464: 1347-1350. 10.1038/nature08939.
Sundararajan P, Atkinson PW, O’Brochta DA: Transposable element interactions in insects: crossmobilization of hobo and Hermes. Insect Mol Biol. 1999, 8: 359-368. 10.1046/j.1365-2583.1999.83128.x.
Feschotte C, Osterlund MT, Peeler R, Wessler SR: DNA-binding specificity of rice mariner-like transposases and interactions with Stowaway MITEs. Nucleic Acids Res. 2005, 33: 2153-2165. 10.1093/nar/gki509.
Loot C, Santiago N, Sanz A, Casacuberta JM: The proteins encoded by the pogo-like Lemi1 element bind the TIRs and subterminal repeated motifs of the Arabidopsis Emigrant MITE: consequences for the transposition mechanism of MITEs. Nucleic Acids Res. 2006, 34: 5238-5246. 10.1093/nar/gkl688.
de Freitas Ortiz M, Loreto EL: Characterization of new hAT transposable elements in 12 Drosophila genomes. Genetica. 2009, 135: 67-75. 10.1007/s10709-008-9259-5.
Feschotte C, Pritham EJ: DNA transposons and the evolution of eukaryotic genomes. Annu Rev Genet. 2007, 41: 331-368. 10.1146/annurev.genet.40.110405.090448.
Grumbling G, Strelets V: FlyBase: anatomical data, images and queries. Nucleic Acids Res. 2006, 34 (Database issue): D484-D488.
Crooks GE, Hon G, Chandonia JM, Brenner SE: WebLogo: a sequence logo generator. Genome Res. 2004, 14: 1188-1190. 10.1101/gr.849004.
Sassi AK, Herédia F, Loreto EL, Valente VLS, Rohde C: Transposable elements P and gypsy in natural populations of Drosophila willistoni. Genet Mol Biol. 2005, 28: 734-739.
Staden R: The Staden sequence analysis package. Mol Biotechnol. 1996, 5: 233-241. 10.1007/BF02900361.
Edgar RC: MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32: 1792-1797. 10.1093/nar/gkh340.
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S: MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011, 28: 2731-2739. 10.1093/molbev/msr121.
Nylander JAA, Ronquist F, Huelsenbeck JP, Nieves-Aldrey JL: Bayesian phylogenetic analysis of combined data. Syst Biol. 2004, 53: 47-67. 10.1080/10635150490264699.
Yao H, Guo L, Fu Y, Borsuk LA, Wen TJ, Skibbe DS, Cui X, Scheffler BE, Cao J, Emrich SJ, Ashlock DA, Schnable PS: Evaluation of fiveab initiogene prediction programs for the discovery of maize genes. Plant Mol Biol. 2005, 57: 445-460. 10.1007/s11103-005-0271-1.
Kohany O, Gentles AJ, Hankus L, Jurka J: Annotation, submission and screening of repetitive elements in repbase: repbasesubmitter and Censor. BMC Bioinforma. 2006, 7: 474-10.1186/1471-2105-7-474.
Acknowledgments
We thank Luís Eduardo Esmanhotto for assistance with English usage. This study was supported by research grants and fellowships from CNPq-Conselho Nacional de Desenvolvimento Científico e Tecnológico, PRONEX-FAPERGS (10/0028-7) and Fapergs (11/0938-0).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
MD performed the experiments and assisted in the experimental design. AL designed the study and carried out the in silico searches. MD and AL carried out the results analysis and wrote the manuscript. ELSL and VLSV assisted in the experimental design, analysis of results and manuscript writing. VLSV and ELSL provided funding and facilities for the study. All authors read and approved the final manuscript.
Maríndia Deprá, Adriana Ludwig contributed equally to this work.
Electronic supplementary material
13100_2012_52_MOESM2_ESM.pdf
Additional file 2:Table describing Mar clones obtained in this work from different Drosophila strains, along with the nomenclature used, size and accession number in GenBank.(PDF 21 KB)
13100_2012_52_MOESM3_ESM.pdf
Additional file 3:Table describing Mar sequences identified in the D. willistoni genome with the nomenclature used in this work and the scaffold position.(PDF 47 KB)
13100_2012_52_MOESM5_ESM.pdf
Additional file 5:Alignment view of the following sequences: canonical Mar, reconstructed full-length Mar (consensus), four D. tropicalis clones, four D. willistoni copies (scaf72, scaf94, scaf95 and scaf96) and the four primers.(PDF 475 KB)
13100_2012_52_MOESM6_ESM.pdf
Additional file 6:Alignment view of the putative Mar transposase and the general transcription factor II-I repeat domain-containing protein 2-like from Xenopus tropicalis.(PDF 347 KB)
13100_2012_52_MOESM7_ESM.txt
Additional file 7:Sequence of the reconstructed full-length Mar , its putative coding region and amino acid sequence.(TXT 5 KB)
13100_2012_52_MOESM8_ESM.pdf
Additional file 8:Comparison of the TSD consensus sequences and TIRs from Mar and five different D. willistoni hAT elements.(PDF 116 KB)
13100_2012_52_MOESM9_ESM.pdf
Additional file 9:Identity and accession number of the sequences used to infer the hAT transposase phylogeny.(PDF 52 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Deprá, M., Ludwig, A., Valente, V.L. et al. Mar, a MITE family of hAT transposons in Drosophila. Mobile DNA 3, 13 (2012). https://doi.org/10.1186/1759-8753-3-13
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1759-8753-3-13