
Abstract
Background
In eukaryotes, long terminal repeat (LTR) retrotransposons such as Copia, BEL and Gypsy integrate their DNA copies into the host genome using a particular type of DDE transposase called integrase (INT). The Gypsy INT-like transposase is also conserved in the Polinton/Maverick self-synthesizing DNA transposons and in the 'cut and paste' DNA transposons known as TDD-4 and TDD-5. Moreover, it is known that INT is similar to bacterial transposases that belong to the IS3, IS481, IS30 and IS630 families. It has been suggested that LTR retrotransposons evolved from a non-LTR retrotransposon fused with a DNA transposon in early eukaryotes. In this paper we analyze a diverse superfamily of eukaryotic cut and paste DNA transposons coding for INT-like transposase and discuss their evolutionary relationship to LTR retrotransposons.
Results
A new diverse eukaryotic superfamily of DNA transposons, named Ginger (for 'Gypsy INteGrasE Related') DNA transposons is defined and analyzed. Analogously to the IS3 and IS481 bacterial transposons, the Ginger termini resemble those of the Gypsy LTR retrotransposons. Currently, Ginger transposons can be divided into two distinct groups named Ginger1 and Ginger2/Tdd. Elements from tshe Ginger1 group are characterized by approximately 40 to 270 base pair (bp) terminal inverted repeats (TIRs), and are flanked by CCGG-specific or CCGT-specific target site duplication (ssTSsD) sequences. The Ginger1-encoded transposases contain an approximate 400 amino acid N-terminal portion sharing high amino acid identity to the entire Gypsy-encoded integrases, including the YPYY motif, zinc finger, DDE domain, and, importantly, the GPY/F motif, a hallmark of Gypsy and endogenous retrovirus (ERV) integrases. Ginger1 transposases also contain additional C-terminal domains: ovarian tumor (OTU)-like protease domain or Ulp1 protease domain. In vertebrate genomes, at least two host genes, which were previously thought to be derived from the Gypsy integrases, apparently have evolved from the Ginger1 transposase genes. We also introduce a second Ginger group, designated Ginger2/Tdd, which includes the previously reported DNA transposon TDD-4.
Conclusions
The Ginger superfamily represents eukaryotic DNA transposons closely related to LTR retrotransposons. Ginger elements provide new insights into the evolution of transposable elements and certain transposable element (TE)-derived genes.
Similar content being viewed by others


Background
All transposable elements (TEs) can be divided into two major classes: retrotransposons and DNA transposons. Based on their transposition mechanisms, eukaryotic retrotransposons can be further divided into non-long terminal repeat (LTR) retrotransposons and LTR retrotransposons [1]. The latter include five clades: Copia, BEL, Gypsy, endogenous retroviruses (ERV) and DIRS. DNA transposons in eukaryotes can be divided into 'cut and paste' transposons, self-replicating transposons (Polinton/Maverick), rolling circle transposons (Helitron), and tyrosine recombinase transposons (Crypton) [2, 3]. Cryptons were originally identified in fungi [4], and recently they were found in sea anemone (Nematostella vectensis), sea urchin (Strongylocentrotus purpuratus) [5] and insects [6, 7].
All known 'cut and paste' DNA transposons consist of 17 superfamilies [2, 3, 8]. Each superfamily encodes a superfamily-specific transposase (TPase), which is generally referred to as DDE transposase for the universally conserved catalytic amino acids it contains: two aspartic acids (D) and one glutamic amino acid (E). The Copia, BEL, Gypsy and ERV LTR retrotransposons also code for DDE transposases responsible for integration of their cDNA copies into the host genome. The LTR retrotransposon-encoded transposases are similar to each other and are conventionally called integrases. The integrases are significantly related to bacterial transposases of IS3 and IS481 insertion sequences, but their relationship to bacterial IS630 insertion sequences and eukaryotic Tc1/Mariner DNA transposons appears to be more distant [9–11]. It has been proposed that integrase-encoding LTR retrotransposons evolved from the combination of a non-LTR retrotransposon and a DNA transposon [1, 10, 12, 13]. Also, Gypsy integrase-like TPases have been found in rare eukaryotic DNA transposons, TDD-4 and TDD-5 [14, 15]. However, it is unclear whether these eukaryotic DNA transposons were derived directly from a Gypsy LTR retrotransposon or an ancestral DNA transposon. Moreover, Polinton DNA transposons also encode a conserved protein similar to the Gypsy integrase, and it was suggested that an ancestral Polinton DNA transposon arose by recruiting either an LTR retrotransposon integrase or DNA transposase by a virus or linear plasmid [16].
In the present work, we describe a superfamily of cut and paste DNA transposons called Ginger (for 'Gypsy INteGrasE Related'), coding for the transposase similar to Gypsy integrases. The Ginger superfamily is composed of two distinctive groups, Ginger1 and Ginger2/Tdd. The Ginger1 group is reported in this paper, and Ginger2/Tdd represents elements phylogenetically related to the previously reported DNA transposon TDD-4 [14].
Results
Ginger1 DNA transposons
A typical autonomous Ginger1 element encodes a single approximately 500 to 800 amino acid long TPase that includes an approximately 400 amino acid N-terminal region highly similar to the integrase (INT) encoded by Gypsy LTR retrotransposon (Figure 1). The homologous regions include the H2C2 zinc finger domain, DDE catalytic domain, and the GPY/F motif that mediates multimerization [17]. The latter is a hallmark of Gypsy and ERV integrases [18]. The amino acid identity between Ginger1 TPases and Gypsy integrases is up to 40% in the approximately 170 amino acid long DDE catalytic region (Figure 1c). Remarkably, in addition to the domains mentioned above, Ginger1 TPases and some Gypsy integrases further share an approximate 40 amino acid motif immediately upstream of the zinc finger domain (Figure 1a, b). This motif is not universal in Gypsy integrases; it is only present in a limited number of integrases belonging to the Athila/Tat group, Cer1 group and a few Athila/Tat related groups containing Gypsy-6-I_HM, Gypsy-15-I_NV, Gypsy-1-I_RO, Gypsy-1-I_DD and DGLT-A1_I (Figures 1b and 2). This motif is designated as YPYY for the four conserved amino acids: Y/F-P-Y/F-Y/F (Figure 1b). So far, we have identified dozens of Ginger1 families in four animal species, including hydra (Hydra magnipapillata), gastropod (Aplysia californica), lancelet (Branchiostoma floridae) and aphid (Acyrthosiphon pisum). The hydra genome harbors the most diverse and abundant Ginger1 elements; a total of 12 Ginger1 families were identified in this species (Table 1). In the Ginger1-2_HM family, the divergence of some elements from the consensus is less than 1%, suggesting that Ginger1-2_HM elements are still active in H. magnipapillata genome. In the current release of B. floridae genome sequences, Ginger-1_BF is found as a single autonomous copy (Table 1), but a few non-autonomous copies carrying the same 5' and 3' end sequences are also found. In the genome of A. pisum, only degenerated Ginger1 elements are recognizable, but complete elements are identified in the remaining three species. The full length Ginger1 elements vary from approximately 2.6 kb to 7 kb, and their terminal inverted repeats (TIRs) are approximately 40 to 270 base pairs (bp) long (Table 1). The target site duplication (TSD) sequences of Ginger1 are 4-bp long and the sequences are highly specific: CCGG (75%) or CCGT/ACGG (24%) (Figure 1a). The 5' ends of Ginger1 elements show the same conserved TGTNR pattern as those of Gypsy LTR retrotransposons.

Ginger1 (for ' Gypsy INteGrasE Related 1') elements and transposases. (a) Schematic features of the Ginger1 DNA transposons. Target site duplications (TSDs) and target sequences preferences of the Ginger1 and Ginger2 groups are compared, which is based on the data of 112 Ginger1 elements from Hydra magnipapillata and 64 Ginger2 elements from Nematostella vectensis. (b-d) Alignment of Ginger1 and Gypsy integrases in the YPYY motif and the H2C2 zinc finger domain (b), the DDE core domain (c), and the GPY/F motif (d). (e) Alignment of the Ulp1 and plant homeodomain (PHD) domains in Ginger1 TPases, Mutator TPases and yeast protein 1EUV_A; the PHD domain is boxed. (f) Alignment of the ovarian tumor (OTU) domains of Ginger TPases, Helitron proteins, and yeast protein 3BY4_A.
In addition to the INT domains, extra domains are also found at the C-terminus of all Ginger1 TPases with the exception of Ginger1-1_BF TPase. These domains include the ovarian tumor (OTU) cysteine protease domain (pfam02338), the C-terminal catalytic domain of Ulp1 protease (pfam02902) and the plant homeodomain (PHD) finger motif (smart00249) from the Conserved Domain Database http://www.ncbi.nlm.nih.gov/sites/entrez?db=cdd (Table 1, Figure 1a, e, f). Either OTU or Ulp1 is present in a particular Ginger1 TPase, but not both. Ginger-1_BF TPase does not contain these extra C-terminal domains, however, it is not clear whether Ginger-1_BF TPase itself lacks these C-terminal domains or this particular single copy element contains an internal deletion. It is also worth noting that some Ginger1 TPase encoding sequences are interrupted by 1 to 4 introns (Table 1). Except for the first intron of Ginger-5_HM (GC-AG) and the first intron of Ginger-1_AC (GT-TG), all these introns conform to the canonical GT-AG intron type [19].
Ginger2 DNA transposons
To our knowledge, only two families of eukaryotic DNA transposons, TDD-4 and TDD-5, have been reported to code for a Gypsy-like integrase. They are present in the protist Dictyostelium discoideum, and are characterized by 5-bp TSD [14, 15]. Their amino acid identity to Ginger1 TPases is only approximately 24% in the DDE catalytic domain. To find out more members of this potentially new group of Ginger elements, we used the DDE core domain of the TDD-4 TPase as a query in Tblastn or Blastp searches against the available National Center for Biotechnology Information (NCBI) databases http://blast.ncbi.nlm.nih.gov/Blast.cgi. As a result, we identified >6 families of homologous DNA transposons from different metazoan species, including hydra (H. magnipapillata), sea anemone (Nematostella vectensis), aphid (A. pisum), nematode (Trichinella spiralis), sea snails (Littorina saxatilis) and lancelet (B. floridae) (Table 2). All these transposons contain approximately 50 to 180 bp long TIRs and produce 4 bp TSDs, instead of the 5 bp TSDs of TDD-4 and TDD-5. However, as shown below, all these elements and TDD-4 or TDD-5 belong to the same group (Figure 2), referred to as Ginger2. In the fungus Malassezia globosa, we also found a protein named XP_001728957.1 closely related to the Ginger2 TPases. Neither of the two 5-kb regions flanking the XP_001728957.1-coding region encodes the ribonuclease H or reverse transcriptase. Therefore, even if the XP_001728957.1-coding region is not flanked by TIRs, we classify it as a Ginger2-1_MG DNA transposon vestige (Figure 2) rather than an LTR retrotransposon. Like Ginger1 and the vast majority of Gypsy LTR retrotransposons, the termini of Ginger2 follow the same conserved pattern, TGTNR. However, in contrast with the GC-rich 4-bp Ginger1 target sequences, Ginger2 elements preferentially target 6-bp AT-rich sequences, RTATAY (Figure 1a). Ginger2 TPases contain the H2C2 zinc finger domain and the DDE catalytic domain, but they lack the GPY/F and YPYY motifs present in Ginger1 TPases. The lowest pairwise amino acid identity in the DDE catalytic core region of the Ginger2 elements is approximately 30%, compared with the 36% identity within the same region of the Ginger1 group. This suggests that the Ginger2 group is more divergent than the Ginger1 group and it is consistent with the observation that Ginger2 elements are present in protists, fungi and animals, whereas Ginger1 elements were identified only in four animal species.

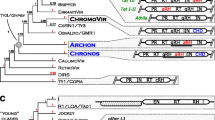
Phylogenetic relationship between the integrases of Ginger1 (for ' Gypsy INteGrasE Related 1'), Ginger2 and Gypsy LTR retrotransposons. The tree, based on the multiple alignments in the zinc finger domain and the DDE domain (see Additional file 1), is constructed by the minimum evolution (ME) method (Poisson correction model, pairwise deletion, gamma parameter = 2, bootstrap replicates = 1,000). The neighbor-joining (NJ) tree is shown in Additional file 2. The ME and NJ bootstrap values of major clades are shown in parenthesis, respectively. Lines in non-black colors differentiate the non-Gypsy integrases. The names of known Gypsy lineages follow previous literatures [18, 20, 43, 44]. The eukaryotic IS481-like integrases in Trichomonas vaginalis are designated as IS418EU. The two distinct Copia clades [45] are named Copia-I and Copia-II, respectively. Integrases containing the YPYY motif are marked with red dots. The clades containing the GPY/F motif are marked with green dots.
Phylogeny of Ginger1, Ginger2 and Gypsy integrases
To better understand the relationships between Ginger1, Ginger2, and the Gypsy lineages, we performed phylogenetic analyses of a wide collection of integrases from Gypsy LTR retrotransposons, exogenous/endogenous retroviruses, and Polinton/Maverick DNA transposons. Copia and BEL integrases and some integrase-like transposases from bacteria and protozoan Trichomonas vaginalis were included as outgroups. These bacterial transposases are from the IS3 and IS481 elements, the protozoan integrase-like TPases are closely related to them. TPase from eukaryotic Mariner/Tc1 and Pogo elements and bacterial IS630 elements are not included for analysis because they are phylogenetically closer to each other [8], and are more distantly related to the integrases encoded by LTR retrotransposons and Ginger than those encoded by IS3 and IS481 elements (data not shown). Although our phylogenetic tree is based on the limited sequence information from the zinc finger domain and the DDE core domain (see Additional file 1), most Gypsy integrases are clustered together away from other older groups, such as IS3/IS481, Copia, BEL and retrovirus [1], with the exception of two lineages ofGypsy-like integrase from fungal species, clustering with Polinton group and retrovirus groups, respectively (Figure 2). Except for the Woot element being separated from the Osvaldo clade, the clades and the polytomy distribution of all known Gypsy lineages are consistent with the other studies based on the analysis of multiple domains [20, 21]. In addition to the known lineages, some extra Gypsy clades also appear in our phylogeny, probably due to a larger data set; some of them might represent new lineages of the Gypsy LTR retrotransposons. Remarkably, the Ginger1 and Ginger2 groups are distinctly separated (Figure 2): Ginger2 integrases tend to group with the integrases of Polinton/Maverick DNA transposons, while Ginger1 are closely grouped with Athila/Tat lineage of Gypsy LTR retrotransposons. Although the YPYY motif is not included in the sequence information used to build the tree, the majority of YPYY motif-containing Gypsy integrases apparently cluster together with the Ginger1 TPases that also contain the YPYY motif (Figure 2), indicating the YPYY motif is genetically significant. However, no Gypsy lineages are found coclustering with Ginger1 or Ginger2 TPases with significant bootstrap values. A similar pattern is also observed in the tree constructed using the different neighbor-joining method (see Additional file 2).
Host genes derived from Ginger1 TPases
Gypsy integrase-1 gene (Gin-1), encoding a Gypsy integrase-like protein, was thought to have evolved from a Gypsy LTR retrotransposon related to the 412/Mdg1 lineage [22]. However, Gin-1 genes and a number of other homologous genes actually evolved from the Ginger1 elements. The most parsimonious scenario is that two independent exaptations of Ginger1 took place during the evolution of vertebrates, which gave rise to two sets of orthologous genes designated here as Gin-1 and Gin-2 (Figure 3). The first exaptation event gave rise to Gin-2 genes and probably happened in the common ancestor of vertebrates, while the second gave rise to Gin-1 genes and happened more recently in the common ancestor of reptiles and mammals. For example, Gin-1 genes are present in lizard Anolis carolinensis (FG759656.1), chicken Gallus gallus (XP_424858.2), opossum Monodelphis domestica (XP_001380076.1) and human (NP_060146.2); Gin-2 genes were found in fish Danio rerio (CAM46974.1), frog Xenopus tropicalis (AAI69154.1), lizard Anolis carolinensis (FG723791.1) and chicken Gallus gallus (XP_415124.1). Gin-2 genes are not found in any currently sequenced mammalian genomes and it is likely that Gin-2 gene was lost in the early stage of mammal evolution. Consistent with this scenario, Gin-1 and Gin-2 encoded proteins form two clusters in the phylogenetic tree, and both cocluster within the Ginger1 families rather than any other Gypsy lineages (Figure 3a, Additional file 3).

Host genes evolved from Ginger1 (for ' Gypsy INteGrasE Related 1') DNA transposon. (a) The phylogenetic relationship between Gin-1, Gin-2, and the Ginger1 elements (in red). The tree is constructed by the minimum evolution (ME) method (Poisson correction model, pairwise deletion, gamma parameter = 2, 1,000 bootstrap replicates). The sequences and alignment are shown in Additional file 3. C. intestinalis_1 gene refers to XM_002130131.1 gene; C. intestinalis_2 gene corresponds to expression sequence tag (EST) named FF869668.1. Integrases from Gypsy 412/Mdg1 lineage are included as outgroups. (b) The alignment of the local sequences around the three conserved introns in Ginger1-5_HM TPase and host proteins. The arrows point to the intron positions, the Arabic numbers (above) indicate the intron phase.
Of the six to eight introns in each of the host genes, only three are universally conserved: they are found at the same positions and have the same intron phases (Figure 3b). Strikingly, all the three conserved introns are found in Ginger1-5_HM TPase gene (Figure 3b), which has four introns in total. In addition, the first conserved intron is also present in the Ginger1-6,7,8_HM TPase genes (data not shown). The data strongly indicate that Gin-1 and Gin-2 genes are derived from a Ginger1-5_HM-like element. In invertebrate tunicate Ciona intestinalis, two genes (XM_002130131.1 and FF869668.1) may also be host genes derived from Ginger1-5_HM-like elements. They also contain the three conserved introns (Figure 3b), and their upstream and downstream genes are only within the range of approximately 0.5 kb to 2.5 kb, within which no flanking TIRs are found.
Discussion
In the present work, we report a new eukaryotic superfamily of DNA transposons, named Ginger, encoding transposases homologous to the integrases of Gypsy LTR retrotransposon. To date, we have identified two distinct Ginger groups, Ginger1 and Ginger2/Tdd. These groups could also be viewed as different superfamilies based on their plausible independent origin. However, further classification is left open here, due to uncertainties inherent in the current phylogenetic data. Previously, DIRS and Ngaro retrotransposons and Crypton DNA transposons were also found to encode the same class of proteins (tyrosine recombinase). However, their evolutionary relationship is not well understood yet due to the scarcity of data [4, 23]. Therefore, the relationship between the Gypsy LTR retrotransposons and the Ginger DNA transposons is of particular interest from the evolutionary perspective.
It is known that transposases from bacterial transposons that belong to the IS3 and IS481 families are significantly similar to the integrases encoded by eukaryotic LTR retrotransposons [9, 11]. Moreover, numerous families of the IS3 and IS481 transposons are characterized by the 3'-TG and CA-5' termini, and by 3 to 5-bp TSDs, analogous to LTR retrotransposons [9, 11]. It has also been proposed that in an early eukaryotic species a first LTR retrotransposon evolved from a non-LTR retrotransposon, which recruited a transposase from a DNA transposon [1, 10, 12, 13]. Given the significant similarity between the integrases encoded by Ginger1, Ginger2 and various LTR retrotransposons, either the Ginger1 or Ginger2 groups of transposons, or both, can be viewed as descendants of the ancestral eukaryotic DNA transposons which provided a TPase transformed into the integrase of the first LTR retrotransposon in early eukaryotes. However, as suggested by the modular evolution model of transposable elements [13], the evolutionary scenarios of Ginger and LTR integrases may be multifold.
If Gypsy was first derived through the fusion of reverse transcriptase with a DNA transposase [1, 10, 12, 13], one possible scenario is a 'reverse evolution' in which Ginger1 elements originated from a Gypsy LTR retrotransposon. This scenario is based on the remarkable similarity between Ginger1 TPase and Gypsy INT, plus the fact that Ginger1 and Ginger2/Tdd are two distinct groups on the phylogenetic tree (Figure 2). Ginger1 TPases not only share up to 40% amino acid sequence identity in the DDE region with some Gypsy integrases, but also contain a GPY/F domain, which has been found only in Gypsy and retroviral integrases so far [18]. Moreover, Ginger1 TPases and a subset of Gypsy integrases contain the same YPYY motif (Figures 1b and 2). The figure of 40% amino acid sequence identity is comparable to the upper similarity level between integrases from different Gypsy lineages (data not shown). In this scenario, the Ginger2 elements may resemble the ancestor element that was recruited into the first LTR retrotransposon.
Alternatively, it is still possible that that Gypsy, Copia and BEL LTR retrotransposons arose independently in early eukaryotes by recruiting three different transposases from the same Ginger-like superfamily. According to this scenario, Ginger1 transposase/transposons may be the 'best preserved' descendants/relatives of the eukaryotic transposon that 'gave birth' to the first Gypsy. Another variant of this scenario is that all LTR retrotransposon-encoded integrases arose from the common ancestor of Ginger1 and Ginger2 TPases. Thus, among numerous hypothetical lineages of this ancestor DNA transposon, Ginger1 and Ginger2 may be the only lineages that survived from the times of early eukaryotic evolution, which took place over 1.6 billion years ago. According to this scenario, the ancient eukaryotic transposase that transformed into the integrase of the first LTR retrotransposon was likely composed of the H2C2 zinc finger, GPY/F motifs and YPYY motifs. During their evolution, the Ginger2, Copia and BEL lineages might have lost the last two motifs.
Of the various Gypsy lineages shown in Figure 2, no lineage clusters with Ginger1 groups with significant bootstrap support. This does not preclude the possibility that Ginger1 groups arose from particular Gypsy lineages. It merely reflects the polytomy of Gypsy LTR retrotransposons [1]. The true ancestral lineages leading to Ginger1 may simply not be present in the available dataset, or they might have been lost in the evolutionary history, for example, due to the recombination process that produces the solo LTRs [24]. The chain of events that could lead to the hypothetical transformation of an LTR retrotransposon to a DNA transposon is also unclear. One simple possibility is that the TIRs of Ginger1 transposons were derived from two LTRs incidentally flanking the integrase region in opposite orientations: one would be the original 3' LTR of an LTR retrotransposon and the other coming from another LTR retrotransposon inserted upstream. The enzymatic mechanism underlying the excision of Ginger1 elements is also unknown (the same applies to Polinton/Maverick transposons). In the life cycle of LTR retrotransposons, integrases normally are responsible only for the integration process. However, in vivo data have shown that retrovirus integrases can reverse the initial strand-transfer reaction at the end of retroviral DNA [25]. Therefore, at least theoretically, Ginger1 DNA can be excised, but the excision process may not be as efficient as the excision of typical 'cut and paste' DNA transposons, since Ginger1 are small families and the copy numbers of individual elements per host are relatively low.
Despite the phylogenetic proximity of Ginger2 and Polinton/Maverick transposons in the phylogenetic tree (Figure 2), Ginger2 is less likely to be derived from Polinton/Maverick integrases, because Polinton/Maverick integrases lack the signature N-terminal C2H2 zinc finger. It cannot be resolved whether Polinton/Maverick integrases arose from Ginger2 or an LTR retrotransposon, because it is unclear whether the two fungal Gypsy-like lineages, clustering with Polinton/Maverick groups and retrovirus groups, respectively (Figure 2), represent some old Gypsy lineages or are just misplaced in the tree.
The Ulp1 protease C-terminal domain and OTU domain of Ginger1 transposases are also found in other transposable elements (Figure 1e, f). For example, Ulp1 domain is encoded in Mutator DNA transposons found in hydra [26], lancelet [27], Arabidopsis thaliana [28, 29], maize [30], rice and Cucumis melo [31]; OTU domains are found in the proteins encoded by some Helitron DNA transposons in animals (Figure 1e) [32]. It has been suggested that Ulp1 proteases are functionally involved in the transposition process [29]. Interestingly, both the OTU and Ulp1 domains belong to the same C-protease family, and have similar functions in hydrolysis of ubiquitin or the small ubiquitin-like modifier (SUMO) protein [33, 34]. Moreover, ubiquitinyl hydrolases are also found associated with other DNA transposases, such as protein XP_001314237.1 in protist T. vaginalis [35], which consist of transposase_11 domain (pfam01609) and Peptidase C19 (cd02657) domain. The latter domain participates in removing ubiquitin molecules from polyubiquinated peptides. These data imply that the ubiquitin pathway may be extensively involved in transposition. One attractive possibility is that both Ulp1 and OTU domains play an active role in the transposition processes. For example, they may activate the downstream factors in the DNA repair system after or during transposition, thereby minimizing the damage in the host genome. Indeed, all the major DNA repair pathways, damage avoidance mechanisms and checking responses are regulated somehow by ubiquitinylation, SUMOylation, or both [36].
Conclusions
Ginger is a new superfamily of cut and paste DNA transposons coding for the transposase homologous to the integrase encoded by Gypsy LTR retrotransposons. The Ginger superfamily contains two distinct groups, Ginger1 and Ginger2/Tdd. Of the two groups, the Ginger1 transposases are more similar to Gypsy integrases. We also describe a number of host genes domesticated from Ginger1 TPase genes. This work takes a step towards finding the direct ancestors of an ancient DNA transposon recruited by a non-LTR retrotransposon to form the first LTR retrotransposon, and also raises the possibility of a new evolutionary pathway transforming a LTR retrotransposon to a cut and paste DNA transposon.
Methods
Data sources
Genomic sequences and mRNA sequences of various species were mainly taken from NCBI GenBank. X. tropicalis genome sequences (release 4.1 assembled scaffolds) were downloaded from Department of Energy (DOE) Joint Genome Institute (JGI) http://www.jgi.doe.gov/. For the phylogenetic analyses, integrase sequences were widely selected from the Gypsy Database (GyDB) http://gydb.uv.es/index.php/Main_Page[20], Repbase Update database http://www.girinst.org/repbase/index.html[37] and GenBank. Other transposable elements without specification of source in this paper are from the Repbase. Additionally, the sequences of TEs reported in this work have been deposited in Repbase with the same family names listed in Table 1 and Table 2.
Sequence analysis
The consensus sequence of each Ginger1 or Ginger2 family, if possible, was rebuilt for analyses. The protein coding region of individual DNA transposons or host genes was either deduced from corresponding mRNA sequences, or manually predicted based on the sequence similarities between homologous proteins; exons and introns were determined accordingly. Multiple protein sequence alignments were carried out using MUSCLE [38] and were adjusted manually. Sequence alignments were edited and illustrated with BioEdit [39]. Logo representation of the TSD sequences was created by the WebLogo [40]. The phylogenetic tree was constructed using neighbor-joining (NJ) method and minimum evolution (ME) method (Poisson correction model, pairwise deletion, 1,000 bootstrap replicates) implemented in the MEGA4 software [41]. The gamma parameter for the phylogenetic tree was estimated using PhyMl [42].
References
Eickbush T, Malik H: Origins and evolution of retrotransposons. Mobile DNA II. Edited by: Craig NL, Craigie R, Gellert M, Lambowitz AM. 2002, Washington, DC, USA: American Society for Microbiology Press, 1111-1144.
Wicker T, Sabot F, Hua-Van A, Bennetzen JL, Capy P, Chalhoub B, Flavell A, Leroy P, Morgante M, Panaud O, Paux E, SanMiguel P, Schulman AH: A unified classification system for eukaryotic transposable elements. Nat Rev Genet. 2007, 8: 973-982. 10.1038/nrg2165.
Kapitonov VV, Jurka J: A universal classification of eukaryotic transposable elements implemented in Repbase. Nat Rev Genet. 2008, 9: 411-412. 10.1038/nrg2165-c1.
Goodwin TJ, Butler MI, Poulter RT: Cryptons: a group of tyrosine-recombinase-encoding DNA transposons from pathogenic fungi. Microbiology. 2003, 149: 3099-3109. 10.1099/mic.0.26529-0.
Jurka J, Kapitonov VV: First cryptons from invertebrates. Repbase Rep. 2008, 8: 232-233.
Jurka J: First cryptons from insects. Repbase Rep. 2009, 9: 480-480.
Jurka J: First cryptons from insects. Repbase Rep. 2009, 9: 673-673.
Bao W, Jurka MG, Kapitonov VV, Jurka J: New superfamilies of eukaryotic DNA transposons and their internal divisions. Mol Biol Evol. 2009, 26: 983-993. 10.1093/molbev/msp013.
Fayet O, Ramond P, Polard P, Prere MF, Chandler M: Functional similarities between retroviruses and the IS3 family of bacterial insertion sequences?. Mol Microbiol. 1990, 4: 1771-1777. 10.1111/j.1365-2958.1990.tb00555.x.
Capy P, Vitalis R, Langin T, Higuet D, Bazin C: Relationships between transposable elements based upon the integrase-transposase domains: is there a common ancestor?. J Mol Evol. 1996, 42: 359-368. 10.1007/BF02337546.
Chandler M, Mahillon J: Insertion sequences revisited. Mobile DNA II. Edited by: Craig NL, Craigie R, Gellert M, Lambowitz AM. 2002, Washington, DC, USA: American Society for Microbiology Press, 305-366.
Capy P, Langin T, Higuet D, Maurer P, Bazin C: Do the integrases of LTR-retrotransposons and class II element transposases have a common ancestor?. Genetica. 1997, 100: 63-72. 10.1023/A:1018300721953.
Capy P, Maisonhaute C: Acquisition/loss of modules: the construction set of transposable elements. Russian J Genet. 2002, 38: 594-601. 10.1023/A:1016027530962.
Wells DJ: Tdd-4, a DNA transposon of Dictyostelium that encodes proteins similar to LTR retroelement integrases. Nucleic Acids Res. 1999, 27: 2408-2415. 10.1093/nar/27.11.2408.
Glockner G, Szafranski K, Winckler T, Dingermann T, Quail MA, Cox E, Eichinger L, Noegel AA, Rosenthal A: The complex repeats of Dictyostelium discoideum. Genome Res. 2001, 11: 585-594. 10.1101/gr.GR-1622RR.
Kapitonov VV, Jurka J: Self-synthesizing DNA transposons in eukaryotes. Proc Natl Acad Sci USA. 2006, 103: 4540-4545. 10.1073/pnas.0600833103.
Ebina H, Chatterjee AG, Judson RL, Levin HL: The GP(Y/F) domain of TF1 integrase multimerizes when present in a fragment, and substitutions in this domain reduce enzymatic activity of the full-length protein. J Biol Chem. 2008, 283: 15965-15974. 10.1074/jbc.M801354200.
Malik HS, Eickbush TH: Modular evolution of the integrase domain in the Ty3/Gypsy class of LTR retrotransposons. J Virol. 1999, 73: 5186-5190.
Burset M, Seledtsov IA, Solovyev VV: Analysis of canonical and non-canonical splice sites in mammalian genomes. Nucleic Acids Res. 2000, 28: 4364-4375. 10.1093/nar/28.21.4364.
Llorens C, Futami R, Bezemer D, Moya A: The Gypsy Database (GyDB) of mobile genetic elements. Nucleic Acids Res. 2008, 36: D38-46. 10.1093/nar/gkm697.
Llorens C, Fares MA, Moya A: Relationships of gag-pol diversity between Ty3/Gypsy and retroviridae LTR retroelements and the three kings hypothesis. BMC Evol Biol. 2008, 8: 276-10.1186/1471-2148-8-276.
Llorens C, Marin I: A mammalian gene evolved from the integrase domain of an LTR retrotransposon. Mol Biol Evol. 2001, 18: 1597-1600.
Poulter RT, Goodwin TJ: DIRS-1 and the other tyrosine recombinase retrotransposons. Cytogenet Genome Res. 2005, 110: 575-588. 10.1159/000084991.
Vitte C, Panaud O: Formation of solo-LTRs through unequal homologous recombination counterbalances amplifications of LTR retrotransposons in rice Oryza sativa L. Mol Biol Evol. 2003, 20: 528-540. 10.1093/molbev/msg055.
Chow SA, Vincent KA, Ellison V, Brown PO: Reversal of integration and DNA splicing mediated by integrase of human immunodeficiency virus. Science. 1992, 255: 723-726. 10.1126/science.1738845.
Bao W, Jurka J: MuDr-type DNA transposons from Hydra magnipapillata. Repbase Rep. 2008, 8: 2075-2075.
Bao W, Jurka J: MuDr-type DNA transposons from Branchiostoma floridae. Repbase Rep. 2009, 9: 683-683.
Kapitonov VV, Jurka J: VANDAL20, a family of monstrous MuDR-like DNA transposons. Repbase Rep. 2001, 1: 31-31.
Hoen DR, Park KC, Elrouby N, Yu Z, Mohabir N, Cowan RK, Bureau TE: Transposon-mediated expansion and diversification of a family of ULP-like genes. Mol Biol Evol. 2006, 23: 1254-1268. 10.1093/molbev/msk015.
Bao W, Jurka J: MuDR-type DNA transposon from maize. Repbase Rep. 2008, 8: 739-739.
van Leeuwen H, Monfort A, Puigdomenech P: Mutator-like elements identified in melon, Arabidopsis and rice contain ULP1 protease domains. Mol Genet Genomics. 2007, 277: 357-364. 10.1007/s00438-006-0194-9.
Kapitonov VV, Jurka J: Helitron-1_SP, a family of autonomous Helitrons in the sea urchin genome. Repbase Rep. 2005, 5: 393-393.
Mossessova E, Lima CD: Ulp1-SUMO crystal structure and genetic analysis reveal conserved interactions and a regulatory element essential for cell growth in yeast. Mol Cell. 2000, 5: 865-876. 10.1016/S1097-2765(00)80326-3.
Messick TE, Russell NS, Iwata AJ, Sarachan KL, Shiekhattar R, Shanks JR, Reyes-Turcu FE, Wilkinson KD, Marmorstein R: Structural basis for ubiquitin recognition by the Otu1 ovarian tumor domain protein. J Biol Chem. 2008, 283: 11038-11049. 10.1074/jbc.M704398200.
Gladyshev EA, Arkhipova IR: A single-copy IS5-like transposon in the genome of a bdelloid rotifer. Mol Biol Evol. 2009, 26: 1921-1929. 10.1093/molbev/msp104.
Bergink S, Jentsch S: Principles of ubiquitin and SUMO modifications in DNA repair. Nature. 2009, 458: 461-467. 10.1038/nature07963.
Jurka J, Kapitonov VV, Pavlicek A, Klonowski P, Kohany O, Walichiewicz J: Repbase update, a database of eukaryotic repetitive elements. Cytogenet Genome Res. 2005, 110: 462-467. 10.1159/000084979.
Edgar RC: MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32: 1792-1797. 10.1093/nar/gkh340.
Hall TA: BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucl Acids Symp Ser. 1999, 41: 95-98.
Crooks GE, Hon G, Chandonia JM, Brenner SE: WebLogo: a sequence logo generator. Genome Res. 2004, 14: 1188-1190. 10.1101/gr.849004.
Tamura K, Dudley J, Nei M, Kumar S: MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol. 2007, 24: 1596-1599. 10.1093/molbev/msm092.
Guindon S, Gascuel O: A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003, 52: 696-704. 10.1080/10635150390235520.
Bae YA, Moon SY, Kong Y, Cho SY, Rhyu MG: CsRn1, a novel active retrotransposon in a parasitic trematode, Clonorchis sinensis, discloses a new phylogenetic clade of Ty3/gypsy-like LTR retrotransposons. Mol Biol Evol. 2001, 18: 1474-1483.
Gladyshev EA, Meselson M, Arkhipova IR: A deep-branching clade of retrovirus-like retrotransposons in bdelloid rotifers. Gene. 2007, 390: 136-145. 10.1016/j.gene.2006.09.025.
Peddigari S, Zhang W, Takechi K, Takano H, Takio S: Two different clades of copia-like retrotransposons in the red alga, Porphyra yezoensis. Gene. 2008, 424: 153-158. 10.1016/j.gene.2008.07.021.
Acknowledgements
This work was supported by the National Institutes of Health grant 5 P41 LM006252. We thank Matthew Jurka for help with editing the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
WB and JJ designed the initial research. WB prepared and processed the data. VVK contributed information on Ginger2 elements and suggested involvement of the ubiquitinylation and SUMOylation pathways in transposition. WB and JJ performed research and drafted the manuscript. All authors edited and approved the manuscript.
Electronic supplementary material
13100_2009_3_MOESM1_ESM.FAS
Additional file 1: Integrase alignments. Integrase sequences alignment in the zinc finger domain and the DDE domain. (FAS 70 KB)
13100_2009_3_MOESM2_ESM.PDF
Additional file 2: Neighbor-joining phylogenetic tree. Neighbor-joining phylogenetic tree is constructed using Poisson correction model, pairwise deletion, gamma parameter = 2, 1,000 bootstrap replicates. (PDF 66 KB)
13100_2009_3_MOESM3_ESM.FAS
Additional file 3: Protein sequence alignments. Sequence alignments between host proteins, Ginger1 Tpases and Gypsy integrases. (FAS 10 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Bao, W., Kapitonov, V.V. & Jurka, J. Ginger DNA transposons in eukaryotes and their evolutionary relationships with long terminal repeat retrotransposons. Mobile DNA 1, 3 (2010). https://doi.org/10.1186/1759-8753-1-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1759-8753-1-3