
Abstract
High plasma uric acid (UA) is a precipitating factor for gout and renal calculi as well as a strong risk factor for Metabolic Syndrome and cardiovascular disease. The main causes for higher plasma UA are either lower excretion, higher synthesis or both. Higher waist circumference and the BMI are associated with higher insulin resistance and leptin production, and both reduce uric acid excretion. The synthesis of fatty acids (tryglicerides) in the liver is associated with the de novo synthesis of purine, accelerating UA production. The role played by diet on hyperuricemia has not yet been fully clarified, but high intake of fructose-rich industrialized food and high alcohol intake (particularly beer) seem to influence uricemia. It is not known whether UA would be a causal factor or an antioxidant protective response. Most authors do not consider the UA as a risk factor, but presenting antioxidant function. UA contributes to > 50% of the antioxidant capacity of the blood. There is still no consensus if UA is a protective or a risk factor, however, it seems that acute elevation is a protective factor, whereas chronic elevation a risk for disease.
Similar content being viewed by others


Introduction
High plasma uric acid (UA) is a prerequisite for gout and is also associated with the Metabolic Syndrome and risk factors for cardiovascular diseases [1–4]. Hyperuricemia increase in 16% all causes of mortality and 39% of total cardiovascular disease [5].
The hyperuricemia is defined as ≥7 mg/dL for men and ≥6.0 mg/dL for women [6], and is found mainly in postmenopausal women, African American, patients with renal disease and alcohol intake [7]. Furthermore, many factors can influence the concentrations of UA, eg. diet, obesity, and Metabolic Syndrome [1, 8–10]. However, we still don't know if UA is a protective factor for the moderate oxidative stress in these situations or if it's a risk factor.
The aim of this review was to discuss the function of the UA in our organism and the main causes and consequences of the higher concentration of UA.
Chemical characterization and biological importance
Uric acid (2,6,8 trioxypurine-C5H4N4O3) is an organic compound that is endogenously produced by animals as a purine metabolite. It is formed by the liver and mainly excreted by the kidneys (65-75%) and intestines (25-35%). UA is the end product of purine metabolism in humans due to the loss of uricase activity, which led to humans having higher UA levels than other mammals [11, 12].
Due to its double bonds, uric acid has excellent antioxidant capacity, and it can be responsible for 2/3 of total plasma antioxidant capacity [13, 14].
Because it is a weak acid that have a high dissociation constant, uric acid circulates in plasma (pH 7.4) predominantly (98%) in the form of a monovalent sodium salt (urate) [15]. It shows low solubility in water (as well as in plasma), and it would theoretically reach plasma saturation in the concentration of 6.4 mg/dL, which may not occur because solubility increase is provided by its binding to proteins, namely albumin, which is its main transporter. Protein-bound uric acid shows plasma solubility that is 70% higher than in its free state [16]. Uric acid pathogenesis is usually associated with gouty arthritis or nephrolithiasis [12].
High uricemia pathogenicity is associated with its low solubility in the extracellular environment leading to crystal formation, low affinity (and deposition) to certain tissues and antigenicity (after crystal phagocytosis). This mixture of quantitative and qualitative etiological hyperuricemia factors is confounding because normouricemic individuals may show symptoms while others with hyperuricemia may not. In the clinical context, hyperuricemia is seen as a prognostic indicator of renal disease, diabetes mellitus, cardiovascular disease and inflammation [7, 17–23], thus being a (modest) risk factor for mortality [1].
Uricemia homeostasis
Physiologically, uric acid plasma concentrations increases with age; they are smaller in women of childbearing age and, in post menopause women, it increase to similar values to those found in males [2, 3].
The balance between uric acid production and excretion in males shows a turnover of 600- 700 mg/day for a pool with a maximum of 1,000 mg, as shown by isotopic studies. Uric acid entry into the pool is controlled by hepatic production, which receives endogenous (nucleoproteins) and exogenous (dietary) precursor proteins. Uric acid exit from the pool in males is mainly controlled by the kidneys and by urine-forming factors, renal plasma flow, glomerular filtration and tubular exchange [24, 25].
Excretion control
In the kidney, uric acid and urate are initially filtered and additionally secreted. However, the largest part (90%) is usually reabsorbed and returns to blood [12].
The factors that can influence the uptake of urate by the kidney are: plasma concentrations, volemia and renal plasma flow modulators. At this point, renal excretion of urate follows the same extra-renal limiting factors as those of glomerular filtration. Kidney participation is associated with glomerular filtration (90% filtrate), proximal reabsorption (by an active process) and post-secretory resorption in the distal tubule, ascending loop and collecting duct. In this phase, the renal excretion of urate follows the limiting factors that accompany renal diseases (glomerular and medullary). The urate renal excretion phase is that of post-secretory resorption [24].
Proximal tubular reabsorption of uric acid is competitive with monocarboxylic organic acids, and it can be inhibited by oxalic acid, lactic acid and ketone bodies (acetoacetic and beta-hydroxybutyric acids). Metabolic situations with higher production and circulation of such acids are accompanied by the hypoexcretion of uric acid in urine [24].
Lactic acid is a product of anaerobic glycolysis (NADH-dependent) in alcoholics, resulting from the metabolism of ethanol (due to higher NADH production), and in inflammation/infection (due to PDH inhibition by pro-inflammatory cytokines) [24].
Ketone bodies are produced in the liver by partial oxidation of acetyl coenzyme A due to excessive peripheral lipolysis. Ketonuria occurs in decompensated diabetic individuals and in acute fasting (weight loss by carbohydrate-restricted diets). Factors that increase uric acid urinary excretion: extracellular volume expansion and tubular resorption inhibition [24].
Acute renal failure can also rise UA by decreased of renal excretion [26]. Furthermore, the consumption of some drugs are associated with the raise of serum UA (ciclosporin, ethambutol, pyrazinamide, cytotoxic chemotherapy). On the other hand, other drugs (benzbromarone, losartan, probenecid, sulfinpyrazon) diminishes the UA concentration mainly by the inhibition of a specific anion transporters (URAT1), that is responsible for the reabsorption of renal uric acid, which explain the uricosuric effect of these drugs [2].
Production control
Purine bases and their nucleotides are rapidly captured by the liver [27] and converted into xanthines, metabolized by uric acid or recycled by salvage pathways or de novo synthesis, and the latter requires a higher energetic cost (ATP consumption) [28]. Uric acid formation by xanthine occurs via hypoxanthine by the action of xanthine oxidase [29]. In most mammals, uric acid is converted into allantoin by uricase and later into urea, which are excreted. For this reason, uricemia in these animals corresponds to 10% of human values [30, 31].
Diseases accompanied by great cell depletion, such as leukemia, leukocytosis and dystrophies can increase the provision of nucleic acids to the liver and result in a larger production of uric acid. Hence, diseases resulting from innate purine metabolic errors may also result in hyperuricemia [2, 3, 12].
Diet
The relation between diet and UA has not yet been fully clarified since most studies have not estimated basal urate concentrations, have not excluded confounding factors or have not even correctly evaluated ingested nutrients [32].
The human diet is very poor in urate, which is produced mainly in the liver and to a less extent in the small intestine. The production of UA depends of purine ingestion [2], however, a purine-rich diet would be responsible for an increasing only in 1 to 2 mg/dL of UA [33, 34].
It is recommended that individuals with hyperuricemia should not ingest a large amount of purine-rich food (for instance, veal, bacon, kid meat, mutton, turkey, pork, duck, goose, etc.) [35], and it is suggested that a high intake of meat and seafood are related to increased UA, but the same relation is not valid for total protein intake [36].
During fructose metabolism, the phosphorylation of such monosaccharide into fructose 1-phosphate occurs by means of enzyme fructokinase. Next, enzyme aldolase B breaks fructose 1-phosphate into dihydroxyacetone phosphate (DHAP) and D-glyceraldehyde. When high fructose intake occurs, fructose phosphorylation into fructose 1-phosphate is fast, but the reaction with aldolase is slow. Hence, fructose 1-phosphate accumulates, and inorganic concentrations of intracellular phosphate also decrease. The low availability of phosphate limits ATP formation (ADP + Pi), and ADP or AMP resulting from such metabolism is catabolized, thus leading to hyperuricemia [37].
There are other ways to increase urate concentrations, such as the intake of sorbitol, sucrose, lactate and methylxanthines [38]. After absorption, sorbitol is converted into fructose by the liver, and thus, it can increase UA production [39, 40].
Fruit and vegetables, in addition to flavonoids, can contain other nutrients that can affect the plasma total antioxidant capacity (TAC). Lotito & Frei (2006) [40] reported that the TAC increase observed after the intake of these flavonoid-rich food types is not specifically caused by flavonoids because such nutrients are probably highly metabolized after ingestion. Possibly, what would increase TAC would be the UA elevation resulting from fructose metabolism. The mechanism for such elevation would be the degradation of purine nucleotides or UA excretion reduction [41–43].
High vitamin-C intake shows an inverse relation with UA [44] as such vitamin has a uricosuric effect due to competition of UA renal resorption through a change in the anion transportation system in the proximal tubule [45, 46].
Beer intake is an independent factor from UA increase [32] both due to its alcoholic content and high-quality purine [47]. Beer intake has a stronger power to increase UA than liqueur, but moderate wine intake does not increase UA [36, 48]. Alcohol intake increases UA concentrations by reducing excretion [49, 50] and increasing urate production [51].
Dairy product intake has an inverse relation with UA [32, 36, 52]. Probably, the mechanism for such association is due to milk-forming proteins (lactalbumin and casein), which have a uricosuric effect [53].
There is an inverse relation between coffee intake and UA concentrations [48], but such association seems to be due to other substances, as it does not seem to result from caffeine. Decreased UA concentrations would result from the improved insulin resistance observed with increased coffee intake, which occurs mainly due to chlorogenic acid, an antioxidant found in that drink [54, 55].
Tsunoda et al. (2002) [56] observed the effects of hypocaloric diets on uric acid in overweight individuals. This type of diet indirectly decreased UA serum concentrations because it improved insulin sensitivity in these individuals, and this occurred regardless of changes in body weight or blood pressure.
Uricemia as a risk factor
Gouty arthritis
Gout is caused by articulation deposition of monosodium urate crystals after chronic hyperuricemia [2]. It affects 1-2% of adults in developed countries [2] and is considered an independent risk factor for cardiovascular disease [3]. Uric acid values higher than 7.0 mg/dl is a risk factor for the development of gout [3].
Renal calculi
Renal calculi are formed by calcium salts deposited on an organic matrix of oxalate or urate. Their formation occurs in the glomerular filtrate in acid (oxalate or urate) or alkaline (carbonates and phosphates) pH in the presence of high calciuria (hyperparathyroidism or bone demineralization). They are usually formed and then dissolved, but under abnormal pH variation or filtrate concentration conditions, they may remain until excretion [30].
The contribution of UA to renal stone formation has an important role in progressive renal failure [57].
Metabolic syndrome and body composition
Meta-analysis has recently shown that UA elevation is related to an increase in (coronary artery disease) infarction and mortality events [1], and epidemiological studies showed that UA is an independent risk factor for cardiovascular diseases [5, 10], particularly in hypertensive and diabetic individuals [21]. It is also speculated that UA is one of the determinants of the metabolic syndrome [22]. Individuals with high UA levels have an odds ratio of 1.6-fold higher for developing MS [58].
UA is associated with Metabolic Syndrome [59] and its components [60], obesity, dyslipidemia, hypertension [18], insulin resistance (metabolic syndrome), increased C-reactive protein (PCR) concentration [23] and endothelial dysfunction [20], or even, to risk factors for cardiovascular diseases [7, 17, 19].
UA increase is observed in individuals with insulin resistance, probably because hyperisulinemia would cause lower renal UA excretion [61]. Additionally, insulin could indirectly act on UA, since there is an association between hyperinsulinemia and hypertriglyceridemia.
Some studies show that high concentrations of plasma triglycerides are related to hyperuricemia [58, 62–64]. There are some explanations for such relation, and one of them is that during triglycerides (TG) synthesis there would be a greater need for NADPH [62]. The synthesis of fatty acids in the liver is associated with the de novo synthesis of purine, thus accelerating UA production [65].
Chen et al. (2007) [58] observed negative correlation between HDL-c and UA. The likely mechanism is the relationship between decreased HDL-C and insulin resistance [66]. Confirming this hypothesis, our research group showed that when adjusted for the other MS components, the relation between UA and HDL-c is lost [67].
In the adipose tissue, there is adipokine production, including that of leptin. One possible explanation for the association between higher waist circumference and hyperuricemia were suggested by Bedir et al. (2003) [68] and Fruehwald-Schultes et al. (1999) [69] which studies found that UA serum concentrations are independently related to leptin concentration, thus suggesting that would be a pathogenic factor responsible for UA increase in obese patients [58].
Some authors report UA to be responsible for blood pressure increase, and others show arterial hypertension (AH) to be a risk factor for hiperuricemia. UA inhibits the bioavailability of nitric oxide, which is a vasodilator [70], and, on the other hand, AH would lead to vascular disease and increase renal vascular resistance. Both of these processes reduce renal flow, thus increasing urate resorption [71].
A recent study showed an inverse relation between muscle mass (MM) and UA in healthy individuals older than 40 years [72]. Chronic elevation of UA concentrations would be a causal factor for sarcopenia, especially through increased inflammation and oxidative stress [72, 73]. The activation of the xanthine oxidase metabolic pathway, which increases UA production and the superoxide radical [74] could elevate the reactive oxygen species (ROS) and it could be the main mechanism for the reduction of MM. Furthermore, UA exerts a pro-inflammatory effect, thus stimulating the production of interleukin-1, interleukin-6 the tumor necrosis factor which also can influence the muscle mass [75].
Uricemia as a protective factor
It is not known whether UA would be a causal factor or an antioxidant protective response against oxidative stress [76]. While chronic high UA concentrations are associated to increased risk for CAD, acute elevations seem to provide antioxidant protection [77]. UA contributes to > 50% of the antioxidant capacity of the blood [78]. Furthermore, the UA has a protective action in vitamins C and E [79] with the stabilizing activities in these vitamins [79, 80] and the presence of ascorbic acid in plasma is required for the antioxidant effect of UA [80]. Also, it has a direct effect on the inhibition of free radicals such as peroxyl radical and peroxynitrite, protecting the cell membrane and DNA [81, 82].
Most authors do not consider UA as a detrimental factor to the body health, because of its antioxidant function [8, 83]. The antioxidant activity of UA also occurs in the brain [84], being a protector for several disease such as multiple sclerosis and neurodegenerative disease. Higher concentration of UA is associated with lower risk of development of Parksons disease and a favorable effect at the disease progression mainly [85].
Conclusions
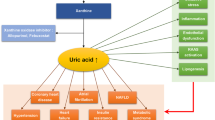
Several factors are associated as cause and consequences of high UA concentration. Higher waist circumference and BMI are associated with higher insulin resistance and leptin production, and both reduce renal uric acid excretion, thus increasing its concentration. HDL-c concentration is negatively associated to insulin resistance, what can influence its negative correlation to uric acid. Obese individuals usually have Metabolic Syndrome diagnostic, which can also increase uric acid serum concentrations due to synthesis increase (triglycerides - TG concentration) and lower excretion (arterial hypertension - AH) (Figure 1).

Determinants of hyperuricemia. UA = Uric Acid, BMI = Body Mass Index, TG = triglycerides.
Additionally, obesity and muscle mass (MM) reduction are associated with low-intensity chronic inflammation, and uric acid levels can increase in order to protect the organism against the moderate oxidative stress resulting from this situation. Low muscle mass (sarcopenia) is negatively associated with uric acid, However, it has not yet been clarified what the cause or effect is. Probably, oxidative stress produced by excessive uric acid can influence muscle mass reduction. The role played by diet on hyperuricemia has not yet been fully clarified, but high intake of fructose-rich industrialized food and high alcohol intake (particularly beer) seem to influence uricemia. The main mechanism of excretion of urate concentrations occurs by means of renal excretion; hence, glomerular function markers (urea and creatinine) are positively associated with uric acid (Figure 1).
Furthermore, there is still no consensus if UA is a protective or a risk factor, however, it seems that the quantity and the duration of the concentration of the uric acid in the blood is essential for this answer. Acute elevation seems to be a protective factor, whereas chronic elevation a risk factor.
References
Kim SY, Guevara JP, Kim KM, Choi HK, Heitjan DF, Albert DA: Hyperuricemia and risk of stroke: a systematic review and meta-analysis. Arthritis Rheum. 2009, 61: 885-892. 10.1002/art.24612.
Richette P, Bardin T: Gout Lancet. 2010, 375: 318-328.
Roddy E, Doherty M: Epidemiology of gout. Arthritis Res Ther. 2010, 12: 223-10.1186/ar3199.
Ruggiero C, Cherubini A, Ble A, Bos AJ, Maggio M, Dixit VD, Lauretani F, Bandinelli S, Senin U, Ferrucci L: Uric acid and inflammatory markers. Eur Heart J. 2006, 27: 1174-1181.
Chen JH, Chuang SY, Chen HJ, Yeh WT, Pan WH: Serum uric acid level as an independent risk factor for all-cause, cardiovascular, and ischemic stroke mortality: a Chinese cohort study. Arthritis Rheum. 2009, 61: 225-232. 10.1002/art.24164.
Hochberg MC, Smolen JS, Weinblatt ME: Rheumatology. 2003, New York: Mosby, 3
Gagliardi AC, Miname MH, Santos RD: Uric acid: A marker of increased cardiovascular risk. Atherosclerosis. 2009, 202: 11-17. 10.1016/j.atherosclerosis.2008.05.022.
Johnson RJ, Kang DH, Feig D, Kivlighn S, Kanellis J, Watanabe S, Tuttle KR, Rodriguez-Iturbe B, Herrera-Acosta J, Mazzali M: Is there a pathogenetic role for uric acid in hypertension and cardiovascular and renal disease?. Hypertension. 2003, 41: 1183-1190. 10.1161/01.HYP.0000069700.62727.C5.
Nakagawa T, Tuttle KR, Short RA, Johnson RJ: Hypothesis: fructose-induced hyperuricemia as a causal mechanism for the epidemic of the metabolic syndrome. Nat Clin Pract Nephrol. 2005, 1: 80-86.
Takahashi MM, de Oliveira EP, de Carvalho AL, Dantas LA, Burini FH, Portero-McLellan KC, Burini RC: Metabolic Syndrome and dietary components are associated with coronary artery disease risk score in free-living adults: a cross-sectional study. Diabetol Metab Syndr. 2011, 3: 7-10.1186/1758-5996-3-7.
Roch-Ramel F, Guisan B: Renal Transport of Urate in Humans. News Physiol Sci. 1999, 14: 80-84.
Alvarez-Lario B, Macarron-Vicente J: Uric acid and evolution. Rheumatology (Oxford). 2010, 49: 2010-2015. 10.1093/rheumatology/keq204.
Maxwell SR, Thomason H, Sandler D, Leguen C, Baxter MA, Thorpe GH, Jones AF, Barnett AH: Antioxidant status in patients with uncomplicated insulin-dependent and non-insulin-dependent diabetes mellitus. Eur J Clin Invest. 1997, 27: 484-490. 10.1046/j.1365-2362.1997.1390687.x.
Sautin YY, Johnson RJ: Uric acid: the oxidant-antioxidant paradox. Nucleosides Nucleotides Nucleic Acids. 2008, 27: 608-619. 10.1080/15257770802138558.
Liebman SE, Taylor JG, Bushinsky DA: Uric acid nephrolithiasis. Curr Rheumatol Rep. 2007, 9: 251-257. 10.1007/s11926-007-0040-z.
Terkeltaub R, Bushinsky DA, Becker MA: Recent developments in our understanding of the renal basis of hyperuricemia and the development of novel antihyperuricemic therapeutics. Arthritis Res Ther. 2006, 8 (Suppl 1): S4-10.1186/ar1909.
Burack RC, Keller JB, Higgins MW: Cardiovascular risk factors and obesity: are baseline levels of blood pressure, glucose, cholesterol and uric acid elevated prior to weight gain?. J Chronic Dis. 1985, 38: 865-872. 10.1016/0021-9681(85)90111-0.
Doehner W, Schoene N, Rauchhaus M, Leyva-Leon F, Pavitt DV, Reaveley DA, Schuler G, Coats AJ, Anker SD, Hambrecht R: Effects of xanthine oxidase inhibition with allopurinol on endothelial function and peripheral blood flow in hyperuricemic patients with chronic heart failure: results from 2 placebo-controlled studies. Circulation. 2002, 105: 2619-2624. 10.1161/01.CIR.0000017502.58595.ED.
Fang J: Alderman MH: Serum uric acid and cardiovascular mortality the NHANES I epidemiologic follow-up study, 1971-1992. National Health and Nutrition Examination Survey. Jama. 2000, 283: 2404-2410. 10.1001/jama.283.18.2404.
Farquharson CA, Butler R, Hill A, Belch JJ, Struthers AD: Allopurinol improves endothelial dysfunction in chronic heart failure. Circulation. 2002, 106: 221-226. 10.1161/01.CIR.0000022140.61460.1D.
Feig DI, Johnson RJ: Hyperuricemia in childhood primary hypertension. Hypertension. 2003, 42: 247-252. 10.1161/01.HYP.0000085858.66548.59.
Onat A, Uyarel H, Hergenc G, Karabulut A, Albayrak S, Sari I, Yazici M, Keles I: Serum uric acid is a determinant of metabolic syndrome in a population-based study. Am J Hypertens. 2006, 19: 1055-1062. 10.1016/j.amjhyper.2006.02.014.
Saito M, Ishimitsu T, Minami J, Ono H, Ohrui M, Matsuoka H: Relations of plasma high-sensitivity C-reactive protein to traditional cardiovascular risk factors. Atherosclerosis. 2003, 167: 73-79. 10.1016/S0021-9150(02)00380-5.
Mount DB, Kwon CY, Zandi-Nejad K: Renal urate transport. Rheum Dis Clin North Am. 2006, 32: 313-331. 10.1016/j.rdc.2006.02.006. vi
Shima Y, Teruya K, Ohta H: Association between intronic SNP in urate-anion exchanger gene, SLC22A12, and serum uric acid levels in Japanese. Life Sci. 2006, 79: 2234-2237. 10.1016/j.lfs.2006.07.030.
Ejaz AA, Mu W, Kang DH, Roncal C, Sautin YY, Henderson G, Tabah-Fisch I, Keller B, Beaver TM, Nakagawa T, Johnson RJ: Could uric acid have a role in acute renal failure?. Clin J Am Soc Nephrol. 2007, 2: 16-21.
Becker BF: Towards the physiological function of uric acid. Free Radic Biol Med. 1993, 14: 615-631. 10.1016/0891-5849(93)90143-I.
Manfredi JP, Holmes EW: Purine salvage pathways in myocardium. Annu Rev Physiol. 1985, 47: 691-705. 10.1146/annurev.ph.47.030185.003355.
Watts RW: Uric acid production with particular reference to the role of xanthine oxidase and its inhibition. Proc R Soc Med. 1966, 59: 287-292.
Hediger MA, Johnson RJ, Miyazaki H, Endou H: Molecular physiology of urate transport. Physiology (Bethesda). 2005, 20: 125-133. 10.1152/physiol.00039.2004.
Wu XW, Muzny DM, Lee CC, Caskey CT: Two independent mutational events in the loss of urate oxidase during hominoid evolution. J Mol Evol. 1992, 34: 78-84. 10.1007/BF00163854.
Yu KH, See LC, Huang YC, Yang CH, Sun JH: Dietary factors associated with hyperuricemia in adults. Semin Arthritis Rheum. 2008, 37: 243-250. 10.1016/j.semarthrit.2007.04.007.
Emmerson BT: The management of gout. N Engl J Med. 1996, 334: 445-451. 10.1056/NEJM199602153340707.
Yu T, Yu TF: Milestones in the treatment of gout. Am J Med. 1974, 56: 676-685. 10.1016/0002-9343(74)90634-2.
Cuppari L: Guia de nutrição: nutrição clínica no adulto. 2005, Barueri: Manole, 474-2
Choi HK, Liu S, Curhan G: Intake of purine-rich foods, protein, and dairy products and relationship to serum levels of uric acid: the Third National Health and Nutrition Examination Survey. Arthritis Rheum. 2005, 52: 283-289. 10.1002/art.20761.
Champe PC, Harvey RA: Biochemistry. 1994, Philadelphia: J.B. Lippincott: Lippincotts' illustrated reviews, 253-255. 2
Wrolstad RE, Shallenberger RS: Free sugars and sorbitol in fruits-a complication from the literature. J Assoc Off Anal Chem. 1981, 64: 91-103.
Bode JC, Zelder O, Rumpelt HJ, Wittkamp U: Depletion of liver adenosine phosphates and metabolic effects of intravenous infusion of fructose or sorbitol in man and in the rat. Eur J Clin Invest. 1973, 3: 436-441. 10.1111/j.1365-2362.1973.tb02211.x.
Lotito SB, Frei B: Consumption of flavonoid-rich foods and increased plasma antioxidant capacity in humans: cause, consequence, or epiphenomenon?. Free Radic Biol Med. 2006, 41: 1727-1746. 10.1016/j.freeradbiomed.2006.04.033.
Hallfrisch J: Metabolic effects of dietary fructose. Faseb J. 1990, 4: 2652-2660.
Mayes PA: Intermediary metabolism of fructose. Am J Clin Nutr. 1993, 58: 754S-765S.
Nakagawa T, Hu H, Zharikov S, Tuttle KR, Short RA, Glushakova O, Ouyang X, Feig DI, Block ER, Herrera-Acosta J, et al: A causal role for uric acid in fructose-induced metabolic syndrome. Am J Physiol Renal Physiol. 2006, 290: F625-F631.
Choi HK, Gao X, Curhan G: Vitamin C intake and the risk of gout in men: a prospective study. Arch Intern Med. 2009, 169: 502-507. 10.1001/archinternmed.2008.606.
Berger L, Gerson CD, Yu TF: The effect of ascorbic acid on uric acid excretion with a commentary on the renal handling of ascorbic acid. Am J Med. 1977, 62: 71-76. 10.1016/0002-9343(77)90351-5.
Stein HB, Hasan A, Fox IH: Ascorbic acid-induced uricosuria. A consequency of megavitamin therapy. Ann Intern Med. 1976, 84: 385-388.
Gibson T, Rodgers AV, Simmonds HA, Toseland P: Beer drinking and its effect on uric acid. Br J Rheumatol. 1984, 23: 203-209. 10.1093/rheumatology/23.3.203.
Choi HK, Curhan G: Coffee, tea, and caffeine consumption and serum uric acid level: the third national health and nutrition examination survey. Arthritis Rheum. 2007, 57: 816-821. 10.1002/art.22762.
Drum DE, Goldman PA, Jankowski CB: Elevation of serum uric acid as a clue to alcohol abuse. Arch Intern Med. 1981, 141: 477-479. 10.1001/archinte.1981.00340040073020.
Eastmond CJ, Garton M, Robins S, Riddoch S: The effects of alcoholic beverages on urate metabolism in gout sufferers. Br J Rheumatol. 1995, 34: 756-759. 10.1093/rheumatology/34.8.756.
Faller J, Fox IH: Ethanol-induced hyperuricemia: evidence for increased urate production by activation of adenine nucleotide turnover. N Engl J Med. 1982, 307: 1598-1602. 10.1056/NEJM198212233072602.
Choi HK, Atkinson K, Karlson EW, Willett W, Curhan G: Purine-rich foods, dairy and protein intake, and the risk of gout in men. N Engl J Med. 2004, 350: 1093-1103. 10.1056/NEJMoa035700.
Ghadirian P, Shatenstein B, Verdy M, Hamet P: The influence of dairy products on plasma uric acid in women. Eur J Epidemiol. 1995, 11: 275-281. 10.1007/BF01719431.
Arion WJ, Canfield WK, Ramos FC, Schindler PW, Burger HJ, Hemmerle H, Schubert G, Below P, Herling AW: Chlorogenic acid and hydroxynitrobenzaldehyde: new inhibitors of hepatic glucose 6-phosphatase. Arch Biochem Biophys. 1997, 339: 315-322. 10.1006/abbi.1996.9874.
Wu T, Giovannucci E, Pischon T, Hankinson SE, Ma J, Rifai N, Rimm EB: Fructose, glycemic load, and quantity and quality of carbohydrate in relation to plasma C-peptide concentrations in US women. Am J Clin Nutr. 2004, 80: 1043-1049.
Tsunoda S, Kamide K, Minami J, Kawano Y: Decreases in serum uric acid by amelioration of insulin resistance in overweight hypertensive patients: effect of a low-energy diet and an insulin-sensitizing agent. Am J Hypertens. 2002, 15: 697-701. 10.1016/S0895-7061(02)02953-9.
Capasso G, Jaeger P, Robertson WG, Unwin RJ: Uric acid and the kidney: urate transport, stone disease and progressive renal failure. Curr Pharm Des. 2005, 11: 4153-4159. 10.2174/138161205774913219.
Chen LY, Zhu WH, Chen ZW, Dai HL, Ren JJ, Chen JH, Chen LQ, Fang LZ: Relationship between hyperuricemia and metabolic syndrome. J Zhejiang Univ Sci B. 2007, 8: 593-598.
Sui X, Church TS, Meriwether RA, Lobelo F, Blair SN: Uric acid and the development of metabolic syndrome in women and men. Metabolism. 2008, 57: 845-852. 10.1016/j.metabol.2008.01.030.
Abdullah AR, Hasan HA, Raigangar VL: Analysis of the Relationship of Leptin, High-Sensitivity C-Reactive Protein, Adiponectin, Insulin, and Uric Acid to Metabolic Syndrome in Lean, Overweight, and Obese Young Females. Metab Syndr Relat Disord. 2009, 7: 17-22. 10.1089/met.2008.0045.
Facchini F, Chen YD, Hollenbeck CB, Reaven GM: Relationship between resistance to insulin-mediated glucose uptake, urinary uric acid clearance, and plasma uric acid concentration. Jama. 1991, 266: 3008-3011. 10.1001/jama.1991.03470210076036.
Clausen JO, Borch-Johnsen K, Ibsen H, Pedersen O: Analysis of the relationship between fasting serum uric acid and the insulin sensitivity index in a population-based sample of 380 young healthy Caucasians. Eur J Endocrinol. 1998, 138: 63-69. 10.1530/eje.0.1380063.
Conen D, Wietlisbach V, Bovet P, Shamlaye C, Riesen W, Paccaud F, Burnier M: Prevalence of hyperuricemia and relation of serum uric acid with cardiovascular risk factors in a developing country. BMC Public Health. 2004, 4: 9-10.1186/1471-2458-4-9.
Schachter M: Uric acid and hypertension. Curr Pharm Des. 2005, 11: 4139-4143. 10.2174/138161205774913246.
Matsuura F, Yamashita S, Nakamura T, Nishida M, Nozaki S, Funahashi T, Matsuzawa Y: Effect of visceral fat accumulation on uric acid metabolism in male obese subjects: visceral fat obesity is linked more closely to overproduction of uric acid than subcutaneous fat obesity. Metabolism. 1998, 47: 929-933. 10.1016/S0026-0495(98)90346-8.
Schmidt MI, Watson RL, Duncan BB, Metcalf P, Brancati FL, Sharrett AR, Davis CE, Heiss G: Clustering of dyslipidemia, hyperuricemia, diabetes, and hypertension and its association with fasting insulin and central and overall obesity in a general population. Atherosclerosis Risk in Communities Study Investigators. Metabolism. 1996, 45: 699-706. 10.1016/S0026-0495(96)90134-1.
de Oliveira EP, Manda RM, Torezan GA, Corrente JE, Burini RC: Dietary, anthropometric, and biochemical determinants of plasma high-density lipoprotein-cholesterol in free-living adults. Cholesterol. 2011, 2011: 851750-
Bedir A, Topbas M, Tanyeri F, Alvur M, Arik N: Leptin might be a regulator of serum uric acid concentrations in humans. Jpn Heart J. 2003, 44: 527-536. 10.1536/jhj.44.527.
Fruehwald-Schultes B, Peters A, Kern W, Beyer J, Pfutzner A: Serum leptin is associated with serum uric acid concentrations in humans. Metabolism. 1999, 48: 677-680. 10.1016/S0026-0495(99)90163-4.
Baldus S, Koster R, Chumley P, Heitzer T, Rudolph V, Ostad MA, Warnholtz A, Staude HJ, Thuneke F, Koss K, et al: Oxypurinol improves coronary and peripheral endothelial function in patients with coronary artery disease. Free Radic Biol Med. 2005, 39: 1184-1190. 10.1016/j.freeradbiomed.2005.06.004.
Messerli FH, Frohlich ED, Dreslinski GR, Suarez DH, Aristimuno GG: Serum uric acid in essential hypertension: an indicator of renal vascular involvement. Ann Intern Med. 1980, 93: 817-821.
Beavers KM, Beavers DP, Serra MC, Bowden RG, Wilson RL: Low relative skeletal muscle mass indicative of sarcopenia is associated with elevations in serum uric acid levels: Findings from NHANES III. Journal of Nutrition, Health and Aging. 2009, 13: 177-182. 10.1007/s12603-009-0054-5.
Pierine DT, Nicola M, de Oliveira EP: Sarcopenia: alterações metabólicas e consequências no envelhecimento. R bras Ci e Mov. 2009, 17: 96-103.
Powers SK, Kavazis AN, DeRuisseau KC: Mechanisms of disuse muscle atrophy: role of oxidative stress. Am J Physiol Regul Integr Comp Physiol. 2005, 288: R337-R344.
Kanellis J, Kang DH: Uric acid as a mediator of endothelial dysfunction, inflammation, and vascular disease. Semin Nephrol. 2005, 25: 39-42. 10.1016/j.semnephrol.2004.09.007.
Baillie JK, Bates MG, Thompson AA, Waring WS, Partridge RW, Schnopp MF, Simpson A, Gulliver-Sloan F, Maxwell SR, Webb DJ: Endogenous urate production augments plasma antioxidant capacity in healthy lowland subjects exposed to high altitude. Chest. 2007, 131: 1473-1478. 10.1378/chest.06-2235.
Yu ZF, Bruce-Keller AJ, Goodman Y, Mattson MP: Uric acid protects neurons against excitotoxic and metabolic insults in cell culture, and against focal ischemic brain injury in vivo. J Neurosci Res. 1998, 53: 613-625. 10.1002/(SICI)1097-4547(19980901)53:5<613::AID-JNR11>3.0.CO;2-1.
Glantzounis GK, Tsimoyiannis EC, Kappas AM, Galaris DA: Uric acid and oxidative stress. Curr Pharm Des. 2005, 11: 4145-4151. 10.2174/138161205774913255.
Ma YS, Stone WL, LeClair IO: The effects of vitamin C and urate on the oxidation kinetics of human low-density lipoprotein. Proc Soc Exp Biol Med. 1994, 206: 53-59.
Frei B, Stocker R, Ames BN: Antioxidant defenses and lipid peroxidation in human blood plasma. Proc Natl Acad Sci USA. 1988, 85: 9748-9752. 10.1073/pnas.85.24.9748.
Hooper DC, Spitsin S, Kean RB, Champion JM, Dickson GM, Chaudhry I, Koprowski H: Uric acid, a natural scavenger of peroxynitrite, in experimental allergic encephalomyelitis and multiple sclerosis. Proc Natl Acad Sci USA. 1998, 95: 675-680. 10.1073/pnas.95.2.675.
Wayner DD, Burton GW, Ingold KU, Barclay LR, Locke SJ: The relative contributions of vitamin E, urate, ascorbate and proteins to the total peroxyl radical-trapping antioxidant activity of human blood plasma. Biochim Biophys Acta. 1987, 924: 408-419. 10.1016/0304-4165(87)90155-3.
Kutzing MK, Firestein BL: Altered uric acid levels and disease states. J Pharmacol Exp Ther. 2008, 324: 1-7.
Scott GS, Hooper DC: The role of uric acid in protection against peroxynitrite-mediated pathology. Med Hypotheses. 2001, 56: 95-100. 10.1054/mehy.2000.1118.
Cipriani S, Chen X, Schwarzschild MA: Urate: a novel biomarker of Parkinson's disease risk, diagnosis and prognosis. Biomark Med. 2010, 4: 701-712. 10.2217/bmm.10.94.
Acknowledgements
CAPES for the financial support
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
EPO wrote the manuscript, RCB read and corrected the final version of the manuscript. All authors read and approved.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
de Oliveira, E.P., Burini, R.C. High plasma uric acid concentration: causes and consequences. Diabetol Metab Syndr 4, 12 (2012). https://doi.org/10.1186/1758-5996-4-12
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1758-5996-4-12