
Abstract
Background
Serratia marcescens is an opportunistic bacterial pathogen with broad range of host ranging from vertebrates, invertebrates and plants. S. marcescens strain W2.3 was isolated from a diseased tilapia fish and it was suspected to be the causal agent for the fish disease as virulence genes were found within its genome. In this study, for the first time, the genome sequences of S. marcescens strain W2.3 were sequenced using the Illumina MiSeq platform.
Result
Several virulent factors of S. marcescens such as serrawettin, a biosurfactant, has been reported to be regulated by N-acyl homoserine lactone (AHL)-based quorum sensing (QS). In our previous studies, an unusual AHL with long acyl side chain was detected from this isolate suggesting the possibility of novel virulence factors regulation. This evokes our interest in the genome of this bacterial strain and hereby we present the draft genome of S. marcescens W2.3, which carries the serrawettin production gene, swrA and the AHL-based QS transcriptional regulator gene, luxR which is an orphan luxR.
Conclusion
With the availability of the whole genome sequences of S. marcescens W2.3, this will pave the way for the study of the QS-mediated genes expression in this bacterium.
Similar content being viewed by others

Background
Serratia marcescens is common microorganism presence in soil and freshwater [1]. However, the emergence of multidrug resistant Serratia has been alarming not only in the medical field but also aquaculture and agriculture sectors [2–4]. In 2009 an endemic disease outbreak in fish farms of Malaysia had killed more than 50% of the tilapia fish. Five bacteria strains including S. marcescens W2.3 have been isolated from the fish samples and been suspected to be the causal agent of the outbreak.
Quorum sensing (QS) describes bacteria community gene regulation by cell-cell communication through the production of QS signalling molecule [5]. S. marcescens which is taxonomically classified as Proteobacteria produces N-acyl homoserine lactone (AHL) as QS signal molecules. Its AHL-based QS system plays a regulatory role in biosurfactant production, biofilm formation, motility, prodigiosin and nuclease production that contribute to the pathogenesis [1, 6]. Unlike most of S. marcescens, S. marcescens W2.3 produce N-dodecanoyl-homoserine lactone (C12-HSL) rather than short chain AHLs. Therefore, this suggests the presence of novel AHLs responding proteins and virulence factors that may be regulated under this long chain AHL-based QS in S. marcescens W2.3.
The rapid maturation of next generation sequencing (NGS) technology coupling with the fast improvement of computing power enable researcher to map bacteria genome within short period of time. Annotating the draft genome with the aid of databases available allow us to look into several genes simultaneously. Here, we present insights of the S. marcescens W2.3 genome, describing the presence of putative QS related genes and the serrawettin coding gene, swrA.
Methods
Bacterial culture
S. marcescens W2.3 is routinely maintained on either LB (Luria- Bertani, BD, USA) agar plates at 37°C or culture for 20 hrs in broth at 28°C with 200 r.p.m shaking.
Genomic DNA extraction
Genomic DNA of the bacteria was extracted with QIAamp DNA Mini kit (Qiagen, USA) and was subjected to RNase (Qiagen, USA) treatment. The DNA was eluted with elution buffer and subjected to DNA quantification with Qubit® 2.0 Fluorometer (dsDNA High Sensitivity Assay Kit) (Invitrogen, USA), and qualification with Nanodrop Spectrophotometer and agarose gel electrophoresis. The genomic DNA was stored in -20°C.
Library preparation and sequencing
DNA sequencing template was prepared with Nextera™ DNA Sample Preparation kit (Nextera, USA). The quality of DNA library was validated by Bioanalyzer 2100 high sensitivity DNA kit prior to sequencing. Upon sequencing, DNA (6 pM) was loaded into the sequencing cartridge and the sequencing was performed on Illumina MiSeq platform.
Read quality assessment
The quality of raw sequences as well as (G + C) content was checked with FastQC. Raw reads were trimmed at Phred 30 and were de novo assembled using CLC Genomic Workbench 5.1 [7]. Trimmed sequences were assembled with length fraction of 0.8 and similarity fraction of 0.8. Contigs with at least 30-fold coverage were subjected to gene prediction using Prodigal 2.6 [8].
Gene annotation was performed using RAST (Rapid Annotation using Subsystem Technology) followed by visualization of the bacterial genome using GeneWiz Browser 0.94 Server [9, 10]. In addition to RAST, Serrawettin genes were annotated by BLAST against NCBInt/nr database with e-value of 0.0001 and aligned with reference genes using LAST [9, 11]. Phylogenetic analysis was performed using MEGA version 5.0 [12].
Quality assurance
The 16S rDNA gene from draft genome was used to check for contamination. RNAmmer 1.2 Server has shown that only a copy of 16S rDNA gene is presence in the draft genome. The contig that carried 16S rDNA gene was annotated by BLAST against NCBI microbial 16S database and confirmed this 16S rDNA gene belongs to Serratia marcescens[13, 14].
Initial findings
This sequencing generated 2,724,434 paired end reads whilst 2,509,440 quality reads were preceded to assembly after trimming. The genome size of Serratia marcescens W2.3 is 5.3 Mbp. The draft genome of S. marcescens W2.3 is made up of 72 contigs with the length of at least 500 bps with the average coverage of 60-folds (Figure 1). N50 of this assembled genome turns out to be 216,941 bp while the (G + C) content of the draft genome is 59.3%. Prodigal shows that this draft genome carried 4891 coding DNA sequence (CDS).

Atlas of Serratia marcescens W2.3. The atlas was constructed by GeneWiz Browser 0.94 and the GC skew was shown in the fifth lane counting from the outer most lane. The genome of Serratia marcescens W2.3 was 5.2 Mbps.
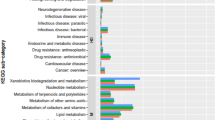
Draft genome was annotated using RAST and the result was shown in Figure 2. Similar with most other bacteria, most of the genes are responsible for carbohydrates and amino acid metabolism with 598 counts. This top hit follows by amino acids and derivatives, cofactors, vitamin, prosthetic groups and pigment production, RNA metabolism and cell wall and capsule synthesis. These genes are responsible for the basic needs in sustaining the life of a bacteria cell. In addition to the necessary genes, there are 105 genes responsible for virulence, disease and defense suggesting S. marcescens W2.3 is a pathogen.

Subsystem category distribution statistics for Serratia marcescens W2.3. The pie chart present the abundance of each subsystem category and the count of each subsystem feature was listed in parentheses at the chart legend.
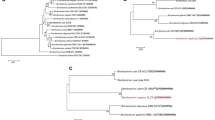
Serratia produces serrawettin that acts as wetting agent to reduce surface tension of the environment [15, 16]. The reduction of host cell surface tension by serrawettin causes the rupture of host cell leading the success of infection by this pathogen [17]. Serrawettin gene, swrA was found within the genome of S. marcescens W2.3. This 2631 bp gene was located at contig number 6. Dotplot (Figure 3) from LAST and BLAST result show that its serrawettin gene has 94% similarity with the serrawettin gene S. liquefaciens (Figure 3A) and 93% similarity with the serrawettin gene from S. marcescens A88copa13 (Figure 3B).

Dotplot of Serratia marcescens W2.3 serrawettin, swrA gene DNA sequence against reference genes. (A) S. marcescens W2.3 serrawettin gene is 94% similar to the swrA of Serratia liquefaciens (gb|AF039572) (B) and 93% similar to the swrA of Serratia marcescens A88copa 13 (gb|JX667980).
In a complete AHL-based QS system, the luxI/R homologs interact with each other where LuxI type protein synthesis AHL and binds to the LuxR-type protein [18]. Subsequently, this AHL-protein complex regulates the expression of certain genes leading to the group behavior of the bacteria [19]. However, luxI and luxR gene do not always occurs in paired in Proteobacteria. For example Pseudomonas aeruginosa and Sinorhizobium meliloti have been reported to carry unpaired luxR gene in their genome [18, 20]. These unpaired receptor protein coding genes does not responsible for any signalling molecule production but they are responsive to the cognate signalling molecules produced by both its existing AHL synthase and the signalling molecules from the environment [21].
A putative luxR gene with the size of 759 bps was identified within contig 7 of S. marcescens W2.3 draft genome. Phylogenetics analysis of the LuxR protein sequence of S. marcescens strain W2.3 and its closely related species shows that it is highly similar to the LuxR protein sequence of S. plymuthica (SptR) (Figure 4). However, no luxI gene was identified within the upstream and downstream of this luxR, thus we hypothesised that this is an orphan luxR. In 1998, Cox et al reported a solo luxR of S. marcescens, carR that regulates its carbapenem production [22]. However, the unpaired luxR gene of S. marcescens W2.3 is not group into the same transcriptional protein family as carR gene; therefore more studies need be conducted in the future for further understanding of the interaction of unusual long AHL with luxR coding in the genome of S. marcescens W2.3 isolated from the diseased tilapia fish and its rule in the pathogenesis.

Phylogenetic analyses of S. marcescens luxR gene. The tree was constructed based on the luxR protein sequences by Neighbor-Joining with bootstraps value of 1000 replicates.
Future directions
The whole genome of S. marcescens W2.3 has shown the presence of virulence factor coding genes and we have found the complete sequence of the serrawettin synthase gene. Since serrawettin production of S. marcescens is reported to be coordinated by QS, our future work will be focusing on the study of the AHLbased QS gene regulation of S. marcescens W2.3.
Availability of supporting data
This whole genome shotgun project has been deposited at DDBJ/EMBL/GenBank under the accession ALOV00000000. The version described in this paper is the first version ALOV01000000.
Abbreviations
- AHL:
-
N-acyl homoserine lactone
- QS:
-
Quorum sensing
- PBS:
-
Phosphate buffer saline
- LB:
-
Luria bertani
- CDS:
-
Coding DNA sequence
- NGS:
-
Next generation sequencing
- RAST:
-
Rapid annotation using subsystem technology
- C12HSL:
-
N- dodecanoyl homoserine lactone.
References
Hejazi A, Falkiner F:Serratia marcescens. J Med Microbiol. 1997, 46 (11): 903-912. 10.1099/00222615-46-11-903.
Kurz CL, Chauvet S, Andrès E, Aurouze M, Vallet I, Michel GP, Uh M, Celli J, Filloux A, De Bentzmann S: Virulence factors of the human opportunistic pathogen Serratia marcescens identified by in vivo screening. The EMBO J. 2003, 22 (7): 1451-10.1093/emboj/cdg159.
Baya A, Toranzo A, Lupiani B, Santos Y, Hetrick F: Serratia marcescens: a potential pathogen for fish. J Fish Dis. 1992, 15 (1): 15-26. 10.1111/j.1365-2761.1992.tb00632.x.
Morohoshi T, Shiono T, Takidouchi K, Kato M, Kato N, Kato J, Ikeda T: Inhibition of quorum sensing in Serratia marcescens AS-1 by synthetic analogs of N-acylhomoserine lactone. Appl Environ Microbiol. 2007, 73 (20): 6339-6344. 10.1128/AEM.00593-07.
Williams P, Winzer K, Chan WC, Camara M, Cámara M: Look who’s talking: communication and quorum sensing in the bacterial world. Phil Trans R Soc B. 2007, 362 (1483): 1119-1134. 10.1098/rstb.2007.2039.
Horng YT, Deng SC, Daykin M, Soo PC, Wei JR, Luh KT, Ho SW, Swift S, Lai HC, Williams P: The LuxR family protein SpnR functions as a negative regulator of N‒acylhomoserine lactone‒dependent quorum sensing in Serratia marcescens. Mol Microbiol. 2002, 45 (6): 1655-1671. 10.1046/j.1365-2958.2002.03117.x.
Chan XY, Chua KH, Puthucheary SD, Yin W-F, Chan K-G: Draft genome sequence of an Aeromonas sp. Strain 159 clinical isolate that shows quorum-sensing activity. J Bacteriol. 2012, 194 (22): 6350-6350. 10.1128/JB.01642-12.
Hyatt D, Chen GL, LoCascio P, Land M, Larimer F, Hauser L: Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics. 2010, 11 (1): 119-10.1186/1471-2105-11-119.
Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, Formsma K, Gerdes S, Glass EM, Kubal M: The RAST Server: rapid annotations using subsystems technology. BMC genomics. 2008, 9 (1): 75-10.1186/1471-2164-9-75.
Hallin PF, Stærfeldt H-H, Rotenberg E, Binnewies TT, Benham CJ, Ussery DW: GeneWiz browser: an interactive tool for visualizing sequenced chromosomes. Stand Genomic Sci. 2009, 1 (2): 204-
Kiełbasa SM, Wan R, Sato K, Horton P, Frith MC: Adaptive seeds tame genomic sequence comparison. Genome Res. 2011, 21 (3): 487-493. 10.1101/gr.113985.110.
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S: MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011, 28 (10): 2731-2739. 10.1093/molbev/msr121.
Lagesen K, Hallin P, Rødland EA, Stærfeldt HH, Rognes T, Ussery DW: RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007, 35 (9): 3100-3108. 10.1093/nar/gkm160.
Wheeler DL, Church DM, Federhen S, Lash AE, Madden TL, Pontius JU, Schuler GD, Schriml LM, Sequeira E, Tatusova TA: Database resources of the national center for biotechnology. Nucleic Acids Res. 2003, 31 (1): 28-33. 10.1093/nar/gkg033.
Eberl L, Molin S, Givskov M: Surface motility of Serratia liquefaciens MG1. J Bacteriol. 1999, 181 (6): 1703-1712.
Zhang L, Sun J, Hao Y, Zhu J, Chu J, Wei D, Shen Y: Microbial production of 2, 3-butanediol by a surfactant (serrawettin)-deficient mutant of Serratia marcescens H30. J Ind Microbiol Biotechnol. 2010, 37 (8): 857-862. 10.1007/s10295-010-0733-6.
Pradel E, Zhang Y, Pujol N, Matsuyama T, Bargmann CI, Ewbank JJ: Detection and avoidance of a natural product from the pathogenic bacterium Serratia marcescens by Caenorhabditis elegans. Proc Natl Acad Sci. 2007, 104 (7): 2295-2300. 10.1073/pnas.0610281104.
Subramoni S, Venturi V: LuxR-family 'solos’: bachelor sensors/regulators of signalling molecules. Microbiology. 2009, 155 (5): 1377-1385. 10.1099/mic.0.026849-0.
Bassler BL: Small talk: cell-to-cell communication in bacteria. Cell. 2002, 109 (4): 421-424. 10.1016/S0092-8674(02)00749-3.
Lee JH, Lequette Y, Greenberg EP: Activity of purified QscR, a Pseudomonas aeruginosa orphan quorum‒sensing transcription factor. Mol Microbiol. 2006, 59 (2): 602-609. 10.1111/j.1365-2958.2005.04960.x.
Patankar AV, González JE: Orphan LuxR regulators of quorum sensing. FEMS Microbiol Rev. 2009, 33 (4): 739-756. 10.1111/j.1574-6976.2009.00163.x.
Cox A, Thomson N, Bycroft B, Stewart G, Williams P, Salmond G: A pheromone-independent CarR protein controls carbapenem antibiotic synthesis in the opportunistic human pathogen Serratia marcescens. Microbiology. 1998, 144 (1): 201-209. 10.1099/00221287-144-1-201.
Acknowledgements
Kok-Gan Chan thanks the University of Malaya for the financial support given under the High Impact Research Grant (UM-MOHE HIR Nature Microbiome Grant No. H-50001-A000027).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
XYC and KWH performed the DNA sequencing assay. XYC, CYC, KWH, WFY and KGC analyzed the sequencing data. XYC, CYC, KKT and KGC contributed to the writing of the manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an open access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Chan, X.Y., Chang, C.Y., Hong, K.W. et al. Insights of biosurfactant producing Serratia marcescens strain W2.3 isolated from diseased tilapia fish: a draft genome analysis. Gut Pathog 5, 29 (2013). https://doi.org/10.1186/1757-4749-5-29
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1757-4749-5-29