
Abstract
The influence of resident gut microbes on xenobiotic metabolism has been investigated at different levels throughout the past five decades. However, with the advance in sequencing and pyrotagging technologies, addressing the influence of microbes on xenobiotics had to evolve from assessing direct metabolic effects on toxins and botanicals by conventional culture-based techniques to elucidating the role of community composition on drugs metabolic profiles through DNA sequence-based phylogeny and metagenomics. Following the completion of the Human Genome Project, the rapid, substantial growth of the Human Microbiome Project (HMP) opens new horizons for studying how microbiome compositional and functional variations affect drug action, fate, and toxicity (pharmacomicrobiomics), notably in the human gut. The HMP continues to characterize the microbial communities associated with the human gut, determine whether there is a common gut microbiome profile shared among healthy humans, and investigate the effect of its alterations on health. Here, we offer a glimpse into the known effects of the gut microbiota on xenobiotic metabolism, with emphasis on cases where microbiome variations lead to different therapeutic outcomes. We discuss a few examples representing how the microbiome interacts with human metabolic enzymes in the liver and intestine. In addition, we attempt to envisage a roadmap for the future implications of the HMP on therapeutics and personalized medicine.
Similar content being viewed by others
Introduction
The gut microbiota is the most predominant and most diverse microbial community residing in the human body[1]. It comprises hundreds of microbial species, together constituting about 10 times the number of body cells[2, 3], and contributes substantially to human metabolic processes to the extent that up to 36 % of small molecules in human blood are contributed by the gut microbiome[4]. The gut microbiota’s impact on drug response and metabolism has been explored since the mid 20th century (reviewed in[5]); however, past studies have mostly focused on assessing the metabolic activity of gut microbial communities on antibiotics and botanicals[6–9]. Meanwhile, the influence of the host genetic makeup on drug response occupied the center stage of personalized medicine research, specifically in the clinical domain, leading to the rise of pharmacogenomic approaches to personalized therapy, while a pivotal player in xenobiotic metabolism, the microbiota, was mostly being overlooked[10, 11].
The various metabolic capabilities of the gut microbiota fueled the study of its effects on drug metabolism[11, 12]. Several approaches were adopted, including comparisons between metabolic patterns of conventional and germfree mice, biochemical assays of microbial metabolic activities in cultures, and mutagenicity tests[5, 6, 13]. Population-based approaches, such as investigating the correlation between compositional variations in gut microbiota and response to a particular drug, e.g., digoxin, were followed as well[9].
The evolution of microbial genomics from culture-based (i.e., sequencing genomes of bacterial species after isolating their colonies) to culture-independent strategies (metagenomics—or shotgun sequencing of microbial and viral communities[14, 15]) has allowed the identification of the molecular signature of the gut microbiome associated with a certain disease or with altered drug response[16]. To describe this new expansion of pharmacogenomics, we suggested the term pharmacomicrobiomics to denote the effect of microbiome variations on drug disposition and response[17, 18]; here, we apply this concept explicitly to the human gut microbiome, the best-studied microbiome for its effect on xenobiotics.
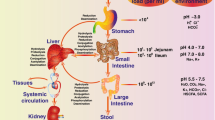
In a broad sense, the term gut pharmacomicrobiomics encompasses the effect of the gut microbiome variations on pharmacokinetic and pharmacodynamic processes[17, 19] (See Section “Term disambiguation”). However, to date, the better-documented effects of the human gut metagenome on drugs are those related to metabolism (i.e., effects on pharmacokinetic), either through: (i) the secretion of enzymes that modify the chemical structure of drug molecules, (ii) the secretion of metabolic products that interfere with drug metabolism, (iii) the modification of the levels and activities of liver and intestinal enzymes, or (iv) the modulation of expression of human metabolic genes[16] (Figure1). Taking into consideration the enormous number of gut-associated microbes, and the extremely large number of diverse genes they encode and pathways they express, understanding the effect of the gut microbiota on human response to drugs is an indispensable step towards providing a comprehensively tailored/personalized therapy that would be more efficient, cost-effective, and with lower adverse drug events[17, 20].

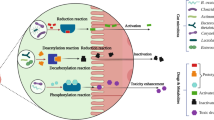
Different ways of interactions between the gut microbiome and drugs, sometimes involving dietary compounds or intestinal and liver enzymes. Liver and intestine cartoons were taken from the publicly available clipart of the University of Wisconsin, Madison. URL:http://www.biochem.wisc.edu/medialab/clipart.aspx.
In this review, we aim at providing an overview of the influence of gut microbiota on drugs, spanning the documented metabolic effects of the microbiota and the different approaches used for their investigation. In addition, we provide an outlook for the future of pharmacomicrobiomics in the context of the Human Microbiome Project (HMP) and for the application of metagenomic approaches as an integral part of pharmacotherapy and personalized medicine.
Term disambiguation
-
Microbiome and microbiomics: To the best of our knowledge, the term ‘microbiome’ was first suggested in 2000 by the Nobel Laureate, Joshua Lederberg, to describe the sum of microbial genomes associated with the human body, which he described as a part of “the human extended genome” (URL:http://www.project-syndicate.org/commentary/microbiology-s-world-wide-web). Soon after, it was used in the same meaning in literature[21, 22]. Currently, however, microbiome is being used to denote two different concepts: (i) the collective microbial genome of a community (i.e., microbial metagenome) or (ii) the sum of all microscopic life forms, viz. microbes, within an environment (i.e., micro.biome). Microbiome was initially confined to host-associated metagenomes, but is now being used interchangeably with microbial metagenome (e.g., the Earth Microbiome Projecthttp://www.earthmicrobiome.org/[23]). The less frequently used term, ‘microbiomics’, describes the study of functional aspects related to the microbiome, including the integration of high-throughput genome-wide data[24].
-
Pharmacogenomics and pharmacomicrobiomics: Pharmacogenomics[25] is a well-established term that describes the effect of human genome variations on drug disposition and action. The term can certainly be applied not just to the human nuclear and mitochondrial genomes, but also to the human extended genome or the genome of the human supraorganism[18]; yet, to specify the impact of the human-associated microbiome on drugs, we have coined the term pharmacomicrobiomics[17, 18], which we consider as a natural expansion of pharmacogenomics, which is likely to spread when more HMP data accrue.
-
Metabolomics and metabonomics: Those two verbally similar terms have been sometimes used interchangeably to describe the high-throughput study of all genome-encoded metabolites produced by a particular organism or a community; however, Nicholson and coworkers carefully denote the difference between the two terms as they use metabolomics to describe the study of genetically controlled metabolites and fluxes produced by one type of cells or tissues, whereas they define metabonomics as the measurement of metabolites produced by a collection of cells/genomes within a multicellular organism or an ecosystem[26] (the latter once described as the ‘meta-metabolome’[27]).
Role of gut microbiota in xenobiotic metabolism
The influence of the gut microbiota on the metabolism of xenobiotics has been regarded extensively as a response-modifying process, and several mechanisms have been proposed and demonstrated[11]. Gut-associated microbes can alter drug metabolism directly by producing enzymes that degrade or activate the drug molecules, or by competing with drug molecules over the metabolizing enzymes[17, 20]. In addition, the gut microbiota may exert its influence by modulating the activity or altering the levels of the host’s drug-metabolizing enzymes or by producing enzyme-inducing metabolites that are originally derived from diet[28, 29] (Figure1). Accordingly, the microbiome’s response-modifying effect has been widely appreciated in nutrition and toxicology, and the role of gut microbiota in metabolism has initially been investigated in terms of the metabolism of compounds of dietary and botanical origin (Table1).
Additionally, the absence of conventional gut microbiota in germfree mice has been correlated with perturbations in levels of liver and intestinal metabolic enzymes in comparison to their corresponding levels in mice with conventional gut ecosystem[29], and conventional gut microbiotas in human and mice were shown to be associated with a modest elevation in the levels of drug-metabolizing enzymes, such as sulfotransferase1 B1 (SULT1B1) and with reduced levels of other enzymes, such as SULT1C1, NAT1 and NAT2[28] (Table2).
Impact of microbiome variations on drug response and toxicity
Most studies on drug-microbe interactions did not take in consideration the microbiome profile/composition of an individual or a population; however, these variations are the basis of pharmacomicrobiomics, and their study has become possible now that the HMP has been established[3, 41], and HMP data have already been made available[1, 42].
Several studies associated a particular 16S rRNA microbial signature with specific biomarker metabolites and clinical outcomes. This association has been extended to encompass several conventional drugs such as digoxin and acetaminophen[9, 43]. Profiling the signatures of the microbial communities in relation to their metabolic effect on drugs among patients is likely to introduce clinical markers that will dictate treatment regimens tailored in accordance with each patient's resident microbiota[43]. Such regimens, in turn, will modify the current treatment strategies that are based on conventional pathologic and pharmacokinetic parameters to take into account the interindividual perturbations in the gut microbiota and the gut ecosystem. These measures are especially true with the evidence of the sym-xenobiotic metabolism that involves both the host and the associated microbiota to biotransform drugs, including first-line therapies[9, 44] (Table3). As a consequence, microbiome-labile medications may be limited, or their dose readjusted, for certain populations or individuals harboring particular gut microbial community profiles. Several drugs that possess structural similarity to microbial products and are thereby potential candidates of microbial metabolism are yet to be studied.
A systems biology view of the host-microbiota metabolome and co-metabolome
Previously reported drug-microbe and drug-microbiome interactions have mostly been described as phenotypic observations of drugs being modified by a microbial species, an entire microbial community, or an even more intricate system consisting of a microbial and a human component. However, in many cases the process, biochemical pathway, or specific reaction remains unknown, which renders the analysis of those interactions by reductionist approaches difficult. Instead, exploring the causality of those interactions might require systems approaches such as the metagenomic analysis of the microbial community followed by the identification of differentially abundant or differentially expressed candidate genes or genomic subsystems[52] involved in those interactions. Yet, metagenomic surveys that determine microbial community profiles, gene presence/absence and abundance, or functional classification of sequence fragments are not sufficient to tell a coherent story about the observed phenotypes since a gene’s presence does not imply its expression or functionality. Consequently, extracting knowledge from those microbiome explorations and translating them into an ultimately tailored therapy requires modeling the human microbiome, variome, and interactions between them via integrating multiple layers of information, including transcriptomic, proteomic, and metabolomic data. Such integration is not always achievable in a system with this complexity. For instance, a statistically sound correlation between mRNA and protein expression levels in mid-log phase Saccharomyces cerevisiae cells has been hindered by technical limitations[53]. If this was the case with a unicellular organism or with relatively uniform cell lines[54], then further levels of complexity are to be expected in the gut microbiome ecosystem, where communities of unicellular organisms coexist in balance with the human multicellular tissues. Systems biology approaches for such complex communities are inevitable but are still in early development[55, 56].
From a holistic perspective, tailoring a pharmacotherapy that accommodates intraindividual and interindividual variations would take into account the variations in the host’s genetic makeup, its associated-microbiome, and metabolomic interactions between the host and its associated microbiota (i.e., co-metabolome). With the recognition of the considerable role of the human microbiome and its variations together with the formerly well-recognized role of the human variome in predicting response to pharmacotherapy, there is a growing demand in both clinical and research domains for proper computational models that are able to comprehensively consider all such aspects of variability[26, 53, 54]. The best-recognized process in altered drug response, controlled by both human genome and microbiome, is the presystemic metabolism or first-pass effect (reviewed in[57]). Since the metabolism of xenobiotics in humas is performed by host and microbial enzymes, the metabolic process is recognized as combinatorial or “sym-xenobiotic” as recently described[10]. Furthermore, a continuous metabolic interaction, termed metabolome-metabolome interaction, exists between the host and its associated microbiota[10]. Modeling the human/microbiome variations and metabolome-metabolome interactions will provide insights into the metabolism of xenobiotics and thereby allow for accurate predictions for drug response[26].
Nicholson and colleagues[26] attempted to visualize the role of both the host and its associated microbiota in xenobiotic metabolism in the gut by proposing an interesting model, assuming six different cell types in both host and microbiota, every type of which has its own transcriptome and metabolome depending on its role. There is a mutual metabolic exchange between the host and microbiota, and the extracellular compartment contains metabolites generated by both of them. Those metabolites are the result of drug and food metabolism, and might lead to metabolic alterations in both the host and its associated microbiota. This probabilistic model of metabolism was introduced in an attempt to tackle the potential interaction between the different host- and microbiome-related factors that would eventually display a certain outcome for metabolism. The model likens the complex process of drug metabolism to a Japanese Pachinko (pinball machine), where pins represent enzymes and transporters involved in metabolism, holes indicate outlets for metabolites, and pathways are represented by the sequence of pins. According to this model, the final outcome is the increment of the probabilities of collisions between pins and balls[26].
Web resources for exploring gut pharmacomicrobiomics
-
Human variome resources:
-
HVP (Human Variome Project):http://www.humanvariomeproject.org[58]
-
HapMap:http://hapmap.ncbi.nlm.nih.gov[59]
-
-
Human microbiome resources:
-
MetaHIT (Metagenomics of the Human Intestinal Tract):http://www.metahit.eu[42]
-
HMP:http://hmpdacc.org[41]
-
-
Tools or databases for browsing the human microbiome:
-
IMG/HMP:http://www.hmpdacc-resources.org/cgi-bin/imgm_hmp/main.cgi[41]
-
MG-RAST:http://metagenomics.anl.gov[60]
-
The SEED Servers:http://www.theseed.org/servers[61]
-
-
Pharmacogenomics/pharmacomicrobiomics databases:
-
PharmGKB (Pharmacogenomics Knowledge Base):http://www.pharmgkb.org[62]
-
PacDB (Pharmacogenetics and Cell Database):http://www.pacdb.org[63]
-
CTDB (Comparative Toxigenomics Database):http://ctdbase.org[64]
-
The PharmacoMicrobiomics Portal:http://www.pharmacomicrobiomics.org[65]
-
-
Enzymes/pathways databases:
-
KEGG (Kyoto Encyclopedia of Genes and Genomes):http://www.genome.jp/kegg/[66]
-
Model SEED:http://seed-viewer.theseed.org/seedviewer.cgi?page=ModelView[67]
-
BRENDA (BRaunschweig ENzyme Database):http://www.brenda-enzymes.org[68]
-
Future anticipations
The current advances in the Human Variome Project[69, 70] and the HMP[3, 41], together with a battery of publicly available web resources (See Section “Web resources for exploring gut pharmacomicrobiomics”) offer a starting point for those interested in drug-microbiome interactions to address several intriguing questions. However, the examples reported previously (e.g., those in Tables1,2 and3) are just the tip of an iceberg of yet-to-be-discovered interactions between the host variome, associated microbiome, their combined metabolome, and chemicals ingested by humans. Ultimately, the study of those interactions in spite of their complexity is driven by the need for devising personalized therapeutic regimens aiming at optimizing drug bioavailability to obtain maximal efficiency and minimal toxicity. Below, we suggest a roadmap of four steps for the development of the nascent field of gut pharmacomicrobiomics and its translation into personalized medicine (Figure2).

A roadmap for the development of the nascent field of gut pharmacomicrobiomics and its translation into the clinical domain.
Capturing current knowledge
The first step is to capture and organize the currently available information on drug-microbiome complex interactions by building databases similar to those built for pharmacogenetics, pharmacogenomics, and drug-drug interactions. Concomitant with building databases is developing tools and resources to support discovery by mining those databases and connecting them to microbial genomic databases (e.g., SEED[61], GOLD[71]) metagenomic/microbiome databases, (e.g., MG-RAST[60], myMGDB, HMP[41], METAHIT[42]), and metabolic pathway databases (e.g., KEGG[66], modelSEED[67], BRENDA[68]).
Developing and performing high-throughput screens for novel drug-microbiome interactions
In parallel with capturing existing knowledge, there is continuous need for digging deeper into the unknown drug-microbiome interaction space. Addressing this need can be achieved via studies involving high-throughput screens of drugs against human microbiota from different individuals looking at the overall action of these microbiotas on representatives of different drug classes, or, reciprocally, via screening individual resident gut microbes against large libraries of drugs or other chemicals.
Developing software and building models for drug response simulation
The accumulated data in literature pointing out to the response variation mediated by mammalian host variome and microbiome calls for the construction of modeling software that considers all such parameters to provide rational hypotheses or accurate predictions for research[26, 55, 72]. Developing such modeling software and using it in building models requires encoding data compiled from the literature regarding the host variome, microbiome and co-metabolome, and incorporating these encoded data into a model capable of retrieving an informative index describing the predicted outcome. For instance, Hlavaty and colleagues[73] used a similar approach to construct a predictive model of an apoptotic pharmacogenetic index for infliximab in treatment of Crohn disease. Following data mining, they used SAS® enterprise miner software to analyze all the genetic variants involved with the apoptotic response of infliximab, they managed to develop a new pharmacogenetic index ranging from 0 which denotes diminished response to 3, indicating a powerful response[73].
Data integration and translation into the clinical domain
The availability of web resources, the generation of more data, and the construction of rigorous models for drug-microbiome interactions will offer a great opportunity to translate this knowledge into diagnostic and clinical measures. In the future, routing clinical practices should include integrating microbiome data and processing them to produce valid assumptions of clinical outcome, based on which the type, dose, and regimen of treatment will be planned for each patient. Accordingly, each case will have its own panel of personalized therapy. For instance, patients harboring gut microbiota known to be associated with elevated levels of metabolic enzymes will be scheduled for higher doses; patients with higher susceptibility to acetaminophen toxicity might either be given a lower dose or an alternative nonsteroidal anti-inflammatory medicine; and patients with a microbiota with higher ability to metabolize digoxin will be scheduled to receive lower dose.
Conclusions
Throughout the past five decades, the study of the effect of gut microbiota went through several phases uncovering its ample significance in drug response. With the continuous growth of the HMP and its expansion to cover diverse human populations, it is anticipated that the primary data concerning the common gut microbiome profile and its diversity among humans will be revealed, enabling to pursue further studies on its effect on drug response among populations. However, several steps are yet to be taken in anticipation of the floods of HMP data, including the construction of databases, software, and models that would provide credible predictions of differential clinical outcome and fuel further hypothesis-driven studies whose findings might be integrated into clinical settings.
Authors’ information
Rama Saad is currently a visiting scholar at Vanderbilt University Medical Center, Nashville, TN, USA.
References
Human Microbiome Project Consortium: Structure, function and diversity of the healthy human microbiome. Nature. 2012, 486: 207-214. 10.1038/nature11234.
Savage DC: Microbial ecology of the gastrointestinal tract. Annu Rev Microbiol. 1977, 31: 107-133. 10.1146/annurev.mi.31.100177.000543.
Peterson J, Garges S, Giovanni M, McInnes P, Wang L, Schloss JA, Bonazzi V, McEwen JE, Wetterstrand KA, Deal C: The NIH Human Microbiome Project. Genome Res. 2009, 19: 2317-2323.
Hood L: Tackling the microbiome. Science. 2012, 336: 1209-10.1126/science.1225475.
Goldman P, Peppercorn MA, Goldin BR: Metabolism of drugs by microorganisms in the intestine. Am J Clin Nutr. 1974, 27: 1348-1355.
Holt R: The bacterial degradation of chloramphenicol. Lancet. 1967, 1: 1259-1260.
Spatz M, Smith DW, McDaniel EG, Laqueur GL: Role of intestinal microorganisms in determining cycasin toxicity. Proc Soc Exp Biol Med. 1967, 124: 691-697.
Lindenbaum J, Rund DG, Butler VP, Tse-Eng D, Saha JR: Inactivation of digoxin by the gut flora: reversal by antibiotic therapy. N Engl J Med. 1981, 305: 789-794. 10.1056/NEJM198110013051403.
Mathan VI, Wiederman J, Dobkin JF, Lindenbaum J: Geographic differences in digoxin inactivation, a metabolic activity of the human anaerobic gut flora. Gut. 1989, 30: 971-977. 10.1136/gut.30.7.971.
Nicholson JK, Holmes E, Wilson ID: Gut microorganisms, mammalian metabolism and personalized health care. Nat Rev Microbiol. 2005, 3: 431-438. 10.1038/nrmicro1152.
Sousa T, Paterson R, Moore V, Carlsson A, Abrahamsson B, Basit AW: The gastrointestinal microbiota as a site for the biotransformation of drugs. Int J Pharm. 2008, 363: 1-25. 10.1016/j.ijpharm.2008.07.009.
Johnson CH, Patterson AD, Idle JR, Gonzalez FJ: Xenobiotic metabolomics: major impact on the metabolome. Annu Rev Pharmacol Toxicol. 2012, 52: 37-56. 10.1146/annurev-pharmtox-010611-134748.
Koch RL, Chrystal EJ, Beaulieu BB, Goldman P: Acetamide–a metabolite of metronidazole formed by the intestinal flora. Biochem Pharmacol. 1979, 28: 3611-3615. 10.1016/0006-2952(79)90407-6.
Riesenfeld CS, Schloss PD, Handelsman J: Metagenomics: genomic analysis of microbial communities. Annu Rev Genet. 2004, 38: 525-552. 10.1146/annurev.genet.38.072902.091216.
Edwards RA, Rohwer F: Viral metagenomics. Nat Rev Microbiol. 2005, 3: 504-510. 10.1038/nrmicro1163.
Haiser HJ, Turnbaugh PJ: Developing a metagenomic view of xenobiotic metabolism. Pharmacol Res. 2012, 10.1016/j.phrs.2012.07.009.
Rizkallah MR, Saad R, Aziz RK: The Human Microbiome Project, personalized medicine and the birth of pharmacomicrobiomics. Curr Pharmacogenomics Person Med. 2010, 8: 182-193.
Aziz RK: Rethinking pharmacogenomics in an ecosystem: drug-microbiome interactions, pharmacomicrobiomics, and personalized medicine for the human supraorganism. Curr Pharmacogenomics Person Med. 2012, 10: 258-261.
Aziz RK, Saad R, Rizkallah MR: PharmacoMicrobiomics or how bugs modulate drugs: an educational initiative to explore the effects of human microbiome on drugs. BMC Bioinforma. 2011, 12: A10-
Wilson ID: Drugs, bugs, and personalized medicine: pharmacometabonomics enters the ring. Proc Natl Acad Sci USA. 2009, 106: 14187-14188.
Relman DA: New technologies, human-microbe interactions, and the search for previously unrecognized pathogens. J Infect Dis. 2002, 186 (Suppl 2): S254-S258.
Shanahan F: The host-microbe interface within the gut. Best Pract Res Clin Gastroenterol. 2002, 16: 915-931. 10.1053/bega.2002.0342.
Gilbert JA, Meyer F, Antonopoulos D, Balaji P, Brown CT, Desai N, Eisen JA, Evers D, Field D, Feng W: Meeting report: the terabase metagenomics workshop and the vision of an Earth microbiome project. Stand Genomic Sci. 2010, 3: 243-248. 10.4056/sigs.1433550.
Egert M, de Graaf AA, Smidt H, de Vos WM, Venema K: Beyond diversity: functional microbiomics of the human colon. Trends Microbiol. 2006, 14: 86-91. 10.1016/j.tim.2005.12.007.
Persidis A: The business of pharmacogenomics. Nat Biotechnol. 1998, 16: 209-210. 10.1038/nbt0298-209.
Nicholson JK, Wilson ID: Opinion: understanding 'global' systems biology: metabonomics and the continuum of metabolism. Nat Rev Drug Discov. 2003, 2: 668-676. 10.1038/nrd1157.
Ibrahim M, Anishetty S: A meta-metabolome network of carbohydrate metabolism: Interactions between gut microbiota and host. Biochem Biophys Res Commun. 2012, 428 (2): 278-284. 10.1016/j.bbrc.2012.10.045.
Meinl W, Sczesny S, Brigelius-Flohe R, Blaut M, Glatt H: Impact of gut microbiota on intestinal and hepatic levels of phase 2 xenobiotic-metabolizing enzymes in the rat. Drug Metab Dispos. 2009, 37: 1179-1186. 10.1124/dmd.108.025916.
Overvik E, Lindeskog P, Midtvedt T, Gustafsson JA: Mutagen excretion and cytochrome P-450-dependent activity in germfree and conventional rats fed a diet containing fried meat. Food Chem Toxicol. 1990, 28: 253-261. 10.1016/0278-6915(90)90037-N.
Sugimura T, Nagao M, Wakabayashi K: Carcinogenicity of food mutagens. Environ Health Perspect. 1996, 104 (Suppl 3): 429-433. 10.1289/ehp.96104s3429.
Rowland IR: Interactions of the gut microflora and the host in toxicology. Toxicol Pathol. 1988, 16: 147-153. 10.1177/019262338801600207.
Lhoste EF, Ouriet V, Bruel S, Flinois JP, Brezillon C, Magdalou J, Cheze C, Nugon-Baudon L: The human colonic microflora influences the alterations of xenobiotic-metabolizing enzymes by catechins in male F344 rats. Food Chem Toxicol. 2003, 41: 695-702. 10.1016/S0278-6915(03)00010-3.
Axelson M, Sjovall J: Formation of catechol estrogens by intestinal bacterial demethylation of 2-methoxyestrone. Biochim Biophys Acta. 1983, 751: 162-165. 10.1016/0005-2760(83)90170-4.
Gonthier MP, Verny MA, Besson C, Remesy C, Scalbert A: Chlorogenic acid bioavailability largely depends on its metabolism by the gut microflora in rats. J Nutr. 2003, 133: 1853-1859.
Gavaghan CL, Nicholson JK, Connor SC, Wilson ID, Wright B, Holmes E: Directly coupled high-performance liquid chromatography and nuclear magnetic resonance spectroscopic with chemometric studies on metabolic variation in Sprague–Dawley rats. Anal Biochem. 2001, 291: 245-252. 10.1006/abio.2000.5034.
Bowey E, Adlercreutz H, Rowland I: Metabolism of isoflavones and lignans by the gut microflora: a study in germ-free and human flora associated rats. Food Chem Toxicol. 2003, 41: 631-636. 10.1016/S0278-6915(02)00324-1.
Delgado S, Ruas-Madiedo P, Suarez A, Mayo B: Interindividual differences in microbial counts and biochemical-associated variables in the feces of healthy Spanish adults. Dig Dis Sci. 2006, 51: 737-743. 10.1007/s10620-006-3200-5.
Akao T, Kawabata K, Yanagisawa E, Ishihara K, Mizuhara Y, Wakui Y, Sakashita Y, Kobashi K: Baicalin, the predominant flavone glucuronide of scutellariae radix, is absorbed from the rat gastrointestinal tract as the aglycone and restored to its original form. J Pharm Pharmacol. 2000, 52: 1563-1568.
Selma MV, Espin JC, Tomas-Barberan FA: Interaction between phenolics and gut microbiota: role in human health. J Agric Food Chem. 2009, 57: 6485-6501. 10.1021/jf902107d.
Bolton EE, Wang Y, Thiessen PA, Bryant SH: PubChem: integrated platform of small molecules and biological activities. Annu Rep Comput Chem. 2008, 4: 217-241.
Proctor LM: The Human Microbiome Project in 2011 and beyond. Cell Host Microbe. 2011, 10: 287-291. 10.1016/j.chom.2011.10.001.
Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T: A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010, 464: 59-65. 10.1038/nature08821.
Clayton TA, Baker D, Lindon JC, Everett JR, Nicholson JK: Pharmacometabonomic identification of a significant host-microbiome metabolic interaction affecting human drug metabolism. Proc Natl Acad Sci USA. 2009, 106: 14728-14733. 10.1073/pnas.0904489106.
Vermes A, Kuijper EJ, Guchelaar HJ, Dankert J: An in vitro study on the active conversion of flucytosine to fluorouracil by microorganisms in the human intestinal microflora. Chemotherapy. 2003, 49: 17-23. 10.1159/000069784.
Grundmann O: The gut microbiome and pre-systemic metabolism: current state and evolving research. J Drug Metab Toxicol. 2010, 1: 105-
Steffens LS, Nicholson S, Paul LV, Nord CE, Patrick S, Abratt VR: Bacteroides fragilis RecA protein overexpression causes resistance to metronidazole. Res Microbiol. 2010, 161: 346-354. 10.1016/j.resmic.2010.04.003.
Strong HA, Renwick AG, George CF, Liu YF, Hill MJ: The reduction of sulphinpyrazone and sulindac by intestinal bacteria. Xenobiotica. 1987, 17: 685-696. 10.3109/00498258709043976.
Strong HA, Warner NJ, Renwick AG, George CF: Sulindac metabolism: the importance of an intact colon. Clin Pharmacol Ther. 1985, 38: 387-393. 10.1038/clpt.1985.192.
Ashida N, Ijichi K, Watanabe Y, Machida H: Metabolism of 5'-ether prodrugs of 1-beta-D-arabinofuranosyl-E-5-(2-bromovinyl)uracil in rats. Biochem Pharmacol. 1993, 46: 2201-2207. 10.1016/0006-2952(93)90610-9.
Nakayama H, Kinouchi T, Kataoka K, Akimoto S, Matsuda Y, Ohnishi Y: Intestinal anaerobic bacteria hydrolyse sorivudine, producing the high blood concentration of 5-(E)-(2-bromovinyl)uracil that increases the level and toxicity of 5-fluorouracil. Pharmacogenetics. 1997, 7: 35-43. 10.1097/00008571-199702000-00005.
Kitamura S, Sugihara K, Kuwasako M, Tatsumi K: The role of mammalian intestinal bacteria in the reductive metabolism of zonisamide. J Pharm Pharmacol. 1997, 49: 253-256.
Overbeek R, Begley T, Butler RM, Choudhuri JV, Chuang HY, Cohoon M, de Crecy-Lagard V, Diaz N, Disz T, Edwards R: The subsystems approach to genome annotation and its use in the project to annotate 1000 genomes. Nucleic Acids Res. 2005, 33: 5691-5702. 10.1093/nar/gki866.
Gygi SP, Rochon Y, Franza BR, Aebersold R: Correlation between protein and mRNA abundance in yeast. Mol Cell Biol. 1999, 19: 1720-1730.
Gry M, Rimini R, Stromberg S, Asplund A, Ponten F, Uhlen M, Nilsson P: Correlations between RNA and protein expression profiles in 23 human cell lines. BMC Genomics. 2009, 10: 365-10.1186/1471-2164-10-365.
Zengler K, Palsson BO: A road map for the development of community systems (CoSy) biology. Nat Rev Microbiol. 2012, 10: 366-372.
Thiele I, Heinken A, Fleming RM: A systems biology approach to studying the role of microbes in human health. Curr Opin Biotechnol. 2012
Doherty MM, Pang KS: First-pass effect: significance of the intestine for absorption and metabolism. Drug Chem Toxicol. 1997, 20: 329-344. 10.3109/01480549709003891.
Howard HJ, Horaitis O, Cotton RG, Vihinen M, Dalgleish R, Robinson P, Brookes AJ, Axton M, Hoffmann R, Tuffery-Giraud S: The Human Variome Project (HVP) 2009 Forum "Towards Establishing Standards". Hum Mutat. 2010, 31: 366-367. 10.1002/humu.21175.
International HapMap Consortium: The International HapMap Project. Nature. 2003, 426: 789-796. 10.1038/nature02168.
Meyer F, Paarmann D, D'Souza M, Olson R, Glass EM, Kubal M, Paczian T, Rodriguez A, Stevens R, Wilke A: The metagenomics RAST server - a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinforma. 2008, 9: 386-10.1186/1471-2105-9-386.
Aziz RK, Devoid S, Disz T, Edwards RA, Henry CS, Olsen GJ, Olson R, Overbeek R, Parrello B, Pusch GD: SEED Servers: high-performance access to the SEED genomes, annotations, and metabolic models. PLoS One. 2012, 7: e48053-10.1371/journal.pone.0048053.
Owen RP, Altman RB, Klein TE: PharmGKB and the International Warfarin Pharmacogenetics Consortium: the changing role for pharmacogenomic databases and single-drug pharmacogenetics. Hum Mutat. 2008, 29: 456-460. 10.1002/humu.20731.
Gamazon ER, Duan S, Zhang W, Huang RS, Kistner EO, Dolan ME, Cox NJ: PACdb: a database for cell-based pharmacogenomics. Pharmacogenet Genomics. 2010, 20: 269-273.
Davis AP, Murphy CG, Rosenstein MC, Wiegers TC, Mattingly CJ: The Comparative Toxicogenomics Database facilitates identification and understanding of chemical-gene-disease associations: arsenic as a case study. BMC Med Genomics. 2008, 1: 48-10.1186/1755-8794-1-48.
Rizkallah MR SG-E, Saad R, Aziz RK: The pharmacomicrobiomics portal: a database for drug-microbiome interactions. Curr Pharmacogenomics Person Med. 2012, 10: 195-203. 10.2174/187569212802510030.
Kanehisa M, Goto S: KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28: 27-30. 10.1093/nar/28.1.27.
Henry CS, DeJongh M, Best AA, Frybarger PM, Linsay B, Stevens RL: High-throughput generation, optimization and analysis of genome-scale metabolic models. Nat Biotechnol. 2010, 28: 977-982. 10.1038/nbt.1672.
Scheer M, Grote A, Chang A, Schomburg I, Munaretto C, Rother M, Sohngen C, Stelzer M, Thiele J, Schomburg D: BRENDA, the enzyme information system in 2011. Nucleic Acids Res. 2011, 39: D670-D676. 10.1093/nar/gkq1089.
Ring HZ, Kwok PY, Cotton RG: Human Variome Project: an international collaboration to catalogue human genetic variation. Pharmacogenomics. 2006, 7: 969-972. 10.2217/14622416.7.7.969.
Ozdemir V, Rosenblatt DS, Warnich L, Srivastava S, Tadmouri GO, Aziz RK, Reddy PJ, Manamperi A, Dove ES, Joly Y: Towards an ecology of collective innovation: Human Variome Project (HVP), Rare Disease Consortium for Autosomal Loci (RaDiCAL) and Data-Enabled Life Sciences Alliance (DELSA). Curr Pharmacogenomics Person Med. 2011, 9: 243-251.
Pagani I, Liolios K, Jansson J, Chen IM, Smirnova T, Nosrat B, Markowitz VM, Kyrpides NC: The Genomes OnLine Database (GOLD) v.4: status of genomic and metagenomic projects and their associated metadata. Nucleic Acids Res. 2012, 40: 571-579. 10.1093/nar/gkr1100.
Heinken A, Sahoo S, Fleming RM, Thiele I: Systems-level characterization of a host-microbe metabolic symbiosis in the mammalian gut. Gut Microbes. 2013, 4: 1-13.
Hlavaty T, Ferrante M, Henckaerts L, Pierik M, Rutgeerts P, Vermeire S: Predictive model for the outcome of infliximab therapy in Crohn's disease based on apoptotic pharmacogenetic index and clinical predictors. Inflamm Bowel Dis. 2007, 13: 372-379. 10.1002/ibd.20024.
Acknowledgements
We thank Ms. Radwa Raed Sharaf for critical reading of the draft and help with proofreading the final version.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no personal or financial competing interests.
Authors’ contributions
RS reviewed literature, collected data, outlined and drafted the manuscript, and participated in writing the final version. MRR collected data and abstracts, and participated in writing the final version. RKA conceived the article, reviewed literature, and wrote the article in its final format. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Saad, R., Rizkallah, M.R. & Aziz, R.K. Gut Pharmacomicrobiomics: the tip of an iceberg of complex interactions between drugs and gut-associated microbes. Gut Pathog 4, 16 (2012). https://doi.org/10.1186/1757-4749-4-16
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1757-4749-4-16