
Background
Helicobacter pylori is a vertically inherited gut commensal that is carcinogenic if it possesses the cag pathogenicity island (cag PaI); infection with H.pylori is the major risk factor for gastric cancer, the second leading cause of death from cancer worldwide (WHO). The cag PaI locus encodes the cagA gene, whose protein product is injected into stomach epithelial cells via a Type IV secretion system, also encoded by the cag PaI. Once there, the cagA protein binds to various cellular proteins, resulting in dysregulation of cell division and carcinogenesis. For this reason, cagA may be described as an oncoprotein. A clear understanding of the mechanism of action of cagA and its benefit to the bacteria is lacking.
Results
Here, we reveal that the cagA gene displays strong signatures of positive selection in bacteria isolated from amerindian populations, using the Ka/Ks ratio. Weaker signatures are also detected in the gene from bacteria isolated from asian populations, using the Ka/Ks ratio and the more sensitive branches-sites model of the PAML package. When the cagA gene isolated from amerindian populations was examined in more detail it was found that the region under positive selection contains the EPIYA domains, which are known to modulate the carcinogenicity of the gene. This means that the carcinogenicity modulating region of the gene is undergoing adaptation. The results are discussed in relation to the high incidences of stomach cancer in some latin american and asian populations.
Conclusion
Positive selection on cagA indicates antagonistic coevolution between host and bacteria, which appears paradoxical given that cagA is detrimental to the human host upon which the bacteria depends. This suggests several non-exclusive possibilities; that gastric cancer has not been a major selective pressure on human populations, that cagA has an undetermined benefit to the human host, or that horizontal transmission of H.pylori between hosts has been more important in the evolution of H.pylori than previously recognized, reducing the selective pressure to lower the pathogenicity of the bacteria. The different patterns of adaptation of the gene in different human populations indicates that there are population specific differences in the human gut environment - due either to differences in host genetics or diet and other lifestyle features.
Similar content being viewed by others

Introduction
Helicobacter pylori is a Gram negative bacterium that lives in the human stomach as part of the normal gastric microbiome [1], and is generally present in the majority of the adult population [2]. The bacterium has co-evolved with human populations [3] and is well adapted and largely specific to the human host. The ancestor of H.pylori was intestinal and during its evolution migrated to the stomach, facilitated by the evolution of a urease that combats the stomach's acid conditions [4, 5]. H.pylori strains may possess a cag pathogenicity island (cag PaI) that contains a cagA gene encoding a 128 kDa protein [6, 7]. The cag PaI seems to have entered the H.pylori genome by lateral gene transfer, after H.pylori differentiated from parental species [2, 8]. Many of the genes of the cag PAI are involved in translocation of the cagA protein into epithelial cells lining the stomach. However, the function of the cagA protein itself is unknown. Infection with cagA+ H.pylori is strongly associated with gastric carcinoma [9–11]; gastric carcinoma is the second leading cause of death from cancer worldwide [12]. In addition, cagA+H.pylori is associated with chronic gastritis and peptic ulcers [13].
The mechanism of pathogenicity of cagA+ H.pylori is as follows. The bacteria attaches to the stomach wall and the cagA protein is injected into an epithelial cell by a bacterial Type IV secretion system, also encoded by the cag PaI locus [14]. Once inside the cell, cagA is phosphorylated on tyrosine residues located within EPIYA domains by members of the src kinases such as c-src, Fyn, Yes [15], Lyn [16] and c-Abl [17]. The cagA protein is membrane associated and interacts with numerous additional cellular proteins, including the oncoprotein Src homology 2 domain containing tyrosine phosphatase (SHP-2 [18]), microtubule affinity-regulating kinase (MARK2 [19]), growth factor receptor-bound protein 2 (Grb-2 [20]), hepatocyte growth factor receptor (c-Met [21]), C-terminal Src kinase (Csk [22]) and p38 (Crk [23]). Tyrosine phosphorylated cagA recruits and activates SHP-2, apparently mimicking the action of Gab1 [24]. Consistent with the mimicry hypothesis, cagA is able to rescue Gab1 deficient Drosophila mutants [25], which is interesting given that cagA has no sequence similarity with Gab1, indeed it has no known homologs. The interaction with SHP-2 causes inhibition of its tumor suppressing activity [18]. Epithelial cells that have been dysregulated adopt the elongated hummingbird phenotype [26]. In addition, cagA activates the transcription factor NF-kB leading to the induction of interleukin 8 (IL-8) and subsequent inflammation [27]. The activation of NF-kB occurs via SHP-2.
Variation in the EPIYA domains of cagA results in variation in the virulences of different cagA+ H.pylori strains [28]. The EPIYA motifs are located in the C-terminal half of the cagA protein and are of types A-D. The EPIYA motifs are the major sites of tyrosine phosphorylation within the cagA protein. The eastern EPIYA-D motif, found in asian populations, is associated with stronger binding to SHP-2, while the western EPIYA-C motif is not. The presence of the EPIYA-D motif in asian cagA sequences may be responsible for the high rates of H.pylori associated disease in asian populations [28].
The study reported here investigates the evolutionary dynamics of the cagA gene from different human populations, and shows that the gene displays varying amounts of positive selection, implying host population genetic differences in the response to H.pylori infection, and indicating the benefit of the gene to H.pylori. The region of the cagA gene under selection contains the EPIYA domains. These observations are an apparent paradox, given the detrimental effects of the oncoprotein on the human host; various scenarios are discussed that may explain the data.
Methods
Sequences and phylogenetic analysis
Complete cagA sequences from different human populations were obtained from the Genbank database (NCBI) and are listed in Table 1. Although isolated from a white american from Tennessee, the USA sequence has an african origin [29], hence it is denoted African(USA). There were two cagA genes in the Peruvian genome, denoted Peru1 and Peru2. There is an additional cagA gene in the Venezuelan genome, however this is likely to be a pseudogene because of a 119 amino acid deletion on the N terminus. Searching of the Genbank database, and other Helicobacter species did not reveal a significant homolog of cagA. DNA alignments were constructed by first aligning the protein sequences, using the MAFFT program [30], and then using this alignment as a template for a DNA alignment, using the PAL2NL program [31]. Bayesian phylogenetic inference of the cagA DNA sequences was conducted using the program MrBayes [32], using a GTR substitution model and a gamma parameter of 0.84, selected using the jModelTest program [33]. The simulation was run for 90000 generations, sampling every 100 generations. A burn-in of 25% was conducted and the consensus tree was constructed from the last 25% of the sampled generations.
Partial rRNA sequences for various Helicobacter species were obtained from Genbank; these were H.fennelliae (GenBank: AF348747), H.acinocychis (GenBank: NR_025940), H.pylori (GenBank: DQ202383), H.nemestrinae (GenBank: AF363064), H.heilmannii (GenBank: AF506794), H.cetorum (GenBank: FN565164), Helicobacter sp. ' solnick 9A1-T71' (GenBank: AF292381), H.bizzozeronii (GenBank: NR026372), H.salomonis (GenBank: NR026065) and H.felis (GenBank: NR025935). The sequences were aligned using the MAFFT program and phylogenetic relationships determined using MrBayes and a HKY model, selected using the jModelTest program. The simulation was run for 10000 generations, sampling every 100 generations. A burn-in of 25% was conducted and the consensus tree was constructed from the last 25% of the sampled generations.
Positive selection analysis
The cagA gene sequences were analyzed for the presence of positive selection by likelihood ratio testing, comparing nested models, null and alternative, using the PAML program [34]. Three tests were performed; the branches test [35, 36], sites test [37] and branches-sites test [38]. An unrooted tree without branch lengths was used for the analysis, generated by the phylogenetic analysis, and the codon frequency table option was utilized in all analyses. Likelihood ratio testing was conducted to determine the signficance of 2Δl, the differences between the log likelihoods of the two models (where l is the log likelihood), using a χ2 distribution with 12 degrees of freedom for the branches model, a χ2 distribution and 2 degrees of freedom for the sites model and a χ2 distribution with 1 degree of freedom for the branches-sites model. The null model used for the branches test was a one-ratio model where Ka/Ks (ω) was the same for all branches, while the alternative model was the free-ratio model where ω was allowed to vary. The null model for the sites test was model 1a (neutral; model = 0, NSsites = 1, fix_omega = 0), and the alternative model was model 2a (selection; model = 0, NSsites = 2, fix_omega = 0). The null model for the branches-sites test was modified according to Yang et al. [39] (neutral; model = 2, NSsites = 2, fix_omega = 1, omega = 1). The alternative model was model A (selection; model = 2, NSsites = 2, fix_omega = 0).
Results and discussion
Positive selection on cagA
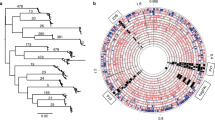
The topology of the phylogenetic tree of the complete H.pylori cagA sequences reproduces the relationships between different human populations around the world (Figure 1), and is consistent with larger scale studies using concatenated sequences that show that H.pylori has co-migrated with humans after their exit from Africa [3]. The reproduction of the evolutionary history of the human populations in the topology of the cagA tree therefore is the result of the tight association of H.pylori with its host [3, 40, 41]. The cagA sequence obtained from an Indian individual is located within the clade formed by european sequences, consistent with results showing that Indian cagA sequences intercalate with european sequences [42] and that most H.pylori from India are related to european strains [43]. The tree also indicates that the Peruvian cagA sequence has undergone a recent gene duplication; this is seen in the operon structure (Figure 2). Strong positive selection on Peru2 indicates that neofunctionalization of the gene is occurring. Presumably, the gene duplication results in gene dosage effects; how this affects the pathogenicity of the strain in unclear. The presence of a pseudogenized cagA gene in the H.pylori genome isolated from a Venezuelan amerindian (see Methods) is interesting; the reason for the disparity between the fates of the duplicated cagA genes in the two related strains is also unclear. The branch lengths on the phylogenetic tree show similarity to each other, with the exception of the Vietnamese lineage; this branch shows considerable accelerated evolution.

Positive selection on cagA from different H.pylori strains. A phylogenetic consensus tree was constructed as described in Methods using complete cagA gene sequences. Numbers above and below branches indicate the values of Ka/Ks calculated for each lineage using the PAML branches test, while numbers after slashes are posterior probabilities of the respective nodes. The scale refers to the average number of substitutions per site.

Diagram of the cag PaI from the Peru strain. Indicated in the figure is the position of the duplicated cagA gene.
2Δl was calculated as 73.6 for the branches test, which was statistically significant. Ka/Ks values of greater than 1 were observed for 5 branches (Figure 1); those leading to the Venezuela (1.56), Peru1 (1.04) and Peru2 (3.10) sequences, to the common ancestor of the amerindian sequences (1.03) and to the lineage leading from the common ancestor of the asian sequences (1.29). These branches are subject to positive selection, while the amerindian common ancestor is neutral over the length of the gene.
2Δl was calculated as 161 between the null and alternative models, for the sites test, which was statistically significant. Estimates of parameters were as follows: p0 = 0.51, p1 = 0.49, ω0 = 0.03, ω1 = 1 (neutral model), p0 = 0.47, p1 = 0.38, p3 = 0.14, ω0 = 0.03, ω1 = 1, ω2 = 3.74 (selection model). Sites identified as being under positive selection, with statistical significance according to the Bayes Empirical Bayes test [39], were: 101, 206, 306, 378, 532, 542, 548, 604, 651, 774, 793, 815, 831, 834, 869, 876, 886, 892, 901, 998, 1004. The numbering was based on the Peru1 sequence.
A branches-sites test was conducted on each branch of the tree. Those lineages found to display positive selection are listed in Table 2. These included the lineages previously identified by the branches test, and additionally the african, Italian, Swedish and Vietnamese lineages. The results showing positive selection in cagA isolated from various populations are consistent with a McDonald-Kreitman test that shows that partial cagA sequences isolated from the Mexican population are under positive selection [44]. Parallel evolution in residues or different regions of the cagA proteins is not observed, although residues in the 900 amino acid region are under stronger diversifying selection, when the Venezuelan and Peru2 genes are examined in a sliding window analysis (Figure 3). This is an interesting result as this region of the cagA gene encodes the EPIYA repeats, which have a role in modulating the carcinogenicity of the cagA gene. Thus, it would appear that the effects of diversifying selection may have a direct role in modulating carcinogenesis.

Sliding window analysis of two cagA genes. Genes from the Venezuela and Peruvian strains (Peru2) were analyzed. Sliding window analysis of a pairwise cagA alignment was conducted using the DNASP5.0 program [82], using the Nei and Gojobori [83] method of calculating Ka/Ks. The alignment was constructed as described in Methods. A sliding window of 100 nucleotides, with a step of 10 was used. Gaps were ignored.
Population specific differences in positive selection
Positive selection on cagA is likely to be due to avoidance of the adaptive immune response, IgG, or to enhance binding to cellular receptors which are antagonistically co-evolving. There is a strong immune response against the cagA protein (cagA is immunodominant); this may have led to an 'arms race' between host and bacteria, and hence the signature of positive selection. This is often the case with extracellular proteins of pathogens, either located on the cell surface or secreted. There is a precedent in bacteria, with the porB porin gene of Neisseria gonorrhoeae and meningitidis[45], and a variety of extracellular proteins from Escherichia coli[46]. Secreted slr proteins from H.pylori also show signatures of positive selection [47]. This scenario would imply that the regions of cagA under positive selection are immunogenic.
H.pylori cagA from a range of populations around the world show evidence of positive selection (using the branches-sites test); these include sequences from Venezuela, Vietnam, Sweden, Peru, Africa and Italy. However, as human and H.pylori strains have co-evolved, cagA genes from some strains have undergone stronger positive selection, particularly the strains with ancestry in the human groups that most recently migrated, the asians and the amerindians [48, 49]. The cause of the differences in strength of selection on the cagA genes presumably lies in genetic differences at the host level, but is also potentially mediated by different responses induced by the cagA protein, resulting from functional differences between different cagA proteins. The intra-population genetic distances are smaller in human groups as they migrated east out from Africa [50]. Host-specific differences may include differences in the immune response, or differences in the activities of cellular cagA binding proteins. Codon usage analysis (Table 3) indicates that the codon adaptation index is similar for different cagA genes, suggesting that there are no strong differences in translational selection between cagA genes from different H.pylori strains, which may indicate no major functional differences between genes or simply reflect the lack of translational selection on highly expressed genes genome-wide [51]. This data helps to inform the sliding window analysis; translational selection has been shown to result in false indications of positive selection [52]: this is not likely to be the case here due to the lack of translational selection on these genes.
Polymorphisms in the IL-1 gene cluster modify gastric cancer risk [53]. The induction of IL-8 secretion by the cag PaI is a major stimulus of the immune response [49]. Thus, differences in host interleukin genotypes may lead to differences in outcome for disease progression and differences in selective pressure on the cagA genes in different populations. Amerindians underwent a population bottleneck during the migration of their ancestors from Asia [48]. Phenotypic evidence of this is the universality of the O blood group amongst amerindians [54], this may have led to a homogeneity of immune response. This may have affected the strains capacity to bind non O human blood antigens; most H.pylori strains are able to bind the A,B and O antigens via the babA adhesin, while amerindian strains from South America bind best to O antigens [55]. It is interesting to note that the east asian population is also relatively genetically homogenous [49].
Both commensal and pathogenic bacteria possess mechanisms for the avoidance of the host immune system. Several mechanisms have been shown to be involved in avoidance of the immune system by H.pylori. However, cagA+ strains elicit a strengthened immune response and increased inflammation [56–58]. Inflammation may be a mechanism to obtain nutrients [59], however if cagA is evolving to avoid the immune system while at the same time stimulating it, then this seems contradictory.
Distribution of gastric cancer worldwide and its relationship with the strength of positive selection on cagA
There are great variations in the incidence of gastric cancer worldwide, with parts of East Asia and Latin America showing high incidences, while other parts of the world such as Africa and parts of Europe showing low incidences (Table 4). The incidence rates do not correlate with rates of infection with H.pylori. For instance, there are high rates of H.pylori associated pathogenicity in Japan, Korea and parts of China, but low in Thailand and Indonesia even though they have high infection rates; this is the 'Asian paradox' [60]. Instead, incidence appears to be linked to the frequency and genotype of cagA[61], while other factors are also likely to play a role such as altitude, diet and host genotype. In addition, recent work shows that recent migrations and population movements have resulted in the introduction of 'non-native' H.pylori strains with different cagA alleles into established human populations [42, 62], this gives an added level of complexity.
Given that amerindian and the ancestral asian cagA sequences show stronger signs of positive selection, and that asian and latin american populations can exhibit high incidences of gastric cancer, this might imply a link between the strength of positive selection on the cagA gene and the oncogenicity of the gene. The results of the sliding window analysis, where the cagA region containing the EPIYA domains is under positive selection, are consistent with this hypothesis. Further work is required. If verified, this form of sequence analysis may help identify at risk populations.
Evolutionary benefit of cagA to H.pylori
The signature of positive selection observed on the cagA gene indicates that the cagA protein is undergoing adaptive evolution in some strains, and is beneficial to the bacteria. Differences in rates of adaptation imply host specific differences. The benefit to the bacteria is mediated via the role of cagA within the pathogenicity island; the specific role of cagA, and that of the PaI, remain to be determined. In general, PaIs have a role in promoting survival of bacterial pathogens [63]. The positive selection observed on the cagA oncogene is unusual as it is the first case observed of positive selection on an oncogene in a vertically transmitted pathogen. Positive selection is a feature of antagonistic coevolution, which implies harmful effects on the host, but also mutualistic coevolution, which implies benefits. Positive selection has been observed on the Epstein Barr Virus - encoded oncogene LMP1 [64] and the human papillomavirus type 16 oncogene [65, 66], however these are horizontally transmitted pathogens where a balance is expected between virulence and transmissibility [67]. This may imply that H.pylori has been horizontally transmitted to a greater extent than previously recognized.
Virulence is a result of enhanced reproduction of a pathogen. Early models proposed that a parasite would be inclined to evolve reduced virulence, given that mortality of host is a disadvantage. However, this view has been criticized as relying on group selection [68]. However, vertically inherited pathogens are expected to become less pathogenic over time; if the pathogen depends on the host for transmission and the transmission is highly efficient then it is not in the interests of the pathogen to significantly reduce the fitness of the host [69]. H.pylori displays two features, in addition to the positive selection observed on cagA, that appear to contradict this paradigm. Firstly, the acquisition of the cag PaI during speciation from related non-pathogenic gut helicobacters (Figure 4a), indicates that H.pylori underwent an initial increase in pathogenicity. Second, the evolution of the more pathogenic EPIYA-D motifs in the cagA gene in some asian strains (Figure 4b), indicates that some cagA+ H.pylori has undergone a more recent additional increase in pathogenicity. To some extent, this contradiction could be explained by the proposal that there is actually a host - beneficial component to cagA, or that it has not exerted a sufficiently deleterious effect on the host. One question that requires answering is whether those strains that are undergoing a greater degree of positive selection are becoming more pathogenic.

Genetic factors leading to an increase in virulence of Helicobacter pylori. a) Small subunit rRNA phylogenetic consensus tree of bacterial species in the human digestive system related to H.pylori, showing the recent acquisition of cagA; b), the EPIYA domains that are present in the cagA gene and the evolution of the EPIYA-D domains in the asian lineages. Tree (a) was constructed as described in Methods, numerals indicate posterior probabilities, tree (b) as in Figure 1.
In addition, potential beneficial effects of cagA at the population level via elimination of the elderly has been suggested [13] (this explanation relies on the theory of inclusive fitness [70]). This essentially views cagA as a gene that enhances intrinsic mortality in old individuals, however it is unclear whether intrinsic mortality in a subgroup of the population has ever been selected for. While H.pylori has largely been considered a pathogen, there is increasing evidence of its positive benefits to human health. For instance, H.pylori has a beneficial role in preventing esophageal cancer, by reducing acid reflux [71, 72], however in the past this has been unlikely to have provided much evolutionary benefit to the human population given that over 90% of patients are over 55 [73], while before the 20th century the average life expectancy of human populations was less than 40. The strongest inverse correlation between esophageal cancer occurrence and infection with H.pylori is in East Asia, attributed to the highly interactive (eastern) form of cagA, which causes pan- and corpus- predominant gastritis and reduces acid production [13]. There is also an inverse relationship between H.pylori and asthma and allergies [74–76], obesity [77] and infant diarrhea [78]. Asthma and obesity are modern illnesses, so are unlikely to have played a role in the evolutionary dynamics of the bacteria.
Ulcers are a modern disease [79], while gastric cancer has been recorded since ancient times. However, it is most prevalent in 55 year olds and over, this indicates that historically it is unlikely to have exerted a strong selective pressure, given that before the 20th century the average life expectancy was considerably lower. These considerations lead to the conclusion that the cagA gene is either insufficiently deleterious to the human host, that the cagA protein has a beneficial component to the host, or that horizontal transmission has been an important feature of H.pylori in the recent past. There is increasing evidence that in developing countries, horizontal transmission of H.pylori occurs due to poor sanitary conditions [80, 81]. If there is (or has been) significant horizontal transmission, then there may be population specific differences in the amount of horizontal transmission which may have led to differences in selective pressures on the pathogen.
H.pylori has been utilized as a model for infective carcinogenesis, and is a model of pathogen evolution. The results of this work suggest that the cagA gene is insufficiently deleterious to the human host, that the cagA protein has a benefit to the host or that horizontal inheritance has affected the evolutionary dynamics of the bacteria more than recognized. The results reported here offer an insight into important aspects of microbe-host coevolution.
References
Bik EM: Molecular analysis of the bacterial microbiota in the human stomach. Proc Natl Acad Sci USA. 2006, 103: 732-737. 10.1073/pnas.0506655103.
Mbulaiteye SM, Hisada M, El-Omar EM: Helicobacter pylori associated global gastric cancer burden. Front Biosci. 2009, 14: 1490-1504.
Linz B: An African origin for the intimate association between humans and Helicobacter pylori. Nature. 2007, 445: 915-918. 10.1038/nature05562.
Gueneau P, Loiseaux-De Goer S: Helicobacter: molecular phylogeny and the origin of gastric colonization in the genus. Infect, Gen and Evol. 2002, 1: 215-223. 10.1016/S1567-1348(02)00025-4.
Weeks DL, Eskandari S, Scott DR, Sachs G: A H+-gated urea channel: the link between Helicobacter pylori urease and gastric colonization. Science. 2000, 287: 482-485. 10.1126/science.287.5452.482.
Censini S, Lange C, Xiang Z, Crabtree JE, Ghiara P, Borodovsky M, Rappuoli R, Covacci A: Cag, a pathogenicity island of Helicobacter pylori, encodes type I-specific and disease-associated virulence factors. Proc Natl Acad Sci USA. 1996, 93: 14648-14653. 10.1073/pnas.93.25.14648.
Tummuru MK, Cover TL, Blaser MJ: Cloning and expression of a high-molecular-mass major antigen of Helicobacter pylori: evidence of linkage to cytotoxin production. Infect Immun. 1993, 61: 1799-1809.
Gressman H, Linz B, Ghai R, Schlapbach R, Yamaoka Y, Kraft C, Suerbaum S, Meyer TF, Achtman M: Gain and loss of multiple genes during the evolution of Helicobacter pylori. PloS Gen. 2005, 1: e43-10.1371/journal.pgen.0010043.
Blaser MJ, Perez-Perez GI, Kleanthous H, Cover TL, Peek RM, Chyou PH, Stemmermann GN, Nomura A: Infection with Helicobacter pylori strains possessing cagA is associated with an increased risk of developing adenocarcinoma of the stomach. Cancer Res. 1995, 55: 2111-2115.
Nomura N, Lee J, Stemmermann GN, Nomura RY, Perez-Perez GI, Blaser MJ: Helicobacter pylori CagA seropositivity and gastric carcinoma risk in a Japanese American population. J Infect Dis. 2002, 186: 1138-1144. 10.1086/343808.
Personnet J, Friedman GD, Orentreich N, Vogelman H: Risk for gastric cancer in people with CagA positive or CagA negative Helicobacter pylori infection. Gut. 1997, 40: 297-301.
Parkin DM, Bray F, Ferlay J, Pisani P: Global cancer statistics, 2002. CA Cancer J Clin. 2005, 55: 74-108. 10.3322/canjclin.55.2.74.
Atherton JC, Blaser MJ: Coadaptation of Helicobacter pylori and humans: ancient history, modern implications. J Clin Invest. 2009, 119: 2475-2487. 10.1172/JCI38605.
Backert S, Selbach M: Role of type IV secretion in Helicobacter pylori pathogenesis. Cell Microbiol. 2008, 10: 1573-1581. 10.1111/j.1462-5822.2008.01156.x.
Selbach M, Moese S, Hauck CR, Meyer TF, Backert S: Src is the kinase of the Helicobacter pylori CagA protein in vitro and in vivo. J Biol Chem. 2002, 277: 6775-6778. 10.1074/jbc.C100754200.
Stein M, Bagnoli F, Halenbeck R, Rappuoli R, Fantl WJ, Covacci A: c-Src/Lyn kinases activate Helicobacter pylori CagA through tyrosine phosphorylation of the EPIYA motifs. Mol Microbiol. 2002, 43: 971-980. 10.1046/j.1365-2958.2002.02781.x.
Poppe M, Feller SM, Römer G, Wessler S: Phosphorylation of Helicobacter pylori CagA by c-Abl leads to cell motility. Oncogene. 2007, 26: 3462-3472. 10.1038/sj.onc.1210139.
Higashi H, Tsutsumi R, Muto S, Sugiyama T, Azuma T, Asaka M, Hatakeyama M: SHP-2 tyrosine phosphatase as an intracellular target of Helicobacter pylori CagA protein. Science. 2002, 295: 683-686. 10.1126/science.1067147.
Saadat I, Higashi H, Obuse C, Umeda M, Murata-Kamiya N, Saito Y, Lu H, Ohnishi N, Azuma T, Suzuki A, Ohno S, Hatakeyama M: Helicobacter pylori CagA targets PAR1/MARK kinase to disrupt epithelial cell polarity. Nature. 2007, 447: 330-333. 10.1038/nature05765.
Mimuro H, Suzuki J, Tanaka J, Asahi M, Haas R, Sasakawa H: Grb2 is a key mediator of Helicobacter pylori CagA protein activities. Mol Cell. 2002, 10: 745-755. 10.1016/S1097-2765(02)00681-0.
Churin Y, Al-Ghoul L, Kepp O, Meyer TF, Birchmeier W, Naumann M: Helicobacter pylori CagA protein targets the c-Met receptor and enhances the motogenic response. J Cell Biol. 2003, 161: 249-255. 10.1083/jcb.200208039.
Tsutsumi R, Higashi H, Higuchi M, Okada M, Hatakeyama M: Attenuation of Helicobacter pylori CagA °-- SHP-2 signaling by interaction between CagA and C-terminal Src kinase. J Biol Chem. 2003, 278: 3664-3670. 10.1074/jbc.M208155200.
Suzuki M, Mimuro H, Suzuki T, Park M, Yamamoto T, Sasakawa C: Interaction of CagA with Crk plays an important role in Helicobacter pylori induced loss of gastric epithelial cell adhesion. J Exp Med. 2005, 202: 1235-1247. 10.1084/jem.20051027.
Hatakeyama M: Helicobacter pylori CagA--a potential bacterial oncoprotein that functionally mimics the mammalian Gab family of adaptor proteins. Microbes and Infections. 2003, 5: 143-150. 10.1016/S1286-4579(02)00085-0.
Botham CM, Wandler AM, Guillemin K: A transgenic Drosophila model demonstrates that the Helicobacter pylori CagA protein functions as a eukaryotic Gab adaptor. PLOS Pathogens. 2008, 4: e1000064-10.1371/journal.ppat.1000064.
Segal ED, Cha J, Lo J, Falkow S, Tompkins LS: Altered states: Involvement of phosphorylated CagA in the induction of host cellular growth changes by Helicobacter pylori. Proc Natl Acad Sci USA. 1999, 96: 14559-14564. 10.1073/pnas.96.25.14559.
Brandt S, Kwok T, Hartig R, Konig W, Backert S: NF-kappaB activation and potentiation of proinflammatory responses by the Helicobacter pylori CagA protein. Proc Natl Acad Sci USA. 2005, 102: 9300-9305. 10.1073/pnas.0409873102.
Higashi H, Tsutsumi R, Fujita A, Yamazaki S, Asaka M, Azuma T, Hatakeyama M: Biological activity of the Helicobacter pylori virulence factor CagA is determined by variation in the tyrosine phosphorylation sites. Proc Natl Acad Sci USA. 2002, 99: 14428-14433. 10.1073/pnas.222375399.
Falush D: Traces of human migrations in Helicobacter pylori populations. Science. 2003, 299: 1582-1585. 10.1126/science.1080857.
Katoh K, Kuma K, Toh H, Miyata T: MAFFT version 5: improvement in accuracy of multiple sequence alignment. Nuc Acid Res. 2005, 33: 511-518. 10.1093/nar/gki198.
Suyama M, Torrents D, Bork P: PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nuc Acids Res. 2006, 34: W609-W612. 10.1093/nar/gkl315.
Ronquist F, Huelsenbeck JP: MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003, 19: 1572-1574. 10.1093/bioinformatics/btg180.
Posada D: jModelTest: Phylogenetic Model Averaging. Mol Biol Evol. 2008, 25: 1253-1256. 10.1093/molbev/msn083.
Yang Z: PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007, 24: 1586-1591. 10.1093/molbev/msm088.
Yang Z: Likelihood ratio tests for detecting positive selection and application to primate lysozyme evolution. Mol Biol Evol. 1998, 15: 568-573.
Nielsen R, Yang Z: Likelihood models for detecting positively selected amino acid sites and applications to the HIV-1 envelope gene. Genetics. 1998, 148: 929-936.
Yang Z, Nielsen R: Codon-substitution models for detecting molecular adaptation at individual sites along specific lineages. Mol Biol Evol. 2002, 19: 908-917.
Anisimova M, Yang Z: Multiple hypothesis testing to detect lineages under positive selection that affects only a few sites. Mol Biol Evol. 2007, 24: 1219-1228. 10.1093/molbev/msm042.
Yang Z, Wong WSW, Nielsen R: Bayes empirical bayes inference of amino acid sites under positive selection. Mol Biol Evol. 2005, 22: 1107-1118. 10.1093/molbev/msi097.
Olberman P, Josenhans C, Moodley Y, Uhr M, Stamer C, Vauterin M, Suerbaum S, Achtman M, Linz B: A global overview of the genetic and functional diversity in the Helicobacter pylori cag pathogenicity island. PloS Genet. 2010, 19: e1001069-
Dominguez-Bello MG, Blaser MJ: The human microbiota as a marker for migrations of individuals and populations. Ann Rev Anthropol. 2011, 40: 451-474. 10.1146/annurev-anthro-081309-145711.
Devi SM, Ahmed I, Francalacci P, Hussain MA, Akhter Y, Alvi A, Sechi LA, Megraud F, Ahmed N: Ancestral European roots of Helicobacter pylori in India. BMC Genomics. 2007, 8: 184-10.1186/1471-2164-8-184.
Breurec S: Evolutionary history of Helicobacter pylori sequences reflect past human migrations in southeast asia. PLoS One. 2011, 6: e22058-10.1371/journal.pone.0022058.
Torres-Morquecho A, Giono-Cerezo S, Camorlinga-Ponce M, Vargas-Mendoza CF, Torres J: Evolution of bacterial genes: evidence of positive Darwinian selection and fixation of base substitutions in virulence genes of Helicobacter pylori. Inf, Genetics and Evol. 2010, 10: 764-776. 10.1016/j.meegid.2010.04.005.
Smith NH, Maynard Smith J, Spratt BG: Sequence Evolution of the porB Gene of Neisseria gonorrhoeae and Neisseria meningitidis: Evidence of Positive Darwinian Selection. Mol Biol Evol. 1995, 12: 363-370.
Petersen L, Bollback JP, Dimmic M, Hubisz M, Nielsen R: Genes under positive selection in Escherichia coli. Genome Res. 2007, 17: 1336-1343. 10.1101/gr.6254707.
Ogura M, Perez JC, Mittl PRE, Dailide G, Tan S, Ito Y, Secka O, Dailidiene D, Putty K, Berg DE, Kalia A: Helicobacter pylori evolution: lineage-specific adaptations in homologs of eukaryotic Sel1-like genes. PloS Comp Biol. 2007, 3: e151-10.1371/journal.pcbi.0030151.
Bonatto SL, Salzano FM: A single and early migration for the peopling of the Americas supported by mitochondrial DNA sequence data. Proc Natl Acad Sci USA. 1997, 94: 1866-1871. 10.1073/pnas.94.5.1866.
Oota H, Kitano T, Jin F, Yuasa I, Wang L, Ueda S, Saitou N, Stoneking M: Extreme mtDNA homogeneity in continental asian populations. Am J Phys Anthropol. 2002, 118: 146-153. 10.1002/ajpa.10056.
Liu H, Prugnolle F, Manica A, Balloux F: A geographically explicit genetic model of worldwide human-settlement history. Am J Hum Genet. 2006, 79: 230-237. 10.1086/505436.
dos Reis M, Savva R, Wernisch L: Solving the riddle of codon usage preferences: a test for translational selection. Nuc Acids Res. 2004, 32: 5036-5044. 10.1093/nar/gkh834.
Parmley JL, Hurst L: How common are intragene windows with Ka > Ks owing to purifying selection on synonymous mutations?. J Mol Evol. 2007, 64: 646-655. 10.1007/s00239-006-0207-7.
El-Omar EM, Carrington M, Chow WH, McColl KE, Bream JH, Young HA, Herrera J, Lissowska J, Yuan CC, Rothman N, Lanyon G, Martin M, Fraumeni JF, Rabkin CS: Interleukin-1 polymorphisms associated with increased risk of gastric cancer. Nature. 2000, 404: 398-402. 10.1038/35006081.
Dominguez-Bello MG, Perez ME, Bortolini MC, Salzano FM, Pericchi LR, Zambrano-Guzman O, Linz B: Amerindian Helicobacter pylori strains go extinct, as European strains expand their host range. PloS One. 2008, 3: e3307-10.1371/journal.pone.0003307.
Aspholm-Hurtig M, Dailide G, Lahmann M, Kalia A, Ilver D: Functional adaptation of BabA, the H pylori ABO blood group antigen binding adhesin. Science. 2004, 305: 519-522. 10.1126/science.1098801.
Crabtree JE, Covacci A, Farmery SM, Xiang Z, Tompkins DS, Perry S, Lindley IJD, Rappuoli R: Helicobacter pylori induced interleukin-8 expression in gastric epithelial cells is associated with CagA positive phenotype. J Clin Pathol. 1995, 48: 41-45. 10.1136/jcp.48.1.41.
Peek RMJ, Miller GG, Tham KT, Perez-Perez GI, Zhao X, Atherton JC, Blaser MJ: Heightened inflammatory response and cytokine expression in vivo to cagA+Helicobacter pylori strains. Lab Invest. 1995, 71: 760-770.
Yamaoka Y, Kita M, Kodama T, Sawai N, Kashima K, Imanishi J: Induction of various cytokines and development of severe mucosal inflammation by cagA gene positive Helicobacter pylori strains. Gut. 1997, 41: 442-451. 10.1136/gut.41.4.442.
Baldary CT, Lanzavecchia A, Telford JL: Immune subversion by Helicobacter pylori. Trends Immun. 2005, 26: 199-207. 10.1016/j.it.2005.01.007.
Prinz C, Schwendy S, Voland P: Hpylori and gastric cancer: shifting the global burden. World J Gastroenterol. 2006, 12: 5458-5464.
Bravo LE, van Doom LJ, Realpe JL, Correa P: Virulence-associated genotypes of Helicobacter pylori: do they explain the African enigma?. Am J Gastroenterol. 2002, 97: 2899-2842.
Breurec S: Expansion of European vacA and cagA alleles to East Asian Helicobacter pylori strains in Cambodia. Infect Genet Evol. 2011,
Juhas M, van der Meer JR, Gaillard M, Harding RM, Hood DW, Crook DW: Genomic islands: tools of bacterial horizontal gene transfer and evolution. FEMS Microbiol Rev. 2009, 33: 376-393. 10.1111/j.1574-6976.2008.00136.x.
Burrows JM, Bromham L, Woolfit M, Piganeau G, Tellam J, Connolly G, Webb N, Poulsen L, Cooper L, Burrows SR, Moss DJ, Haryana SM, Ng M, Nicholls JM, Khanna R: Selection pressure-driven evolution of the Epstein-Barr virus encoded oncogene LMP1 in virus isolates from Southeast Asia. J Virol. 2004, 78: 7131-7137. 10.1128/JVI.78.13.7131-7137.2004.
DeFilippis VR, Ayala FJ, Villarreal LP: Evidence of diversifying selection in human papillomavirus type 16 E6 but not E7 oncogenes. J Mol Evol. 2002, 55: 491-499. 10.1007/s00239-002-2344-y.
Chen Z, Terai M, Fu L, Herrero R, DeSalle R, Burk RD: Diversifying selection in human papillomavirus type 16 lineages based on complete genome analyses. J Virol. 2005, 79: 7014-7023. 10.1128/JVI.79.11.7014-7023.2005.
Anderson RM, May RM: Coevolution of hosts and parasites. Parasitology. 1982, 85: 411-426. 10.1017/S0031182000055360.
Lenski RE, May RM: The evolution of virulence in parasites and pathogens: reconciliation between two competing hypotheses. J Theor Biol. 1994, 169: 253-265. 10.1006/jtbi.1994.1146.
Fine PEM: Vectors and vertical transmission: an epidemiologic perspective. Ann NY Acad Sci. 1975, 266: 173-194. 10.1111/j.1749-6632.1975.tb35099.x.
Hamilton WD: The genetical evolution of social behaviour I and II --. Journal of Theoretical Biology. 1964, 7: 1-16. 10.1016/0022-5193(64)90038-4. 17-52
Blaser MJ: Disappearing microbiota Helicobacter pylori protection against esophageal adenocarcinoma. Cancer Prev Res (Phila Pa). 2008, 1: 308-311. 10.1158/1940-6207.CAPR-08-0170.
Islami F, Kamangar F: Helicobacter pylori and esophageal cancer risk: a meta-analysis. Cancer Prev Res (Phila Pa). 2008, 1: 329-338. 10.1158/1940-6207.CAPR-08-0109.
Christie J, Shepherd N, Codling B, Valori R: Gastric cancer below the age of 55: implications for screening patients with uncomplicated dyspepsia. Gut. 1997, 41: 513-517. 10.1136/gut.41.4.513.
Chen Y, Blaser MJ: Inverse associations of Helicobacter pylori with asthma and allergy. Arch Intern Med. 2007, 167: 821-827. 10.1001/archinte.167.8.821.
Chen Y, Blaser MJ: Helicobacter pylori colonization is inversely associated with childhood asthma. J Infect Dis. 2008, 198: 553-560. 10.1086/590158.
Codolo G: The neutrophil-activating protein of Helicobacter pylori down-modulates Th2 inflammation in ovalbumin-induced allergic asthma. Cell Microbiol. 2008, 10: 2355-2363. 10.1111/j.1462-5822.2008.01217.x.
Kamada T, Hata J, Kusonoki H, Ito M, Tanaka S, Kawamura Y, Chayama K, Haruma K: Eradication of Helicobacter pylori increases the incidence of hyperlipidaemia and obesity in peptic ulcer patients. Dig Liver Dis. 2005, 37: 39-43. 10.1016/j.dld.2004.07.017.
Rothenbascher D, Blaser MJ, Bode G, Brenner H: Inverse relationship between gastric colonization of Helicobacter pylori and diarrheal illnesses in children: results of a population-based cross-sectional study. J Infect Dis. 2000, 182: 1446-1449. 10.1086/315887.
Baron JH, Sonnenberg A: Period- and cohort-age contours of deaths from gastric and duodenal ulcer in New York 1804-1998. Am J Gastroenterol. 2001, 96: 2887-2891.
Herrera PM, Mendez M, Velapatio B, Santivaez L, Balqui J, Finger SA, Sherman J, Zimic M, Cabrera L, Watanabe J, Rodriguez C, Gilman RH, Berg DE: DNA-level diversity and relatedness of Helicobacter pylori strains in shantytown families in Peru and transmission in a developing country setting. J Clin Microbiol. 2008, 46: 3912-3918. 10.1128/JCM.01453-08.
Schwarz S, Morelli G, Kusecek B, Manica A, Balloux F, Owen RJ, Graham DY, van der Merwe S, Achtman M, Suerbaum S: Horizontal versus familial transmission of Helicobacter pylori. PloS Pathogens. 2008, 4: e1000180-10.1371/journal.ppat.1000180.
Librado P, Rozas J: DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009, 25: 1451-1452. 10.1093/bioinformatics/btp187.
Nei M, Gojobori T: Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol Biol Evol. 1986, 3: 418-426.
Acknowledgements
This work was supported by the Biology Department, University of Puerto Rico - Rio Piedras.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
GD and SM conducted the analyses, MD and SM designed the study. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Delgado-Rosado, G., Dominguez-Bello, M.G. & Massey, S.E. Positive selection on a bacterial oncoprotein associated with gastric cancer. Gut Pathog 3, 18 (2011). https://doi.org/10.1186/1757-4749-3-18
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1757-4749-3-18