
Abstract
Regulatory DNA elements such as enhancers, silencers and insulators are embedded in metazoan genomes, and they control gene expression during development. Although they fulfil different roles, they share specific properties. Herein we discuss some examples and a parsimonious model for their function is proposed. All are transcription units that tether their target promoters close to, or distant from, transcriptional hot spots (or 'factories').
Similar content being viewed by others
Introduction
The complex linear organisation [1] of many metazoan genomes encodes regulatory sequences that can be categorised into two major groups: enhancers and silencers. Enhancers are short motifs that contain binding sites for transcription factors; they activate their target genes without regard to orientation and often over great separations in cis or in trans[2]. Silencers suppress gene expression [3] and/or confine it within specific chromatin boundaries (and thus are also called 'insulators') [4]. The interplay between these contrasting regulatory elements, their target promoters and epigenetic modifications at all levels of three-dimensional organisation (that is, nucleosomes, chromatin fibres, loops, rosettes, chromosomes and chromosome location) [5–9] fine-tune expression during development and differentiation. However, the mechanisms involved in this interplay remain elusive, although some can be computationally predicted [10]. Although enhancers and silencers have apparently opposite effects, accumulating evidence suggests they share more properties than intuition would suggest [11]. Herein we try to reconcile their apparently disparate modes of action. We suggest they act by tethering their target promoters close to, or distant from, hot spots of nucleoplasmic transcription (known as 'transcription factories') as they produce noncoding transcripts (ncRNAs) [12–15].
Enhancers
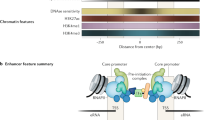
Enhancers were characterised almost 30 years ago [16], but their functional definitions vary because of their flexibility of action (whether in cis or in trans) [17, 18], position (relative orientation and/or distance) and genomic location (in gene deserts, introns and/or untranslated regions) [2]. Although sequence conservation between species can, in some cases, be an efficient predictor of enhancer identity, there are examples where genes with identical expression patterns in different species rely on enhancers that bear no similarities [19]. Within a single genome, however, sensitivity to DNase I and characteristic modifications of histone tails provide a more reliable means of identification. They typically occupy approximately 200 bp of 'open' chromatin (making them DNase-sensitive) [20], are flanked by regions rich in mono- and/or dimethylated lysine 4 of histone H3 (H3K4me1/H3K4me2) and acetylated lysine 27 of histone H3 (H3K27ac) and, generally, bind p300 [21]. Attempts have been made to classify enhancers into subclasses that are differentially used during development. Comparison between mouse embryonic stem (ES) cells, their differentiated derivatives and terminally differentiated murine cells allow distinctions between 'active', 'intermediate' and 'poised' enhancers (here additional marks are used, for example, H3K27me3 or H3K36me3) [21]. These accessible DNA stretches are often bound (and can thus be identified) by acetyltransferase p300, Mediator subunits, chromodomain helicase DNA binding protein 7, cohesin and/or CCCTC-binding factor (CTCF) [21, 22]. Most importantly, canonical enhancers are characterised by the presence of bound RNA polymerase II (RNAPII) [23, 24].
The first and most studied example of gene regulation by an enhancer is provided by the β-globin locus; here, the locus control region (LCR) is located 40 to 60 kb upstream from the promoter it regulates. The two interact when the chromatin fibre forms new, or rearranges preexisting, loops [17, 25]. All other cis-regulatory elements in this locus are also in close proximity, where they form an 'active chromatin hub' [12, 26]. An active chromatin hub, as defined in the β-globin locus paradigm, arises from the three-dimensional clustering of DNA-hypersensitive sites, depends on specific DNA-protein interactions and brings together all essential components for transcriptional activation [17]. Similarly, in a comprehensive study of the immunoglobulin heavy-chain locus [6] (and many other loci), the multitude of preexisting loops and connecting regulatory elements are rearranged to form new ones that interact upon activation. Obviously, most of these conformations (and in fact most seen using chromosome conformation capture (3C)) concern a population of cells and will not be refined until single-cell 3C is developed and implemented.
Enhancers are transcribed into RNAs (eRNAs) that do not encode proteins, run the length of the enhancer sequence and appear to stabilise enhancer-promoter interactions [11, 24, 27–29]. eRNAs derived from elements upstream of the Arc promoter depend on the activity of that promoter, as removing the promoter abolishes eRNA production [28]. β-globin-associated ncRNAs are still produced in the absence of the β-globin promoter [28, 30, 31]. However, the rate at which eRNAs are turned over, the exact mechanism by which they function and their abundance (relative to the mRNAs they regulate) all remain to be determined.
An additional class of ncRNAs longer than 200 nucleotides (long intergenic ncRNAs (lincRNAs)) were found in a survey of human transcripts, and some exhibited enhancer function [27]. In different human cells, more than 3,000 lincRNAs have now been identified [32, 33]. Some seem essential for the activation of the thymidine kinase promoter, as well as for the expression of neighbouring protein-coding genes (although not all act as bona fide enhancers) [34]. For example, HOTTIP (a lincRNA transcribed from the 5' end of the HOXA locus) coordinates the activation of several HOXA genes; chromatin looping brings HOTTIP close to its targets, and this drives H3K4 trimethylation and transcription [35].
Silencers
At the opposite functional extreme lie silencers. They prevent gene expression during differentiation and progression through the cell cycle [36]. This again correlates with RNA production (in some cases, through the generation of RNA duplexes that underlie the methylation of DNA at the promoter [37, 38]).
Accumulating evidence supports a broad and general role of both long and short RNA molecules in transcriptional inhibition. Antigene RNAs (agRNAs) are small RNAs that target promoters and downstream regions [37]. The expression of genes encoding progesterone, low-density lipoprotein, the androgen receptor, cyclooxygenase-2, the major vault protein and huntingtin is inhibited by agRNAs [37, 39]. Similarly, miRNAs, which are 20 to 22 nucleotides long, regulate gene expression post-transcriptionally [40], and they may also act at the level of transcriptional initiation or elongation. This is now supported by deep sequencing of nuclear and cytoplasmic small RNA libraries, where the majority of mature miRNAs localise in the nucleus (and not only in the cytoplasm) [41]. For instance, introduction of miRNA mimics that target the progesterone gene promoter decreases RNAPII occupancy. It also increases H3K9me2 levels in an Argonaute 2 (Ago-2)-dependent manner and leads to gene silencing [42]. Note that mature miRNAs in the nucleus can also act as 'enhancers' [43].
Polycomb complexes PRC1 and PRC2 rely on noncoding transcripts from silencing elements for recruitment to target sites. A range of examples are available: for instance, repression in cis in CD4+ T-cells and ES cells (where PRC2-catalysed H3K27 trimethylation recruits PRC1 to prevent chromatin remodelling of targeted loci [44]) and the PRC2-HOTAIR interaction (where transcripts produced from the XOXC locus establish repression of XOXD[33]). In human breast cancer cells, overexpression of HOTAIR results in the promiscuous association of PRC2 with more than 850 targets, which are in turn silenced [45]. Furthermore, in the well-studied cascade of X chromosome inactivation, the ncRNA Xist binds PRC2, which in turn drives H3K27 trimethylation [46, 47] and propagation of PRC1's binding to multiple sites along the silenced allele [48]. Here the three-dimensional conformation is also critical for efficient silencing and results in chromatin compaction and/or rearrangement [49]. Such equilibria may, however, be shifted by the eviction of Polycomb proteins to restore an active state [47].
Insulators
Functionally autonomous domains are strung along the chromatin fibre, and these need to be insulated from their neighbours to prevent the action of irrelevant enhancers and silencers. Insulator or boundary elements perform this task. These can be further categorised as enhancer blockers (when the insulator is located between a promoter and a cognate enhancer) and barriers (when located between a promoter and a silencer) [50]. Mutating or deleting insulators alters the pattern of gene expression and leads to developmental defects [51].
It has been suggested that insulators evolved from a class of promoters binding a specific subset of transcription factors that drive chromatin remodelling and long-range interactions [11]. Many are marked by DNase I hypersensitivity [52] and/or the presence of bound RNAPII. Specifically, in the Drosophila Hox gene cluster, stalled polymerases, in conjunction with elongation factors DISF and NELF, insulate four of eight promoters from Hox enhancers, and this correlates with the rearrangement and/or de novo formation of chromatin loops [53].
Perhaps the most abundant protein associated with insulator activity is CTCF. In the well-studied example of the Igf2-H19 imprinted locus, CTCF prevents activation of the maternal Igf2 allele by a distal enhancer. When its cognate binding site is lost, the gene is reactivated [54]. However, in this locus, CTCF is a positive regulator of the H19 gene [45]. Moreover, CTCF mediates enhancer-promoter, insulator-insulator and insulator-promoter interactions [11]. The insulator function of CTCF is regulated by cohesins [55, 56]. Their respective binding sites coincide in various cell types, including the IL-3 and granulocyte-macrophage colony-stimulating factor loci [57], as well as the renin, ETNK2[58], CFTR[50, 52] and c-Myc genes [59].
However, the CTCF-cohesin duplet is characteristic of only one type of insulator or boundary. In a comprehensive mapping of such Drosophila elements, additional factors, such as boundary element associated factor, GAGA and CP190, were used to identify and classify domain boundaries [60]. Again, DNase I hypersensitivity characterises many of these elements, and examples exist where their function is Ago-2-dependent (and so transcription-dependent, but RNAi-independent) [61].
A model
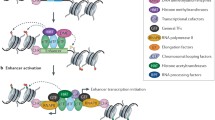
The following four models have been proposed to describe gene regulation by enhancers (Figure 1). (1) According to the tracking model, a protein loads onto the enhancer and tracks along the chromatin fibre towards the promoter, where it stimulates transcription [62]. (2) The linking model is similar, but here the loaded protein drives polymerisation of proteins in the direction of the promoter [63]. (3) In the relocation model, a given gene relocates to compartments in the nucleus where enhancer-promoter interactions (and so transcription) are favoured [64, 65]. (4) The looping model (which shares features with the relocation model) predicts a direct contact between an enhancer and a relevant promoter that loops out the intervening DNA [12, 65, 66] and thus is closely linked to the three-dimensional genome architecture [1, 7, 65]. Next, activators bound to the enhancer interact with the mediator complex, which recruits RNAPII and general transcription factors to the promoter [34, 67]. This last model is now favoured, as it readily explains enhancer-promoter interactions in trans[18, 68] and is supported by a wealth of experimental data derived from 3C [69] and modelling [1, 6–10, 15].

Existing models for the function of enhancers. The four existing models describing gene regulation by enhancers are depicted. (A) The tracking model, where a transcription factor (purple hexagon) loads onto the enhancer and tracks along the chromatin fibre towards the promoter, where it stimulates transcription by association with the polymerase (pink oval). (B) The linking model, where the loaded transcription factor drives polymerization of proteins in the direction of the promoter. (C) The relocation model, where a gene relocates to nuclear subcompartments (pink halo) favouring enhancer-promoter interactions, and so transcription. (D) The looping model, where the enhancer comes into proximity with the relevant promoter due to protein-protein interactions. This loops out the intervening chromatin and triggers transcriptional activation.
Similarly, among the three major models proposed for insulator function (roadblock, sink/decoy and topological loop models), the topological loop model is best supported by experimental data: Rearrangement and/or de novo formation of appropriately oriented loops efficiently insulate promoters from enhancer elements [70]. Note also that recent data show how gene repression dependent on gypsy insulators in Drosophila propagates between distant loci to be repressed via the organisation of local loops [71].
Gene regulation from distal regulatory elements via local looping or broader rearrangements in three-dimensional organisation is now widely accepted. For example, we have seen that the β-globin LCR loops back to its target promoter to activate it [17] through an active chromatin hub [12, 26], whereas Gata-1 represses the Kit gene locus via specific loop formation and exchange with Gata-2 reforms the enhancer-promoter loop and reactivates expression [72]. The IgH locus is another example of how this might occur, because its approximately 2.7-Mbp region is reorganised spatially during activation [6]. Similarly, various transcription factors have been implicated in forming regulatory chromatin loops, including EKLF [26]; Gata-1, Gata-2 and Gata-3 [72]; CTCF [73, 74]; Ldb1 [75]; and cohesin [56, 76]. Knocking them out or down results in loss of looping and changes in transcriptional state [26, 77, 78].
On a broader scale, the genome is organised nonrandomly in three-dimensional space [1, 6–10, 15] as a result of a variety of chromatin loops and rosettes [15, 64, 79], and the idea that transcription is also architecturally organised is gradually gaining ground [13–15]. It has been proposed that the transcription of protein-coding genes occurs in nucleoplasmic hot spots (that is, transcription factories) where a high local concentration of the required molecular machinery renders the whole process more efficient [14, 15]. By definition, these harbour at least two RNA polymerases, each transcribing a different template. The β-globin active chromatin hub can be classified as a transcription factory, as it contains at least two polymerases: one transcribing the enhancer and another transcribing a protein-coding gene. Not only do active genes tend to colocalise in the nucleus to be transcribed [80, 81], but different types of genes seem to cluster in 'specialised' transcription factories, where they are coregulated and expressed. For example, RNAPII genes are transcribed in separate factories from RNAPIIIgenes, whereas erythropoietic genes and TNFα-responsive genes are copied at sites distinct from those of constitutive and/or nonresponsive ones [75, 82–88]. Although factories with different polymerising activities can now be isolated and their proteins characterised using mass spectrometry [89], the mechanism by which factories are 'marked' by specific transcription factors and the relative representation of different subtypes of factories remain undetermined.
How can these ideas be extended to explain the function of enhancers and silencers and/or insulators? As we have established, all share common features (for example, DNase I hypersensitivity, active chromatin marks and interaction with transcription factors and RNAPII); therefore, we propose that canonical regulatory elements are primarily transcription units (Table 1) and that, in order for them to be functional, they need to be transcribed (and so associated with a transcription factory). This hypothesis defines two key aspects of chromatin structure: proximity between distant DNA sequences due to looping and tethering of active genes to a factory.
Does the number of factories in a given cell suffice to accommodate all transcription units, including enhancers and/or silencers? To date, the lowest estimate of about 200 factories concerns murine primary cells and comes from RNAPII immunostaining ex vivo[81]. This suggests that about 80 transcription units would share a factory (assuming 16,000 active transcription units, as in HeLa cells) [86] or that a number of them are transcribed outside a factory. Other approaches in HeLa cells return a number that is an order of magnitude higher: approximately 2,000 factories, each hosting an average of 8 transcription units [97, 98]. Moreover, the density and diameter of these transcriptional hot spots appear to be constant between cell types, suggesting an underlying topology accessible to transcription units in different nuclear neighbourhoods [86, 99]. The difference between these numbers may be explained by a difference in sensitivity of detection [86, 98]. But does most transcription occur in factories? It seems it may, as some estimates indicate that more than 95% of nascent nucleoplasmic RNA is found in factories (assessed using incorporation of various precursors in a variety of cell types) [13, 97–99]. Nonetheless, these issues will probably be resolved only by imaging factories in different types of living cells.
Now consider that an enhancer (transcription unit 1) (Figure 2A) tethers its target promoter (in unit 2) close to factory or hub A that contains the necessary machinery. As a result, the target promoter 2 will diffuse through the nucleoplasm and frequently collide with a polymerase in factory A to initiate transcription. Although promoter 3 is also tethered close to the same factory, it will initiate rarely (because factory A lacks the necessary transcription factors required by this particular promoter). Although promoter 3 can initiate in factory B (which contains high concentrations of the relevant factors), it will do so rarely, simply because it is tethered close to factory A and far from B. Next, transcription unit 1 acts as an enhancer of unit 2 and as a silencer of unit 3. The addition of histone modifications that mark the various units as active or inactive will now reinforce the status quo. After that, once unit 1 has been transcribed, these marks will make it more likely that unit 1 or unit 2 will reinitiate in factory A to create a virtuous cycle. Similarly, at another developmental stage, when a different set of transcription factors are expressed (Figure 2B), unit 1 might be transcribed in factory C. It is again flanked by units 2 and 3, but these can now be transcribed efficiently only in factory B (which is rich in the necessary factors). As units 2 and 3 cannot stably interact with each other by binding to factory C, unit 1 now acts as an insulator or barrier. As before, histone marks will reinforce this different (virtuous) cycle.

A simple model for the function of regulatory elements. Spheres A, B and C represent factories rich in different sets of transcription factors and associated halos indicate the probability that promoter 1, 2 or 3 will collide with a factory (red indicates high probability). The low-probability zone immediately around the factory arises because the intrinsic stiffness of the chromatin fibre restricts the formation of very small loops). Curved black arrow indicates collision between promoter and factory that yields a productive initiation. Dashed grey arrows indicate the preferred site of initiation (as factory B is rich in the relevant transcription factors). Blocked red arrows indicate unproductive collisions (as the factory contains few of the relevant factors). (A) Enhancers and silencers. Transcription unit 1 is being transcribed by a polymerase in factory A. This tethers unit 2 in a 'hot zone', where it has a high probability of colliding with a polymerase in factory A (which contains high local concentrations of factors necessary for initiation by promoters 1 and 2). As a result, unit 1 acts as an enhancer for unit 2. At the same time, unit 3 is tethered far from factory B (which is rich in the factors required for its initiation). Here unit 1 acts as a silencer of unit 3. (B) Insulator. At a different stage in development, a different constellation of transcription factors are expressed. Chromatin domains containing units 2 and 3 are separated by unit 1 (now transcribed in factory C, which contains low concentrations of the factors required by units 2 and 3), so they rarely bind to factory A and interact. Here unit 1 acts as an insulator or barrier.
Conclusions
The model we propose here (Figure 2) illustrates a case where nongenic transcription unit 1, in its normal genomic location, acts as an enhancer, silencer or insulator or barrier, depending on the target and developmental stage. We imagine that most regulatory motifs normally act in only one way; however, when moved out of their normal context (usually the case in the assays used to test for the action of these motifs), they will act differently, depending on the new context (which includes proximity to an appropriate factory). This model encapsulates notions of transcriptional activity, epigenetic marks and three-dimensional architecture, which, in combination, provide the context that determines promoter activity.
Abbreviations
- bp:
-
base pair
- kb:
-
kilobase
- IL:
-
interleukin
- Mbp:
-
megabase pair
- miRNA:
-
microRNA
- RNAi:
-
RNA interference
- TNF:
-
tumour necrosis factor.
References
Knoch TA, Göker M, Lohner R, Abuseiris A, Grosveld FG: Fine-structured multi-scaling long-range correlations in completely sequenced genomes: features, origin, and classification. Eur Biophys J. 2009, 38: 757-779.
Bulger M, Groudine M: Functional and mechanistic diversity of distal transcription enhancers. Cell. 2011, 144: 327-339. A published erratum appears in Cell 2011, 144:825
Maeda RK, Karch F: Gene expression in time and space: additive vs hierarchical organization of cis-regulatory regions. Curr Opin Genet Dev. 2011, 21: 187-193.
Yang J, Corces VG: Chromatin insulators: a role in nuclear organization and gene expression. Adv Cancer Res. 2011, 110: 43-76.
Cremer T, Cremer C: Chromosome territories, nuclear architecture and gene regulation in mammalian cells. Nat Rev Genet. 2001, 2: 292-301.
Jhunjhunwala S, van Zelm MC, Peak MM, Cutchin S, Riblet R, van Dongen JJM, Grosveld FG, Knoch TA, Murre C: The three-dimensional structure of the immunoglobulin heavy-chain locus: implications for long-range genomic interactions. Cell. 2008, 133: 265-279.
Knoch TA: Towards a holistic understanding of the human genome by determination and integration of its sequential and three-dimensional organization. High Performance Computing in Science and Engineering 2003. Edited by: Krause E, Jäger W, Resch M. 2003, Berlin: Springer, 421-440.
Rauch J, Knoch TA, Solovei I, Teller K, Stein S, Buiting K, Horsthemke B, Langowski J, Cremer T, Hausmann M, Cremer C: Light optical precision measurements of the active and inactive Prader-Willi syndrome imprinted regions in human cell nuclei. Differentiation. 2008, 76: 66-82.
Knoch TA, Münkel C, Langowski J: Three-dimensional organization of chromosome territo-ries in the human interphase nucleus. High Performance Computing in Science and Engineering 1999. Edited by: Krause E, Jäger W. 2000, Berlin: Springer, 229-238.
Knoch TA: Approaching the Three-Dimensional Organization of the Human Genome. 2002, Mannheim, Germany: TAK Press, [http://archiv.ub.uni-heidelberg.de/volltextserver/frontdoor.php?source_opus=3105]3
Raab JR, Kamakaka RT: Insulators and promoters: closer than we think. Nat Rev Genet. 2010, 11: 439-446.
de Laat W, Grosveld F: Spatial organization of gene expression: the active chromatin hub. Chromosome Res. 2003, 11: 447-459.
Sutherland H, Bickmore WA: Transcription factories: gene expression in unions?. Nat Rev Genet. 2009, 10: 457-466.
Chakalova L, Fraser P: Organization of transcription. Cold Spring Harb Perspect Biol. 2010, 2: a000729.
Cook PR: A model for all genomes: the role of transcription factories. J Mol Biol. 2010, 395: 1-10.
Banerji J, Rusconi S, Schaffner W: Expression of a β-globin gene is enhanced by remote SV40 DNA sequences. Cell. 1981, 27: 299-308.
Tolhuis B, Palstra RJ, Splinter E, Grosveld F, de Laat W: Looping and interaction between hypersensitive sites in the active β-globin locus. Mol Cell. 2002, 10: 1453-1465.
Lomvardas S, Barnea G, Pisapia DJ, Mendelsohn M, Kirkland J, Axel R: Interchromosomal interactions and olfactory receptor choice. Cell. 2006, 126: 403-413.
Hare EE, Peterson BK, Iyer VN, Meier R, Eisen MB: Sepsid even-skipped enhancers are functionally conserved in Drosophila despite lack of sequence conservation. PLoS Genet. 2008, 4: e1000106.
He HH, Meyer CA, Shin H, Bailey ST, Wei G, Wang Q, Zhang Y, Xu K, Ni M, Lupien M, Mieczkowski P, Lieb JD, Zhao K, Brown M, Liu XS: Nucleosome dynamics define transcriptional enhancers. Nat Genet. 2010, 42: 343-347.
Zentner GE, Tesar PJ, Scacheri PC: Epigenetic signatures distinguish multiple classes of enhancers with distinct cellular functions. Genome Res. 2011, 21: 1273-1283.
Heintzman ND, Hon GC, Hawkins RD, Kheradpour P, Stark A, Harp LF, Ye Z, Lee LK, Stuart RK, Ching CW, Ching KA, Antosiewicz-Bourget JE, Liu H, Zhang X, Green RD, Lobanenkov VV, Stewart R, Thomson JA, Crawford GE, Kellis M, Ren B: Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature. 2009, 459: 108-112.
De Santa F, Barozzi I, Mietton F, Ghisletti S, Polletti S, Tusi BK, Muller H, Ragoussis J, Wei CL, Natoli G: A large fraction of extragenic RNA pol II transcription sites overlap enhancers. PLoS Biol. 2010, 8: e1000384.
Kim TK, Hemberg M, Gray JM, Costa AM, Bear DM, Wu J, Harmin DA, Laptewicz M, Barbara-Haley K, Kuersten S, Markenscoff-Papadimitriou E, Kuhl D, Bito H, Worley PF, Kreiman G, Greenberg ME: Widespread transcription at neuronal activity-regulated enhancers. Nature. 2010, 465: 182-187.
Carter D, Chakalova L, Osborne CS, Dai YF, Fraser P: Long-range chromatin regulatory interactions in vivo. Nat Genet. 2002, 32: 623-626.
Drissen R, Palstra RJ, Gillemans N, Splinter E, Grosveld F, Philipsen S, de Laat W: The active spatial organization of the β-globin locus requires the transcription factor EKLF. Genes Dev. 2004, 18: 2485-2490.
Ørom UA, Derrien T, Beringer M, Gumireddy K, Gardini A, Bussotti G, Lai F, Zytnicki M, Notredame C, Huang Q, Guigo R, Shiekhattar R: Long noncoding RNAs with enhancer-like function in human cells. Cell. 2010, 143: 46-58.
Ørom UA, Shiekhattar R: Long non-coding RNAs and enhancers. Curr Opin Genet Dev. 2011, 21: 194-198.
Wang D, Garcia-Bassets I, Benner C, Li W, Su X, Zhou Y, Qiu J, Liu W, Kaikkonen MU, Ohgi KA, Glass CK, Rosenfeld MG, Fu XD: Reprogramming transcription by distinct classes of enhancers functionally defined by eRNA. Nature. 2011, 474: 390-394.
Ling J, Ainol L, Zhang L, Yu X, Pi W, Tuan D: HS2 enhancer function is blocked by a transcriptional terminator inserted between the enhancer and the promoter. J Biol Chem. 2004, 279: 51704-51713.
Ling J, Baibakov B, Pi W, Emerson BM, Tuan D: The HS2 enhancer of the β-globin locus control region initiates synthesis of non-coding, polyadenylated RNAs independent of a cis-linked globin promoter. J Mol Biol. 2005, 350: 883-896.
Rinn JL, Kertesz M, Wang JK, Squazzo SL, Xu X, Brugmann SA, Goodnough LH, Helms JA, Farnham PJ, Segal E, Chang HY: Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell. 2007, 129: 1311-1323.
Khalil AM, Guttman M, Huarte M, Garber M, Raj A, Rivea Morales D, Thomas K, Presser A, Bernstein BE, van Oudenaarden A, Regev A, Lander ES, Rinn JL: Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc Natl Acad Sci USA. 2009, 106: 11667-11672.
Ong CT, Corces VG: Enhancer function: new insights into the regulation of tissue-specific gene expression. Nat Rev Genet. 2011, 12: 283-293.
Wang KC, Yang YW, Liu B, Sanyal A, Corces-Zimmerman R, Chen Y, Lajoie BR, Protacio A, Flynn RA, Gupta RA, Wysocka J, Lei M, Dekker J, Helms JA, Chang HY: A long noncoding RNA maintains active chromatin to coordinate homeotic gene expression. Nature. 2011, 472: 120-124.
Li LM, Arnosti DN: Long- and short-range transcriptional repressors induce distinct chromatin states on repressed genes. Curr Biol. 2011, 21: 406-412.
Janowski BA, Corey DR: Minireview: Switching on progesterone receptor expression with duplex RNA. Mol Endocrinol. 2010, 24: 2243-2252.
Morris KV, Chan SW, Jacobsen SE, Looney DJ: Small interfering RNA-induced transcriptional gene silencing in human cells. Science. 2004, 305: 1289-1292.
Janowski BA, Huffman KE, Schwartz JC, Ram R, Hardy D, Shames DS, Minna JD, Corey DR: Inhibiting gene expression at transcription start sites in chromosomal DNA with antigene RNAs. Nat Chem Biol. 2005, 1: 216-222.
Bartel DP: MicroRNAs: target recognition and regulatory functions. Cell. 2009, 136: 215-233.
Liao JY, Ma LM, Guo YH, Zhang YC, Zhou H, Shao P, Chen YQ, Qu LH: Deep sequencing of human nuclear and cytoplasmic small RNAs reveals an unexpectedly complex subcellular distribution of miRNAs and tRNA 3' trailers. PLoS One. 2010, 5: e10563.
Younger ST, Corey DR: Transcriptional gene silencing in mammalian cells by miRNA mimics that target gene promoters. Nucleic Acids Res. 2011, 39: 5682-5691.
Pawlicki JM, Steitz JA: Primary microRNA transcript retention at sites of transcription leads to enhanced microRNA production. J Cell Biol. 2008, 182: 61-76.
Kanhere A, Viiri K, Araújo CC, Rasaiyaah J, Bouwman RD, Whyte WA, Pereira CF, Brookes E, Walker K, Bell GW, Pombo A, Fisher AG, Young RA, Jenner RG: Short RNAs are transcribed from repressed polycomb target genes and interact with polycomb repressive complex-2. Mol Cell. 2010, 38: 675-688.
Schoenherr CJ, Levorse JM, Tilghman SM: CTCF maintains differential methylation at the Igf2/H19 locus. Nat Genet. 2003, 33: 66-69.
Zhao J, Sun BK, Erwin JA, Song JJ, Lee JT: Polycomb proteins targeted by a short repeat RNA to the mouse X chromosome. Science. 2008, 322: 750-756.
Vernimmen D, Lynch MD, De Gobbi M, Garrick D, Sharpe JA, Sloane-Stanley JA, Smith AJ, Higgs DR: Polycomb eviction as a new distant enhancer function. Genes Dev. 2011, 25: 1583-1588.
Bernstein E, Duncan EM, Masui O, Gil J, Heard E, Allis CD: Mouse polycomb proteins bind differentially to methylated histone H3 and RNA and are enriched in facultative heterochromatin. Mol Cell Biol. 2006, 26: 2560-2569.
Splinter E, de Wit E, Nora EP, Klous P, van de Werken HJ, Zhu Y, Kaaij LJ, van Ijcken W, Gribnau J, Heard E, de Laat W: The inactive X chromosome adopts a unique three-dimensional conformation that is dependent on Xist RNA. Genes Dev. 2011, 25: 1371-1383.
Gaszner M, Felsenfeld G: Insulators: exploiting transcriptional and epigenetic mechanisms. Nat Rev Genet. 2006, 7: 703-713.
Feinberg AP: Phenotypic plasticity and the epigenetics of human disease. Nature. 2007, 447: 433-440.
Valenzuela L, Kamakaka RT: Chromatin insulators. Annu Rev Genet. 2006, 40: 107-138.
Chopra VS, Cande J, Hong JW, Levine M: Stalled Hox promoters as chromosomal boundaries. Genes Dev. 2009, 23: 1505-1509.
Hark AT, Schoenherr CJ, Katz DJ, Ingram RS, Levorse JM, Tilghman SM: CTCF mediates methylation-sensitive enhancer-blocking activity at the H19/Igf2 locus. Nature. 2000, 405: 486-489.
Parelho V, Hadjur S, Spivakov M, Leleu M, Sauer S, Gregson HC, Jarmuz A, Canzonetta C, Webster Z, Nesterova T, Cobb BS, Yokomori K, Dillon N, Aragon L, Fisher AG, Merkenschlager M: Cohesins functionally associate with CTCF on mammalian chromosome arms. Cell. 2008, 132: 422-433.
Wendt KS, Yoshida K, Itoh T, Bando M, Koch B, Schirghuber E, Tsutsumi S, Nagae G, Ishihara K, Mishiro T, Yahata K, Imamoto F, Aburatani H, Nakao M, Imamoto N, Maeshima K, Shirahige K, Peters JM: Cohesin mediates transcriptional insulation by CCCTC-binding factor. Nature. 2008, 451: 796-801.
Bowers SR, Mirabella F, Calero-Nieto FJ, Valeaux S, Hadjur S, Baxter EW, Merkenschlager M, Cockerill PN: A conserved insulator that recruits CTCF and cohesin exists between the closely related but divergently regulated interleukin-3 and granulocyte-macrophage colony-stimulating factor genes. Mol Cell Biol. 2009, 29: 1682-1693.
Zhou X, Weatherford ET, Liu X, Born E, Keen HL, Sigmund CD: Dysregulated human renin expression in transgenic mice carrying truncated genomic constructs: evidence supporting the presence of insulators at the renin locus. Am J Physiol Renal Physiol. 2008, 295: F642-653.
Komura J, Ikehata H, Ono T: Chromatin fine structure of the c-MYC insulator element/DNase I-hypersensitive site I is not preserved during mitosis. Proc Natl Acad Sci USA. 2007, 104: 15741-15746.
Nègre N, Brown CD, Shah PK, Kheradpour P, Morrison CA, Henikoff JG, Feng X, Ahmad K, Russell S, White RA, Stein L, Henikoff S, Kellis M, White KP: A comprehensive map of insulator elements for the Drosophila genome. PLoS Genet. 2010, 6: e1000814.
Moshkovich N, Nisha P, Boyle PJ, Thompson BA, Dale RK, Lei EP: RNAi-independent role for Argonaute2 in CTCF/CP190 chromatin insulator function. Genes Dev. 2011, 25: 1686-1701.
Kolesky SE, Ouhammouch M, Geiduschek EP: The mechanism of transcriptional activation by the topologically DNA-linked sliding clamp of bacteriophage T4. J Mol Biol. 2002, 321: 767-784.
Bulger M, Groudine M: Looping versus linking: toward a model for long-distance gene activation. Genes Dev. 1999, 13: 2465-2477.
Lanctôt C, Cheutin T, Cremer M, Cavalli G, Cremer T: Dynamic genome architecture in the nuclear space: regulation of gene expression in three dimensions. Nat Rev Genet. 2007, 8: 104-115.
Cook PR: Nongenic transcription, gene regulation and action at a distance. J Cell Sci. 2003, 116: 4483-4491.
Ptashne M, Gann A: Transcriptional activation by recruitment. Nature. 1997, 386: 569-577.
Malik S, Roeder RG: The metazoan Mediator co-activator complex as an integrative hub for transcriptional regulation. Nat Rev Genet. 2010, 11: 761-772.
Apostolou E, Thanos D: Virus Infection Induces NF-κB-dependent interchromosomal associations mediating monoallelic IFN-β gene expression. Cell. 2008, 134: 85-96.
Simonis M, Kooren J, de Laat W: An evaluation of 3C-based methods to capture DNA interactions. Nat Methods. 2007, 4: 895-901.
Gohl D, Aoki T, Blanton J, Shanower G, Kappes G, Schedl P: Mechanism of chromosomal boundary action: roadblock, sink, or loop?. Genetics. 2011, 187: 731-748.
Comet I, Schuettengruber B, Sexton T, Cavalli G: A chromatin insulator driving three-dimensional Polycomb response element (PRE) contacts and Polycomb association with the chromatin fiber. Proc Natl Acad Sci USA. 2011, 108: 2294-2299.
Jing H, Vakoc CR, Ying L, Mandat S, Wang H, Zheng X, Blobel GA: Exchange of GATA factors mediates transitions in looped chromatin organization at a developmentally regulated gene locus. Mol Cell. 2008, 29: 232-242.
Splinter E, Heath H, Kooren J, Palstra RJ, Klous P, Grosveld F, Galjart N, de Laat W: CTCF mediates long-range chromatin looping and local histone modification in the β-globin locus. Genes Dev. 2006, 20: 2349-2354.
Majumder P, Gomez JA, Chadwick BP, Boss JM: The insulator factor CTCF controls MHC class II gene expression and is required for the formation of long-distance chromatin interactions. J Exp Med. 2008, 205: 785-798.
Soler E, Andrieu-Soler C, de Boer E, Bryne JC, Thongjuea S, Stadhouders R, Palstra RJ, Stevens M, Kockx C, van Ijcken W, Hou J, Steinhoff C, Rijkers E, Lenhard B, Grosveld F: The genome-wide dynamics of the binding of Ldb1 complexes during erythroid differentiation. Genes Dev. 2010, 24: 277-289.
Nativio R, Wendt KS, Ito Y, Huddleston JE, Uribe-Lewis S, Woodfine K, Krueger C, Reik W, Peters JM, Murrell A: Cohesin is required for higher-order chromatin conformation at the imprinted IGF2-H19 locus. PLoS Genet. 2009, 5: e1000739.
Spilianakis CG, Flavell RA: Long-range intrachromosomal interactions in the T helper type 2 cytokine locus. Nat Immunol. 2004, 5: 1017-1027.
Vakoc CR, Letting DL, Gheldof N, Sawado T, Bender MA, Groudine M, Weiss MJ, Dekker J, Blobel GA: Proximity among distant regulatory elements at the β-globin locus requires GATA-1 and FOG-1. Mol Cell. 2005, 17: 453-462.
Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, Sandstrom R, Bernstein B, Bender MA, Groudine M, Gnirke A, Stamatoyannopoulos J, Mirny LA, Lander ES, Dekker J: Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009, 326: 289-293.
Simonis M, Klous P, Splinter E, Moshkin Y, Willemsen R, de Wit E, van Steensel B, de Laat W: Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture-on-chip (4C). Nat Genet. 2006, 38: 1348-1354.
Osborne CS, Chakalova L, Brown KE, Carter D, Horton A, Debrand E, Goyenechea B, Mitchell JA, Lopes S, Reik W, Fraser P: Active genes dynamically colocalize to shared sites of ongoing transcription. Nat Genet. 2004, 36: 1065-1071.
Schoenfelder S, Sexton T, Chakalova L, Cope NF, Horton A, Andrews S, Kurukuti S, Mitchell JA, Umlauf D, Dimitrova DS, Eskiw CH, Luo Y, Wei CL, Ruan Y, Bieker JJ, Fraser P: Preferential associations between co-regulated genes reveal a transcriptional interactome in erythroid cells. Nat Genet. 2010, 42: 53-61.
Baù D, Sanyal A, Lajoie BR, Capriotti E, Byron M, Lawrence JB, Dekker J, Marti-Renom MA: The three-dimensional folding of the α-globin gene domain reveals formation of chromatin globules. Nat Struct Mol Biol. 2011, 18: 107-114.
Xu M, Cook PR: The role of specialized transcription factories in chromosome pairing. Biochim Biophys Acta. 2008, 1783: 2155-2160.
Papantonis A, Cook PR: Genome architecture and the role of transcription. Curr Opin Cell Biol. 2010, 22: 271-276.
Pombo A, Cuello P, Schul W, Yoon JB, Roeder RG, Cook PR, Murphy S: Regional and temporal specialization in the nucleus: a transcriptionally-active nuclear domain rich in PTF, Oct1 and PIKA antigens associates with specific chromosomes early in the cell cycle. EMBO J. 1998, 17: 1768-1778.
Pombo A, Jackson DA, Hollinshead M, Wang Z, Roeder RG, Cook PR: Regional specialization in human nuclei: visualization of discrete sites of transcription by RNA polymerase III. EMBO J. 1999, 18: 2241-2253.
Papantonis A, Larkin JD, Wada Y, Ohta Y, Ihara S, Kodama T, Cook PR: Active RNA polymerases: mobile or immobile molecular machines?. PLoS Biol. 2010, 8: e1000419.
Melnik S, Deng B, Papantonis A, Baboo S, Carr IM, Cook PR: The proteomes of transcription factories containing RNA polymerases I, II or III. Nat Methods. 2011, 8: 963-968.
Sikorska M, Sandhu JK, Deb-Rinker P, Jezierski A, LeBlanc J, Charlebois C, Ribecco-Lutkiewicz M, Bani-Yaghoub M, Walker PR: Epigenetic modifications of SOX2 enhancers, SRR1 and SRR2, correlate with in vitro neural differentiation. J Neurosci Res. 2008, 86: 1680-1693.
Deshane J, Kim J, Bolisetty S, Hock TD, Hill-Kapturczak N, Agarwal A: Sp1 regulates chromatin looping between an intronic enhancer and distal promoter of the human heme oxygenase-1 gene in renal cells. J Biol Chem. 2010, 285: 16476-16486.
Chung JH, Whiteley M, Felsenfeld G: A 5' element of the chicken β-globin domain serves as an insulator in human erythroid cells and protects against position effect in Drosophila. Cell. 1993, 74: 505-514.
Antes TJ, Namciu SJ, Fournier RE, Levy-Wilson B: The 5' boundary of the human apolipoprotein B chromatin domain in intestinal cells. Biochemistry. 2001, 40: 6731-6742.
Filippova GN, Thienes CP, Penn BH, Cho DH, Hu YJ, Moore JM, Klesert TR, Lobanenkov VV, Tapscott SJ: CTCF-binding sites flank CTG/CAG repeats and form a methylation-sensitive insulator at the DM1 locus. Nat Genet. 2001, 28: 335-343.
Petrykowska HM, Vockley CM, Elnitski L: Detection and characterization of silencers and enhancer-blockers in the greater CFTR locus. Genome Res. 2008, 18: 1238-1246.
Valadez-Graham V, Razin SV, Recillas-Targa F: CTCF-dependent enhancer blockers at the upstream region of the chicken α-globin gene domain. Nucleic Acids Res. 2004, 32: 1354-1362.
Iborra FJ, Pombo A, McManus J, Jackson DA, Cook PR: The topology of transcription by immobilized polymerases. Exp Cell Res. 1996, 229: 167-173.
Pombo A, Hollinshead M, Cook PR: Bridging the resolution gap: imaging the same transcription factories in cryosections by light and electron microscopy. J Histochem Cytochem. 1999, 47: 471-480.
Faro-Trindade I, Cook PR: A conserved organization of transcription during embryonic stem cell differentiation and in cells with high C value. Mol Biol Cell. 2006, 17: 2910-2920.
Kheradmand Kia S, Solaimani Kartalaei P, Farahbakhshian E, Pourfarzad F, von Lindern M, Verrijzer CP: EZH2-dependent chromatin looping controls INK4a and INK4b, but not ARF, during human progenitor cell differentiation and cellular senescence. Epigenetics Chromatin. 2009, 2: 16.
Acknowledgements
We thank Eric Soler, Pavlos Fanis and members of our laboratories, as well as members of the virtual EpiGenSys laboratories, for their comments. This work was supported by ERASysBio+/FP7 and the ERASysBio+ local funding organisations: the Dutch Ministry for Science and Education, the Netherlands Science Organization and the UK Biotechnology and Biological Sciences Research Council. TAK thanks the High-Performance Computing Center Stuttgart (HumNuc), the Supercomputing Center Karlsruhe (ChromDyn), the Computing Facility of the German Cancer Research Center (DKFZ), the Erasmus Computing Grid, the German Ministry for Science & Technology (BMBF 01 KW 9602/2, German Human Genome Project grant 01AK803A and German MediGRID), and grant 01IG07015G (German Services@MediGRID) and the European Commission (FP7 EDGEs).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
All authors structured, wrote and proofread the manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Kolovos, P., Knoch, T.A., Grosveld, F.G. et al. Enhancers and silencers: an integrated and simple model for their function. Epigenetics & Chromatin 5, 1 (2012). https://doi.org/10.1186/1756-8935-5-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1756-8935-5-1