
Abstract
Treatment of myelofibrosis (MF), a BCR-ABL–negative myeloproliferative neoplasm, is challenging. The only current potentially curative option, allogeneic hematopoietic stem cell transplant, is recommended for few patients. The remaining patients are treated with palliative therapies to manage MF-related anemia and splenomegaly. Identification of a mutation in the Janus kinase 2 (JAK2) gene (JAK2 V617F) in more than half of all patients with MF has prompted the discovery and clinical development of inhibitors that target JAK2. Although treatment with JAK2 inhibitors has been shown to improve symptom response and quality of life in patients with MF, these drugs do not alter the underlying disease; therefore, novel therapies are needed. The hedgehog (Hh) signaling pathway has been shown to play a role in normal hematopoiesis and in the tumorigenesis of hematologic malignancies. Moreover, inhibitors of the Hh pathway have been shown to inhibit growth and self-renewal capacity in preclinical models of MF. In a mouse model of MF, combined inhibition of the Hh and JAK pathways reduced JAK2 mutant allele burden, reduced bone marrow fibrosis, and reduced white blood cell and platelet counts. Preliminary clinical data also suggest that inhibition of the Hh pathway, alone or in combination with JAK2 inhibition, may enable disease modification in patients with MF. Future studies, including one combining the Hh pathway inhibitor sonidegib and the JAK2 inhibitor ruxolitinib, are underway in patients with MF and will inform whether this combination approach can lead to true disease modification.
Similar content being viewed by others


Myelofibrosis
Myelofibrosis (MF) is one of several BCR-ABL–negative myeloproliferative neoplasms (MPNs), which are derived from multipotent, hematopoietic myeloid progenitors[1, 2]. MF can be primary (PMF) or secondary to the MPNs polycythemia vera (PV) or essential thrombocythemia (ET) (post-PV or post-ET, respectively)[2]. MF is characterized by stem cell–derived clonal myeloproliferation, abnormal cytokine expression, bone marrow fibrosis, cytopenias, extramedullary hematopoiesis (eg, splenomegaly), cachexia, and constitutional symptoms including night sweats, fevers, weight loss, and fatigue[3–5]. Disease complications also include infections, portal hypertension, bleeding, extremity pain, and progression of disease with blastic transformation, resembling acute leukemia[5–8].
MF is most commonly characterized by a mutation in Janus kinase 2 (JAK2 V617F), which is present in approximately 96%, 55%, and 65% of patients with PV, ET, and PMF, respectively[5, 9]. The JAK2 V617F gain-of-function mutation leads to constitutive activation of the JAK/signal transducer and activation of transcription pathway, which regulates the expression of genes involved in proliferation, survival, and resistance to apoptosis (B-cell CLL/lymphoma 2-like 1, myeloid cell leukemia sequence 1, pim-1 oncogene, and cyclin D1; Figure 1A)[10]. Recently, a high frequency of calreticulin mutations has been found in JAK2 nonmutated MF[11, 12]. Mutations in other genes, including myeloproliferative leukemia virus oncogene, SH2B adaptor protein 3, tet methylcytosine dioxygenase 2, additional sex combs like 1 homolog (ASXL1), isocitrate dehydrogenase (IDH), enhancer of zeste homolog 2 (EZH2), DNA (cytosine-5-)-methyltransferase 3 α, casitas B-lineage lymphoma proto-oncogene, TP53, splicing factor 3b subunit 1, and serine/arginine-rich splicing factor 2 (SRSF2), have been found less frequently in patients with MF[5]. Some of these mutations have been linked with poor survival (ASXL1, EZH2, and SRSF2) and/or leukemic transformation (ASXL1, SRSF2, and IDH) in patients with PMF[13].

Janus kinase 2 (JAK2)/signal transducer and activation of transcription (STAT) and hedgehog (Hh) signaling pathways in normal development (A) and mechanisms of Hh signaling in cancer (B). (A) JAK/STAT signaling: the JAK2/STAT signaling pathway is activated upon binding of a cytokine to its receptor, causing phosphorylation and activation of JAK2, which then recruits and phosphorylates STATs. STATs dimerize, translocate to the nucleus, and activate target gene transcription. Hh signaling: in the absence of Hh ligand, patched (PTCH) inhibits smoothened (SMO). Glioma-associated oncogene homolog 1/2 (GLI1/2) transcription factors are sequestered in the cytoplasm by a repressor complex containing suppressor of fused (SUFU) and degraded. GLI3 is released from SUFU, processed into a repressor form (GLI3R), and translocates to the nucleus to inhibit transcription of Hh pathway target genes. Hh signaling is activated upon binding of Hh to PTCH. PTCH-mediated inhibition of SMO is relieved, and SMO activates release of GLIs from the SUFU complex. Activated GLIs (GLIA) then translocate to the nucleus to regulate target gene transcription. (B) Several mechanisms of Hh pathway activation in cancer have been proposed, including ligand independent (mutation driven) and ligand dependent (autocrine or paracrine) signaling. During autocrine signaling, Hh ligands produced in the tumor activate Hh signaling in the same tumor cells. Paracrine signaling can involve tumor-to-stroma signaling or stroma-to-tumor signaling (reverse paracrine). During tumor-to-stroma signaling, Hh ligands produced in the tumor activate Hh signaling in surrounding stromal cells, which release growth hormones that in turn feed tumor growth. In the reverse model (stroma-to-tumor), which has been observed in hematologic malignancies (lymphoma, myeloid neoplasms, and multiple myeloma), Hh ligands produced in stromal cells activate Hh signaling in the tumor. BCL2, B-cell CLL/lymphoma 2; BCL2L1, BCL2-like 1; BMP, bone morphogenetic protein; CCND1, cyclin D1; MCL1, myeloid cell leukemia sequence 1; PIM1, pim-1 oncogene.
According to the Dynamic International Prognosis Scoring System Plus (DIPSS Plus), patients with MF are assigned to one of 4 risk groups—low, intermediate-1, intermediate-2, and high. These risk groups are based on 8 factors independently associated with decreased survival: age > 65 years, hemoglobin levels < 10 g/dL, leukocyte count > 25 × 109/L, circulating blood blasts ≥ 1%, constitutional symptoms, red blood cell transfusion, platelet count < 100 × 109/L, and unfavorable karyotype[14]. Median survival varies considerably according to risk group, ranging in one study from 16 to 185 months for high- and low-risk patients, respectively[14].
Current treatment strategies
The DIPSS Plus and Myeloproliferative Neoplasm Symptom Assessment Form are used to inform treatment regimen decisions[7, 15]. For patients with asymptomatic low-risk or intermediate-1 disease, observation is generally recommended[5, 16]. For symptomatic patients, current therapies include allogeneic hematopoietic stem cell transplant (HSCT) and palliative treatments that help alleviate disease symptoms such as anemia and splenomegaly[5, 16, 17]. Allogeneic HSCT is associated with significant risk of morbidity and mortality due to relapse, infection, and graft-versus-host disease, and therefore is recommended only for patients aged < 65 years with intermediate- or high-risk disease[18]. Reduced-intensity conditioning regimens have shown more favorable outcomes but still pose a high risk for patients aged > 55 years and patients with mismatched donors[19].
Therapies intended to treat MF-associated anemia include growth factors (eg, erythropoietin), androgens (eg, danazol), and the immunomodulatory drugs (IMiDs) thalidomide (± prednisone), lenalidomide (± prednisone), and pomalidomide (± prednisone)[20–26]. IMiDs have also been shown to improve splenomegaly[27–29]. Other agents used to treat MF-associated splenomegaly include the nonspecific oral myelosuppressive agent hydroxyurea, the oral alkylators melphalan and busulfan, and the purine nucleoside analog 2-CdA[30–32]. Hydroxyurea is a choice for splenomegaly in patients with MF as well[5]. Although generally well tolerated, hydroxyurea can lead to myelosuppression, which can exacerbate MF-associated anemia[14, 16].
Based on the finding that the majority of patients with MF have a mutation in JAK2, numerous inhibitors of JAK2 (ruxolitinib [INCB018424], fedratinib [SAR302503; TG101348], lestaurtinib [CEP-701], momelotinib [CYT387], pacritinib [SB1518], AZD1480, BMS-911543, gandotinib [LY2784544], AT9283, and XL019) have been developed and are being evaluated in clinical trials. Of note, JAK inhibitors also have activity in JAK2 nonmutated MF/PMF[33, 34]. Ruxolitinib, an inhibitor of JAK1 and JAK2, was approved in 2011 by the US Food and Drug Administration (FDA) for use in patients with intermediate- or high-risk MF (PMF, post-PV MF, and post-ET MF) and in 2012 by Health Canada and the European Medicines Agency for the treatment of MF-related splenomegaly and symptoms[35–37]. JAK2 inhibitors differ according to their specificity for JAK2 and have variable efficacy and toxicity profiles[5, 17].
Unmet need in the treatment of MF
Currently, the only potentially curative therapy for patients with MF is allogeneic HSCT[16, 38]. Due to treatment-related morbidity and mortality, HSCT is recommended for patients with intermediate-2– or high-risk disease who are fit enough to undergo the procedure. The majority of patients with MF are treated with palliative therapies, which improve disease symptoms rather than altering the natural history of disease[17]. The discovery of the JAK2 gain-of-function mutation, JAK2 V617F[39–42], followed by the development and approval of ruxolitinib has marked a new era in the treatment of MF, providing improved symptomatic responses and quality of life in comparison with traditional therapies[36, 37, 43–45]. However, treatment with JAK2 inhibitors has shown only limited evidence of disease modification–JAK2 inhibitors do not improve bone marrow fibrosis and most provide limited reduction of JAK2 V617F allelic burden[16, 17]. Ruxolitinib appears to block inflammatory cytokine activity rather than stem cell–derived clonal myeloproliferation, which is the primary driver of the disease[46]. Therefore, disease resistance can ensue following an initial response to JAK2 inhibition[16, 46]. In addition, treatment-related anemia may exacerbate preexisting MF-related anemia[33, 43, 44].
To further improve the responses to JAK2 inhibitors, various combinations have been clinically tested. For example, combination of JAK2 inhibitors with agents that improve anemia (eg, IMiDs) or target signaling pathways involved in proliferation, survival, and self-renewal may further improve the outcome of patients with MF[26, 47–49]. Combinations of JAK2 inhibitors with inhibitors of the hedgehog (Hh) pathway, which plays a role in the maintenance of cancer stem cells[50], could provide an avenue of targeting stem cell–derived clonal myeloproliferation (which evades JAK2-targeted monotherapy)[51]. Other combination partners, including hypomethylating agents (Tibes, unpublished observation) and Aurora-kinase inhibtors have also been proposed[52]. The preclinical rationale and current clinical evidence supporting use of Hh pathway–targeted therapies in the treatment of patients with MF will be discussed herein.
Rationale for targeting the Hh pathway in MF
The Hh pathway and its role in hematopoiesis
The Hh signaling pathway plays a role in proliferation, differentiation, and survival during embryonic development and in tissue and stem cell maintenance in the adult[50, 53]. Hh signaling is initiated when one of 3 ligands–sonic hedgehog (SHH), Indian hedgehog (IHH), or desert hedgehog (DHH)–binds to patched (PTCH), a 12-transmembrane receptor, relieving its inhibition of smoothened (SMO), a 7-transmembrane G-like protein–coupled receptor (Figure 1A). SMO then translocates to the primary cilium and activates the glioma-associated oncogene homolog (GLI) transcription factors, a process that involves their release from a repressor complex including suppressor of fused. Once released, GLIs translocate to the nucleus to regulate the transcription of target genes including GLI1/2, PTCH, cyclin D1, and B-cell CLL/lymphoma 2.
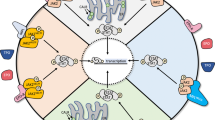
Hh signaling is required during hematopoiesis (Figure 2); however, its exact role is not completely understood and may differ depending on the stage of hematopoiesis, cell type (stem, primitive, or differentiated cell), and physiological state[54]. During primitive hematopoiesis, when embryonic mesoderm is committed to becoming hematopoietic precursors (eg, erythrocytes) and blood islands begin to form[55, 56], Ihh is expressed in the visceral endoderm surrounding the epiblast and in the endodermal layer of the mature yolk sac and induces the expression of Ptch1, Smo, and Gli1 within these tissues[57]. Murine Ihh knockout mice and in vitro studies in Ihh- deficient embryonic stem cell lines suggest that Ihh is required for hematopoiesis and vasculogenesis[57–60]. Survival of half of Ihh−/− mice and the observation that Smo−/− mice die earlier suggest that Dhh and/or Shh may also play a role in primitive hematopoiesis and vasculogenesis[57, 61].

Role of hedgehog (Hh) signaling in hematopoiesis. Preclinical studies suggest that the Hh signaling pathway may be involved in numerous stages and processes of hematopoiesis, including primitive hematopoiesis, definitive hematopoiesis—establishment, proliferation, and differentiation (lymphoid and myeloid lineages)—and maintenance of HSCs. The exact role for Hh signaling at each of these stages/processes is not clear. HSC, hematopoietic stem cell.
Preclinical studies also suggest that Hh plays a role not only in establishing definitive hematopoiesis, which is characterized by formation of multipotent hematopoietic stem cells (HSCs), but also in the proliferation and differentiation of HSCs (Table 1)[62–70]. Activated Hh signaling through loss of Ptch leads to increased HSC formation and activity[64, 66], enhanced recoverability following treatment with 5-fluorouracil[65, 66], and increased regeneration capacity[65, 66]. Conversely, loss of pathway activity through mutation of the downstream effector, Gli1, in mice leads to decreased proliferation of long-term HSCs and myeloid progenitors, reduced myeloid differentiation, and delayed recovery following 5-fluorouracil treatment[69]. Interestingly, reduced HSC activity (through loss of Gli1) led to increased engraftment. Together, these studies suggest that inhibition of the Hh pathway at different nodes (ie, Smo vs Gli1) affects hematopoiesis differently.
The role of Hh signaling in long-term HSCs is not well understood—several groups have reported conflicting results (Table 1); however, in each study, activated Hh signaling led to aberrant hematopoiesis[65–67]. There have also been some discrepancies in studies involving deletion of Smo, based on the temporal expression pattern of the experimental driver used (embryogenesis vs adulthood) and its specificity (hematopoietic and endothelial tissue vs HSCs, lymphocytes, and liver cells)[65, 68, 73, 74]. Disruption of Hh signaling earlier and in more tissues affected HSC function, whereas disruption of Hh signaling in adult HSCs had no effect, suggesting that Hh signaling may be important during early definitive hematopoiesis.
Numerous studies have also presented evidence implicating the Hh pathway in the maintenance or homeostasis of hematopoietic precursors[72, 75–79]. Activated Hh signaling in nonhematopoietic cells (ie, epithelial cells or marrow niche cells) led to apoptosis of lymphoid progenitors or an increase in the number of lineage-negative bone marrow cells and increased mobilization of myeloid progenitors[67]. Inhibition of Hh signaling in marrow stromal cells led to impaired differentiation of B-lymphoid cells from hematopoietic progenitors—the number of myeloid progenitors was increased at the expense of lymphoid progenitors[72]. These and several other studies suggest that Hh signaling may be required in a noncell autonomous manner where Hh signaling functions in the nonhematopoietic bone marrow cells (ie, stroma or epithelial cells) surrounding HSCs to maintain, particularly myeloid, hematopoietic precursors (Figure 2)[67, 72, 76–79].
The Hh pathway in MF and other hematologic malignancies
To date, preclinical data on the potential role of the Hh pathway in MF are limited. However, in one study, expression of GLI1 and PTCH1 were shown to be increased up to 100-fold in granulocytes isolated from patients with MPNs compared with control granulocytes[51]. The Hh pathway was also shown to be up-regulated in a mouse bone marrow transplant model[51]. In this same model, mice were treated with vehicle, ruxolitinib, or a combination of ruxolitinib and the SMO inhibitor sonidegib (LDE225), for 28 days[51]. Combination therapy resulted in increased efficacy in MPNs—causing a greater reduction of mutant allele burden in the bone marrow, reduced bone marrow fibrosis, lower white blood cell count, and lower platelet count than treatment with vehicle or ruxolitinib alone (Table 2). Moreover, in the Gata1low mouse model of MF, gene expression analysis of the spleen and bone marrow identified alterations in the expression of bone morphogenetic protein 4, an indirect target of the Hh pathway, further supporting a role for Hh signaling in MF[80, 81].
There are many preclinical studies implicating the Hh pathway in the pathogenesis of other hematologic malignancies and solid tumors[92]. Aberrant Hh signaling in cancer is postulated to occur through ligand-independent and ligand-dependent mechanisms (Figure 1B)[93]. Ligand-independent or mutation-driven signaling occurs when mutations in Hh pathway components—loss-of-function mutations in the negative regulators PTCH and SUFU (suppressor of fused), or gain-of-function mutations in the positive regulator SMO— lead to constitutive pathway activation within tumor cells. This type of signaling has been observed in basal cell carcinoma (PTCH and SMO mutations)[94, 95], medulloblastoma (PTCH and SUFU mutations)[96], and rhabdomyosarcoma (PTCH and SUFU loss of heterozygosity)[97].
Ligand-dependent mechanisms involve autocrine or paracrine Hh signaling[93]. During autocrine Hh signaling, tumor cells both secrete and respond to Hh—this type of Hh signaling has been identified in chronic myeloid leukemia (CML), small cell lung cancer, pancreatic cancer, breast cancer, and glioma[93]. Paracrine Hh signaling involves tumor-to-stroma or stroma-to-tumor (reverse paracrine) signaling. During tumor-to-stroma paracrine signaling, tumor cells produce and secrete Hh ligands which activate Hh signaling in surrounding stromal cells. Activated stromal cells release growth hormones which in turn stimulate tumor cell proliferation. Evidence for tumor-to-stroma paracrine signaling has been observed in pancreatic, colon, and prostate cancers[93]. Evidence for reverse paracrine signaling (stroma-to-tumor) in which Hh ligand produced in bone marrow stromal cells activates Hh signaling in surrounding tumor cells, has been reported for hematologic malignancies such as lymphoma, myeloid neoplasms, and multiple myeloma (MM)[91, 98]. In addition, the Hh pathway has been implicated in the maintenance and differentiation of cancer stem cells in CML, B-cell acute lymphocytic leukemia (B-ALL), and MM[50, 99, 100]. Moreover, up-regulation of Hh pathway components has been observed in the tumor stem cells of numerous hematologic malignancies, including BCR-ABL+ leukemic stem cells (LSCs)[65, 68], clonogenic B-ALL cells[87], CD34+ acute myeloid leukemia (AML)– and myelodysplastic syndromes (MDS)–derived cells[77], and MM CD138− tumor stem cells[91]. Pharmacologic inhibition of SMO has been shown to inhibit leukemogenesis through inhibition of LSC cell growth, self-renewal, and secondary transplantation capacity and induction of cell death in CML, AML, and ALL models (Table 2)[65, 68, 82–88]. Hh signaling has also been implicated in the progression of CML in mouse bone marrow transplant models[65, 68]. Constitutively active Smo was shown to increase the frequency of CML stem cells and accelerate disease progression[68]. Conversely, genetic loss or pharmacologic inhibition of Smo significantly impaired CML progression and prolonged survival[65, 68]. These data suggest that the Hh signaling pathway plays a role in numerous hematologic malignancies, including MF, and its inhibition may block tumor stem cell growth and disease progression.
Clinical studies of HH pathway inhibitors in patients with MF and other hematologic malignancies
Several Hh pathway inhibitors that target SMO have demonstrated single-agent efficacy in patients with ligand-independent tumors[101–105], including vismodegib, which was approved by the FDA in 2012 for the treatment of patients with locally advanced or metastatic basal cell carcinoma[101, 106]. Patients with Hh-activated medulloblastoma have also responded to treatment with vismodegib and the SMO inhibitor sonidegib[102, 104, 105]. Conversely, limited single-agent activity has been observed in ligand-dependent solid tumors—this lack of activity may be due in part to the contributions of other signaling pathways and stromal factors[107]. To date, saridegib (IPI-926), sonidegib, and PF-04449913 are the only SMO inhibitors that have been or are being tested in patients with MF (NCT01371617, NCT01787552, and NCT00953758, respectively) (Table 3). A phase 2 study of saridegib in patients with MF (NCT01371617) was halted following evaluation of an initial cohort of 12 patients—the level of clinical activity observed with saridegib did not meet the prespecified expansion criteria[108]. No further data have been reported. Data from a phase 1 trial of single-agent PF-04449913 presented at the American Society of Hematology in 2011 showed that PF-04449913 demonstrated activity in patients with refractory, resistant, or intolerant select hematologic malignancies, including MF (NCT00953758)[109]. The dose-limiting toxicity at 80 mg once daily was grade 3 hypoxia and pleural effusion. Of 6 patients with MF treated with PF-04449913, 5 achieved stable disease and 1 achieved clinical improvement with > 50% reduction in extramedullary disease. This patient remained on the study after 385 days and showed a spleen reduction from 10 to 3.5 cm over 8 weeks. Another patient achieved a marked reduction in bone marrow fibrosis.
Sonidegib is currently being investigated in combination with ruxolitinib in patients with MF in a phase 1/2 study (NCT01787552). Patients with PMF, post-PV MF, or post-ET MF are eligible. Primary endpoints include determination of dose-limiting toxicities, maximum tolerated dose and/or recommended phase 2 dose of the combination, and proportion of patients achieving ≥ 35% decrease in spleen volume. Secondary endpoints include safety, pharmacokinetics, improvement in bone marrow fibrosis, and change in total symptom score (modified Myelofibrosis Symptom Assessment Form v 2.0), JAK2 V617F allele burden, cytokine levels, and European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire 30.
These inhibitors, as well as the SMO inhibitors vismodegib (first in class) and BMS-833923, are being investigated in other hematologic malignancies, including ALL, AML/MDS, CML, and MM (Table 3)[111].
Assessment of Hh pathway inhibition in the clinic
For maximization of the potential of Hh pathway inhibitor therapy in patients with MF and related myeloid malignancies such as MDS and AML, and demonstration of a benefit over current therapies, it will be important to develop a method to assess the association of Hh pathway inhibitor activity with efficacy. In other tumor types, GLI1 expression has been used to determine changes in Hh pathway activity and confirm targeted inhibition in patients treated with SMO inhibitors[99, 103, 104, 112, 113]. In patients with MF, AML, or CML, one study showed that gene expression analysis of bone marrow CD34+ LSCs before and after treatment with PF-04449913 showed up-regulation of growth arrest specific 1 and kinesin family member 27, 2 negative regulators of the Hh signaling pathway[113]. Although changes in the expression of downstream Hh pathway components can be used to detect Hh pathway repression, a more appropriate measure of Hh pathway inhibitor activity in patients with MF is evidence of disease modification through histopathologic (bone marrow fibrosis) and cytogenetic (JAK2 V617F allele burden) remission. In patients with MF with JAK2 V617F mutations, change in allele burden following treatment with a Hh pathway inhibitor would be an appropriate marker for stem cell inhibition. Similarly, for patients with MDS or AML disease-initiating mutations, reduction in allele burden would indicate a possible on-target effect. In patients without mutations, identification of an appropriate marker is yet to be accomplished. Sustained responses following treatment discontinuation may also reflect disease modification. Ultimately, in order to assess the efficacy of future targeted therapies, a combination of endpoints, including disease-specific histopathologic (ie, reduction of fibrosis) and molecular (ie, allele burden reduction) changes and clinical efficacy (ie, improvement in blood counts), should be considered. Future preclinical studies in JAK2 V617F–negative MF and correlative data from the ongoing trials of Hh pathway inhibitors in patients with MF may better define the optimal method for determination of efficacy and identification of predictive and pharmacodynamic biomarkers in patients treated with Hh pathway inhibitors.
Conclusions
Despite recent advances in the treatment of MF, lack of true disease modification following treatment with current therapies warrants the identification of novel therapies. Inhibitors of the Hh signaling pathway, which has been implicated in the maintenance of HSCs, have shown preliminary activity as single agents or in combination with ruxolitinib in preclinical and clinical studies in MF. A clinical study combining the Hh pathway inhibitor sonidegib with the JAK2 inhibitor ruxolitinib in patients with MF is currently underway. In addition, we are currently working on preclinical studies and the development of a clinical trial to test the combination of Hh pathway inhibitors with the hypomethylating agent 5-azacitidine (Tibes, personal communication). These and future studies will test the hypothesis that targeting pathways involved in stem cell maintenance will not only extend the duration of benefit but will also lead to true disease modification in patients with MF treated with JAK2 inhibitors, as well as test their activity in other hematologic malignancies.
Authors’ information
RT: A physician-scientist conducting early clinical trials with novel molecular-targeted agents in patients with myeloid malignancies, including MDS, AML and MPNs/MF. Performing laboratory research to develop new rational therapeutic combinations in acute and chronic leukemias and MF. Involved in early stages of the development of several SMO (Hedgehog pathway) inhibitors including the first-in-class agent vismodegib.
RAM: An accomplished investigator leading clinical developmental efforts and large trials for new agents and therapies in MPNs and MF. Involved in pivotal trials for JAK2 inhibitors.
Abbreviations
- AML:
-
Acute myeloid leukemia
- ASXL1:
-
Additional sex combs like 1 homolog
- B-ALL:
-
B-cell acute lymphocytic leukemia
- CML:
-
Chronic myeloid leukemia
- DHH:
-
Desert hedgehog
- DIPSS:
-
Dynamic International Prognosis Scoring System
- ET:
-
Essential thrombocythemia
- EZH2:
-
Enhancer of zeste homolog 2
- GLI:
-
Glioma-associated oncogene homolog
- Hh:
-
Hedgehog
- HSC:
-
Hematopoietic stem cell
- HSCT:
-
Hematopoietic stem cell transplant
- IDH:
-
Isocitrate dehydrogenase
- IHH:
-
Indian hedgehog
- IMiD:
-
Immunomodulatory drug
- JAK2:
-
Janus kinase 2
- LSC:
-
Leukemic stem cell
- MDS:
-
Myelodysplastic syndromes
- MF:
-
Myelofibrosis
- MM:
-
Multiple myeloma
- MPN:
-
Myeloproliferative neoplasm
- PMF:
-
Primary myelofibrosis
- PTCH:
-
Patched
- PV:
-
Polycythemia vera
- SHH:
-
Sonic hedgehog
- SMO:
-
Smoothened
- SRSF2:
-
Serine/arginine-rich splicing factor 2
- STAT:
-
Signal transducer and activation of transcription
- SUFU:
-
Suppressor of fused.
References
Levine RL, Gilliland DG: Myeloproliferative disorders. Blood. 2008, 112: 2190-2198. 10.1182/blood-2008-03-077966.
Barosi G, Mesa RA, Thiele J, Cervantes F, Campbell PJ, Verstovsek S, Dupriez B, Levine RL, Passamonti F, Gotlib J, Reilly JT, Vannucchi AM, Hanson CA, Solberg LA, Orazi A, Tefferi A, International Working Group for Myelofibrosis Research and Treatment (IWG-MRT): Proposed criteria for the diagnosis of post-polycythemia vera and post-essential thrombocythemia myelofibrosis: a consensus statement from the International Working Group for Myelofibrosis Research and Treatment. Leukemia. 2008, 22: 437-438. 10.1038/sj.leu.2404914.
Tibes R, Bogenberger JM, Benson KL, Mesa RA: Current outlook on molecular pathogenesis and treatment of myeloproliferative neoplasms. Mol Diagn Ther. 2012, 16: 269-283. 10.1007/s40291-012-0006-3.
Mesa RA, Niblack J, Wadleigh M, Verstovsek S, Camoriano J, Barnes S, Tan AD, Atherton PJ, Sloan JA, Tefferi A: The burden of fatigue and quality of life in myeloproliferative disorders (MPDs): an international Internet-based survey of 1179 MPD patients. Cancer. 2007, 109: 68-76. 10.1002/cncr.22365.
Tefferi A: Primary myelofibrosis: 2013 update on diagnosis, risk-stratification, and management. Am J Hematol. 2013, 88: 141-150. 10.1002/ajh.23384.
Cervantes F, Dupriez B, Pereira A, Passamonti F, Reilly JT, Morra E, Vannucchi AM, Mesa RA, Demory JL, Barosi G, Rumi E, Tefferi A: New prognostic scoring system for primary myelofibrosis based on a study of the International Working Group for Myelofibrosis Research and Treatment. Blood. 2009, 113: 2895-2901. 10.1182/blood-2008-07-170449.
Scherber R, Dueck AC, Johansson P, Barbui T, Barosi G, Vannucchi AM, Passamonti F, Andreasson B, Ferarri ML, Rambaldi A, Samuelsson J, Birgegard G, Tefferi A, Harrison CN, Radia D, Mesa RA: The Myeloproliferative Neoplasm Symptom Assessment Form (MPN-SAF): international prospective validation and reliability trial in 402 patients. Blood. 2011, 118: 401-408. 10.1182/blood-2011-01-328955.
Mesa RA, Li CY, Ketterling RP, Schroeder GS, Knudson RA, Tefferi A: Leukemic transformation in myelofibrosis with myeloid metaplasia: a single-institution experience with 91 cases. Blood. 2005, 105: 973-977.
Tefferi A: Novel mutations and their functional and clinical relevance in myeloproliferative neoplasms: JAK2, MPL, TET2, ASXL1, CBL, IDH and IKZF1. Leukemia. 2010, 24: 1128-1138. 10.1038/leu.2010.69.
Fiskus W, Ganguly S, Kambhampati S, Bhalla KN: Role of additional novel therapies in myeloproliferative neoplasms. Hematol Oncol Clin North Am. 2012, 26: 959-980. 10.1016/j.hoc.2012.07.001.
Klampfl T, Gisslinger H, Harutyunyan AS, Nivarthi H, Rumi E, Milosevic JD, Them NC, Berg T, Gisslinger B, Pietra D, Chen D, Vladimer GI, Bagienski K, Milanesi C, Casetti IC, Sant’antonio E, Ferretti V, Elena C, Schischlik F, Cleary C, Six M, Schalling M, Schonegger A, Bock C, Malcovati L, Pascutto C, Superti-Furga G, Cazzola M, Kralovics R: Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med. 2013, 369: 2379-2390. 10.1056/NEJMoa1311347.
Nangalia J, Massie CE, Baxter EJ, Nice FL, Gundem G, Wedge DC, Avezov E, Li J, Kollmann K, Kent DG, Aziz A, Godfrey AL, Hinton J, Martincorena I, Van Loo P, Jones AV, Guglielmelli P, Tarpey P, Harding HP, Fitzpatrick JD, Goudie CT, Ortmann CA, Loughran SJ, Raine K, Jones DR, Butler AP, Teague JW, O’Meara S, McLaren S, Bianchi M: Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med. 2013, 369: 2391-2405. 10.1056/NEJMoa1312542.
Vannucchi AM, Lasho TL, Guglielmelli P, Biamonte F, Pardanani A, Pereira A, Finke C, Score J, Gangat N, Mannarelli C, Ketterling RP, Rotunno G, Knudson RA, Susini MC, Laborde RR, Spolverini A, Pancrazzi A, Pieri L, Manfredini R, Tagliafico E, Zini R, Jones A, Zoi K, Reiter A, Duncombe A, Pietra D, Rumi E, Cervantes F, Barosi G, Cazzola M: Mutations and prognosis in primary myelofibrosis. Leukemia. 2013, 27: 1861-1869. 10.1038/leu.2013.119.
Gangat N, Caramazza D, Vaidya R, George G, Begna K, Schwager S, Van Dyke D, Hanson C, Wu W, Pardanani A, Cervantes F, Passamonti F, Tefferi A: Dipss plus: a refined dynamic international prognostic scoring system for primary myelofibrosis that incorporates prognostic information from karyotype, platelet count, and transfusion status. J Clin Oncol. 2011, 29: 392-397. 10.1200/JCO.2010.32.2446.
Mesa RA, Schwager S, Radia D, Cheville A, Hussein K, Niblack J, Pardanani AD, Steensma DP, Litzow MR, Rivera CE, Camoriano J, Verstovsek S, Sloan J, Harrison C, Kantarjian H, Tefferi A: The Myelofibrosis Symptom Assessment Form (MFSAF): an evidence-based brief inventory to measure quality of life and symptomatic response to treatment in myelofibrosis. Leuk Res. 2009, 33: 1199-1203. 10.1016/j.leukres.2009.01.035.
Tibes R, Bogenberger JM, Mesa RA: Recent developments in myelofibrosis. Blood Lymphat Cancer: Targets Ther. 2012, 2: 125-136.
Tibes R, Bogenberger JM, Geyer HL, Mesa RA: JAK2 inhibitors in the treatment of myeloproliferative neoplasms. Expert Opin Investig Drugs. 2012, 21: 1755-1774. 10.1517/13543784.2012.721352.
Ballen KK, Shrestha S, Sobocinski KA, Zhang MJ, Bashey A, Bolwell BJ, Cervantes F, Devine SM, Gale RP, Gupta V, Hahn TE, Hogan WJ, Kroger N, Litzow MR, Marks DI, Maziarz RT, McCarthy PL, Schiller G, Schouten HC, Roy V, Wiernik PH, Horowitz MM, Giralt SA, Arora M: Outcome of transplantation for myelofibrosis. Biol Blood Marrow Transplant. 2010, 16: 358-367. 10.1016/j.bbmt.2009.10.025.
Kröger N, Holler E, Kobbe G, Bornhäuser M, Schwerdtfeger R, Baurmann H, Nagler A, Bethge W, Stelljes M, Uharek L, Wandt H, Burchert A, Corradini P, Schubert J, Kaufmann M, Dreger P, Wulf GG, Einsele H, Zabelina T, Kvasnicka HM, Thiele J, Brand R, Zander AR, Niederwieser D, de Witte TM: Allogeneic stem cell transplantation after reduced-intensity conditioning in patients with myelofibrosis: a prospective, multicenter study of the chronic leukemia working party of the European group for blood and marrow transplantation. Blood. 2009, 114: 5264-5270. 10.1182/blood-2009-07-234880.
Cervantes F, Alvarez-Larrán A, Hernández-Boluda JC, Sureda A, Torrebadell M, Montserrat E: Erythropoietin treatment of the anaemia of myelofibrosis with myeloid metaplasia: results in 20 patients and review of the literature. Br J Haematol. 2004, 127: 399-403. 10.1111/j.1365-2141.2004.05229.x.
Barosi G, Elliott M, Canepa L, Ballerini F, Piccaluga PP, Visani G, Marchetti M, Pozzato G, Zorat F, Tefferi A: Thalidomide in myelofibrosis with myeloid metaplasia: a pooled-analysis of individual patient data from five studies. Leuk Lymphoma. 2002, 43: 2301-2307. 10.1080/1042819021000040008.
Mesa RA, Steensma DP, Pardanani A, Li CY, Elliott M, Kaufmann SH, Wiseman G, Gray LA, Schroeder G, Reeder T, Zeldis JB, Tefferi A: A phase 2 trial of combination low-dose thalidomide and prednisone for the treatment of myelofibrosis with myeloid metaplasia. Blood. 2003, 101: 2534-2541. 10.1182/blood-2002-09-2928.
Tefferi A, Cortes J, Verstovsek S, Mesa RA, Thomas D, Lasho TL, Hogan WJ, Litzow MR, Allred JB, Jones D, Byrne C, Zeldis JB, Ketterling RP, McClure RF, Giles F, Kantarjian HM: Lenalidomide therapy in myelofibrosis with myeloid metaplasia. Blood. 2006, 108: 1158-1164. 10.1182/blood-2006-02-004572.
Cervantes F, Hernández-Boluda JC, Alvarez A, Nadal E, Montserrat E: Danazol treatment of idiopathic myelofibrosis with severe anemia. Haematologica. 2000, 85: 595-599.
Mesa RA, Yao X, Cripe LD, Li CY, Tefferi A, Tallman MS: Lenalidomide and prednisone for primary and post polycythemia vera/essential thrombocythemia myelofibrosis (MF): an Eastern Cooperative Oncology Group (ECOG) phase II trial [abstract]. Blood. 2008, 112: s1753-
Begna KH, Pardanani A, Mesa R, Litzow MR, Hogan WJ, Hanson CA, Tefferi A: Long-term outcome of pomalidomide therapy in myelofibrosis. Am J Hematol. 2012, 87: 66-68. 10.1002/ajh.22233.
Thapaliya P, Tefferi A, Pardanani A, Steensma DP, Camoriano J, Wu W, Geyer S, Mesa RA: International Working Group for Myelofibrosis Research and Treatment response assessment and long-term follow-up of 50 myelofibrosis patients treated with thalidomide-prednisone based regimens. Am J Hematol. 2011, 86: 96-98. 10.1002/ajh.21892.
Quintás-Cardama A, Kantarjian HM, Manshouri T, Thomas D, Cortes J, Ravandi F, Garcia-Manero G, Ferrajoli A, Bueso-Ramos C, Verstovsek S: Lenalidomide plus prednisone results in durable clinical, histopathologic, and molecular responses in patients with myelofibrosis. J Clin Oncol. 2009, 27: 4760-4766. 10.1200/JCO.2009.22.6548.
Jabbour E, Thomas D, Kantarjian H, Zhou L, Pierce S, Cortes J, Verstovsek S: Comparison of thalidomide and lenalidomide as therapy for myelofibrosis. Blood. 2011, 118: 899-902. 10.1182/blood-2010-12-325589.
Lofvenberg E, Wahlin A: Management of polycythaemia vera, essential thrombocythaemia and myelofibrosis with hydroxyurea. Eur J Haematol. 1988, 41: 375-381.
Manoharan A, Pitney WR: Chemotherapy resolves symptoms and reverses marrow fibrosis in myelofibrosis. Scand J Haematol. 1984, 33: 453-459.
Petti MC, Latagliata R, Spadea T, Spadea A, Montefusco E, Aloe Spiriti MA, Avvisati G, Breccia M, Pescarmona E, Mandelli F: Melphalan treatment in patients with myelofibrosis with myeloid metaplasia. Br J Haematol. 2002, 116: 576-581. 10.1046/j.0007-1048.2001.03331.x.
Verstovsek S, Kantarjian H, Mesa RA, Pardanani AD, Cortes-Franco J, Thomas DA, Estrov Z, Fridman JS, Bradley EC, Erickson-Viitanen S, Vaddi K, Levy R, Tefferi A: Safety and efficacy of INCB018424, a JAK1 and JAK2 inhibitor, in myelofibrosis. N Engl J Med. 2010, 363: 1117-1127. 10.1056/NEJMoa1002028.
Pardanani AD, Caramazza D, George G, Lasho TL, Hogan WJ, Litzow MR, Begna K, Hanson CA, McClure RF, Bavisotto LM, Smith G, Kowalski M, Sirhan S, Roberts AW, Gupta V, Gotlib J, Tefferi A: Safety and efficacy of CYT387, a JAK-1/2 inhibitor, for the treatment of myelofibrosis [abstract]. J Clin Oncol. 2011, 29: s6514-
JAKAFI prescribing information.http://www.accessdata.fda.gov/drugsatfda_docs/label/2013/202192s003lbl.pdf,
European Medicines Agency: Committee for Medicinal Products for Human Use:Jakavi assessment report.http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002464/WC500133226.pdf,
Novartis Pharmaceuticals Canada Inc: JAKAVI Product Monograph. 2013, Dorval, Quebec: Health Canada,http://hc-sc.gc.ca,
Scott BL, Gooley TA, Sorror ML, Rezvani AR, Linenberger ML, Grim J, Sandmaier BM, Myerson D, Chauncey TR, Storb R, Buxhofer-Ausch V, Radich JP, Appelbaum FR, Deeg HJ: The Dynamic International Prognostic Scoring System for myelofibrosis predicts outcomes after hematopoietic cell transplantation. Blood. 2012, 119: 2657-2664. 10.1182/blood-2011-08-372904.
Baxter EJ, Scott LM, Campbell PJ, East C, Fourouclas N, Swanton S, Vassiliou GS, Bench AJ, Boyd EM, Curtin N, Scott MA, Erber WN, Green AR: Cancer Genome Project: Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005, 365: 1054-1061. 10.1016/S0140-6736(05)71142-9.
Kralovics R, Passamonti F, Buser AS, Teo SS, Tiedt R, Passweg JR, Tichelli A, Cazzola M, Skoda RC: A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005, 352: 1779-1790. 10.1056/NEJMoa051113.
Levine RL, Wadleigh M, Cools J, Ebert BL, Wernig G, Huntly BJ, Boggon TJ, Wlodarska I, Clark JJ, Moore S, Adelsperger J, Koo S, Lee JC, Gabriel S, Mercher T, D’Andrea A, Frohling S, Dohner K, Marynen P, Vandenberghe P, Mesa RA, Tefferi A, Griffin JD, Eck MJ, Sellers WR, Meyerson M, Golub TR, Lee SJ, Gilliland DG: Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 2005, 7: 387-397. 10.1016/j.ccr.2005.03.023.
James C, Ugo V, Le Couédic JP, Staerk J, Delhommeau F, Lacout C, Garçon L, Raslova H, Berger R, Bennaceur-Griscelli A, Villeval JL, Constantinescu SN, Casadevall N, Vainchenker W: A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005, 434: 1144-1148. 10.1038/nature03546.
Verstovsek S, Mesa RA, Gotlib J, Levy RS, Gupta V, DiPersio JF, Catalano JV, Deininger M, Miller C, Silver RT, Talpaz M, Winton EF, Harvey JH, Arcasoy MO, Hexner E, Lyons RM, Paquette R, Raza A, Vaddi K, Erickson-Viitanen S, Koumenis IL, Sun W, Sandor V, Kantarjian HM: A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N Engl J Med. 2012, 366: 799-807. 10.1056/NEJMoa1110557.
Harrison C, Kiladjian JJ, Al-Ali HK, Gisslinger H, Waltzman R, Stalbovskaya V, McQuitty M, Hunter DS, Levy R, Knoops L, Cervantes F, Vannucchi AM, Barbui T, Barosi G: JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N Engl J Med. 2012, 366: 787-798. 10.1056/NEJMoa1110556.
Mesa RA, Gotlib J, Gupta V, DiPersio JF, Catalano J, Deininger MW, Shields A, Miller CB, Silver RT, Talpaz M, Winton EF, Harvey JH, Hare T, Erickson-Viitanen S, Sun W, Sandor VA, Levy RS, Kantarjian HM, Verstovskey S: Associations between improvements in myelofibrosis (MF) symptoms and quality of life measures with splenomegaly reduction in COMFORT-I: a randomized, double-blind, phase III trial of the JAK1 and JAK2 inhibitor ruxolitinib versus placebo in patients with MF [abstract]. Blood. 2011, 118: s3842-10.1182/blood-2010-12-327908.
Tefferi A: JAK inhibitors for myeloproliferative neoplasms: clarifying facts from myths. Blood. 2012, 119: 2721-2730. 10.1182/blood-2011-11-395228.
Guglielmelli P, Barosi G, Rambaldi A, Marchioli R, Masciulli A, Tozzi L, Biamonte F, Bartalucci N, Gattoni E, Lupo ML, Finazzi G, Pancrazzi A, Antonioli E, Susini MC, Pieri L, Malevolti E, Usala E, Occhini U, Grossi A, Caglio S, Paratore S, Bosi A, Barbui T, Vannucchi AM, AIRC-Gruppo Italiano Malattie Mieloproliferative (AGIMM) investigators: Safety and efficacy of everolimus, a mTOR inhibitor, as single agent in a phase 1/2 study in patients with myelofibrosis. Blood. 2011, 118: 2069-2076. 10.1182/blood-2011-01-330563.
Mascarenhas J, Lu M, Li T, Petersen B, Hochman T, Najfeld V, Goldberg JD, Hoffman R: A phase I study of panobinostat (LBH589) in patients with primary myelofibrosis (PMF) and post-polycythaemia vera/essential thrombocythaemia myelofibrosis (post-PV/ET MF). Br J Haematol. 2013, 161: 68-75. 10.1111/bjh.12220.
Rambaldi A, Finazzi G, Vannucchi AM, Martinelli V, Rodeghiero F, Nobile F, Specchia G, Pogliani EM, Olimpieri OM, Fioritoni G, Musolino C, Saglio G, Sivera P, Barosi G, Di Tollo S, Barbui T: A phase II study of the HDAC inhibitor givinostat in combination with hydroxyurea in patients with polycythemia vera resistant to hydroxyurea monotherapy [abstract]. Blood. 2011, 118: s 1748-
Merchant AA, Matsui W: Targeting hedgehog—a cancer stem cell pathway. Clin Cancer Res. 2010, 16: 3130-3140. 10.1158/1078-0432.CCR-09-2846.
Bhagwat N, Keller MD, Rampal R, Koppikar P, Shank K, De Stanchina E, Rose K, Amakye D, Levine RL: Improved efficacy of combination of JAK2 and hedgehog inhibitors in myelofibrosis [abstract]. Blood. 2013, 122: s666-10.1182/blood-2012-10-461830.
Goldenson B, Malinge S, Stein BL, Lasho TL, Breyfogle L, Schultz R, Yang Q, Gilles-Gendre L, Koppikar P, Abdel-Wahab O, Ebert BL, Pardanani A, Gurbuxani S, Levine RS, Mullally A, Tefferi A, Crispino JD: Aurora A kinase is a novel therapeutic target in the myeloproliferative neoplasms [abstract]. Blood. 2013, 122: s109-10.1182/blood-2013-03-494039.
McMahon AP, Ingham PW, Tabin CJ: Developmental roles and clinical significance of hedgehog signaling. Curr Top Dev Biol. 2003, 53: 1-114.
Mar BG, Amakye D, Aifantis I, Buonamici S: The controversial role of the hedgehog pathway in normal and malignant hematopoiesis. Leukemia. 2011, 25: 1665-1673. 10.1038/leu.2011.143.
Palis J, Yoder MC: Yolk-sac hematopoiesis: the first blood cells of mouse and man. Exp Hematol. 2001, 29: 927-936. 10.1016/S0301-472X(01)00669-5.
Wong PM, Chung SW, Chui DH, Eaves CJ: Properties of the earliest clonogenic hemopoietic precursors to appear in the developing murine yolk sac. Proc Natl Acad Sci USA. 1986, 83: 3851-3854. 10.1073/pnas.83.11.3851.
Dyer MA, Farrington SM, Mohn D, Munday JR, Baron MH: Indian hedgehog activates hematopoiesis and vasculogenesis and can respecify prospective neurectodermal cell fate in the mouse embryo. Development. 2001, 128: 1717-1730.
St-Jacques B, Hammerschmidt M, McMahon AP: Indian hedgehog signaling regulates proliferation and differentiation of chondrocytes and is essential for bone formation. Genes Dev. 1999, 13: 2072-2086. 10.1101/gad.13.16.2072.
Maye P, Becker S, Kasameyer E, Byrd N, Grabel L: Indian hedgehog signaling in extraembryonic endoderm and ectoderm differentiation in ES embryoid bodies. Mech Dev. 2000, 94: 117-132. 10.1016/S0925-4773(00)00304-X.
Byrd N, Becker S, Maye P, Narasimhaiah R, St-Jacques B, Zhang X, McMahon J, McMahon A, Grabel L: Hedgehog is required for murine yolk sac angiogenesis. Development. 2002, 129: 361-372.
Zhang XM, Ramalho-Santos M, McMahon AP: Smoothened mutants reveal redundant roles for Shh and Ihh signaling including regulation of L/R symmetry by the mouse node. Cell. 2001, 106: 781-792.
Cridland SO, Keys JR, Papathanasiou P, Perkins AC: Indian hedgehog supports definitive erythropoiesis. Blood Cells Mol Dis. 2009, 43: 149-155. 10.1016/j.bcmd.2009.04.004.
Gering M, Patient R: Hedgehog signaling is required for adult blood stem cell formation in zebrafish embryos. Dev Cell. 2005, 8: 389-400. 10.1016/j.devcel.2005.01.010.
Peeters M, Ottersbach K, Bollerot K, Orelio C, de Bruijn M, Wijgerde M, Dzierzak E: Ventral embryonic tissues and hedgehog proteins induce early AGM hematopoietic stem cell development. Development. 2009, 136: 2613-2621. 10.1242/dev.034728.
Dierks C, Beigi R, Guo GR, Zirlik K, Stegert MR, Manley P, Trussell C, Schmitt-Graeff A, Landwerlin K, Veelken H, Warmuth M: Expansion of Bcr-Abl-positive leukemic stem cells is dependent on hedgehog pathway activation. Cancer Cell. 2008, 14: 238-249. 10.1016/j.ccr.2008.08.003.
Trowbridge JJ, Scott MP, Bhatia M: Hedgehog modulates cell cycle regulators in stem cells to control hematopoietic regeneration. Proc Natl Acad Sci USA. 2006, 103: 14134-14139. 10.1073/pnas.0604568103.
Siggins SL, Nguyen NY, McCormack MP, Vasudevan S, Villani R, Jane SM, Wainwright BJ, Curtis DJ: The hedgehog receptor patched1 regulates myeloid and lymphoid progenitors by distinct cell-extrinsic mechanisms. Blood. 2009, 114: 995-1004. 10.1182/blood-2009-03-208330.
Zhao C, Chen A, Jamieson CH, Fereshteh M, Abrahamsson A, Blum J, Kwon HY, Kim J, Chute JP, Rizzieri D, Munchhof M, VanArsdale T, Beachy PA, Reya T: Hedgehog signalling is essential for maintenance of cancer stem cells in myeloid leukaemia. Nature. 2009, 458: 776-779. 10.1038/nature07737.
Merchant A, Joseph G, Wang Q, Brennan S, Matsui W: Gli1 regulates the proliferation and differentiation of HSCs and myeloid progenitors. Blood. 2010, 115: 2391-2396. 10.1182/blood-2009-09-241703.
McIntyre BA, Ramos-Mejia V, Rampalli S, Mechael R, Lee JH, Alev C, Sheng G, Bhatia M: Gli3-mediated hedgehog inhibition in human pluripotent stem cells initiates and augments developmental programming of adult hematopoiesis. Blood. 2013, 121: 1543-1552. 10.1182/blood-2012-09-457747.
Lau CI, Outram SV, Saldana JI, Furmanski AL, Dessens JT, Crompton T: Regulation of murine normal and stress-induced erythropoiesis by desert hedgehog. Blood. 2012, 119: 4741-4751. 10.1182/blood-2011-10-387266.
Cooper CL, Hardy RR, Reth M, Desiderio S: Non-cell-autonomous hedgehog signaling promotes murine B lymphopoiesis from hematopoietic progenitors. Blood. 2012, 119: 5438-5448. 10.1182/blood-2011-12-397976.
Gao J, Graves S, Koch U, Liu S, Jankovic V, Buonamici S, El Andaloussi A, Nimer SD, Kee BL, Taichman R, Radtke F, Aifantis I: Hedgehog signaling is dispensable for adult hematopoietic stem cell function. Cell Stem Cell. 2009, 4: 548-558. 10.1016/j.stem.2009.03.015.
Hofmann I, Stover EH, Cullen DE, Mao J, Morgan KJ, Lee BH, Kharas MG, Miller PG, Cornejo MG, Okabe R, Armstrong SA, Ghilardi N, Gould S, de Sauvage FJ, McMahon AP, Gilliland DG: Hedgehog signaling is dispensable for adult murine hematopoietic stem cell function and hematopoiesis. Cell Stem Cell. 2009, 4: 559-567. 10.1016/j.stem.2009.03.016.
Chaklader M, Das P, Pereira JA, Chaudhuri S, Law S: Altered canonical hedgehog-gli signalling axis in pesticide-induced bone marrow aplasia mouse model. Arh Hig Rada Toksikol. 2012, 63: 271-282.
Mandal L, Martinez-Agosto JA, Evans CJ, Hartenstein V, Banerjee U: A hedgehog- and antennapedia-dependent niche maintains Drosophila haematopoietic precursors. Nature. 2007, 446: 320-324. 10.1038/nature05585.
Kobune M, Ito Y, Kawano Y, Sasaki K, Uchida H, Nakamura K, Dehari H, Chiba H, Takimoto R, Matsunaga T, Terui T, Kato J, Niitsu Y, Hamada H: Indian hedgehog gene transfer augments hematopoietic support of human stromal cells including NOD/SCID-β2m−/− repopulating cells. Blood. 2004, 104: 1002-1009. 10.1182/blood-2003-09-3347.
Tokusumi Y, Tokusumi T, Shoue DA, Schulz RA: Gene regulatory networks controlling hematopoietic progenitor niche cell production and differentiation in the Drosophila lymph gland. PLoS One. 2012, 7: e41604-10.1371/journal.pone.0041604.
Boyd AL, Salci KR, Shapovalova Z, McIntyre BA, Bhatia M: Nonhematopoietic cells represent a more rational target of in vivo hedgehog signaling affecting normal or acute myeloid leukemia progenitors. Exp Hematol. 2013, 41: 858-869. 10.1016/j.exphem.2013.05.287.
Zingariello M, Martelli F, Ciaffoni F, Masiello F, Ghinassi B, D’Amore E, Massa M, Barosi G, Sancillo L, Li X, Goldberg JD, Rana RA, Migliaccio AR: Characterization of the TGF-β1 signaling abnormalities in the Gata1low mouse model of myelofibrosis. Blood. 2013, 121: 3345-3363. 10.1182/blood-2012-06-439661.
Astorga J, Carlsson P: Hedgehog induction of murine vasculogenesis is mediated by Foxf1 and Bmp4. Development. 2007, 134: 3753-3761. 10.1242/dev.004432.
Okabe S, Tauchi T, Tanaka Y, Katagiri S, Ohyashiki K: Effects of the hedgehog inhibitor GDC-0449, alone or in combination with dasatinib, on BCR-ABL-positive leukemia cells. Stem Cells Dev. 2012, 21: 2939-2948. 10.1089/scd.2012.0016.
Katagiri S, Tauchi T, Okabe S, Minami Y, Kimura S, Maekawa T, Naoe T, Ohyashiki K: Combination of ponatinib with hedgehog antagonist vismodegib for therapy-resistant BCR-ABL1-positive leukemia. Clin Cancer Res. 2013, 19: 1422-1432. 10.1158/1078-0432.CCR-12-1777.
Tauchi T, Katagiri S, Okabe S, Minami Y, Naoe T, Ohyashiki K: Targeting the hedgehog signaling pathway limits the self-renewal of BCR-ABL1 positive leukemia vells: molecular mechanisms [abstract]. Blood. 2012, 120: s531-
David A, Zhang B, Ho Y, Buonamici S, Manley P, Holyoake T, Bhatia R, Copland M: Combination of the hedgehog pathway inhibitor LDE225 and nilotinib targets the leukemic stem cell population in chronic myeloid leukaemia [abstract]. Haematologica. 2011, 96: s0521-
Minami Y, Fukushima N, Naoe T: Effect of treatment with hedgehog inhibitor, PF-04449913, on leukemia-initiation potential in acute myeloid leukemia cells [abstract]. J Clin Oncol. 2011, 31: se13520-
Lin TL, Wang QH, Brown P, Peacock C, Merchant AA, Brennan S, Jones E, McGovern K, Watkins DN, Sakamoto KM, Matsui W: Self-renewal of acute lymphocytic leukemia cells is limited by the hedgehog pathway inhibitors cyclopamine and IPI-926. PLoS One. 2010, 5: e15262-10.1371/journal.pone.0015262.
Lang F, Badura S, Ruthardt M, Rieger MA, Ottmann OG: Modulation of leukemic stem cell self-renewal and cell fate decisions by inhibition of hedgehog signalling in human acute lymphoblastic leukemia (ALL) [abstract]. Blood. 2012, 120: s2578-
Trudel GC, Paliwal P, Lainas I: Dasatinib plus SMO antagonist versus dasatinib alone for treating patients (pts) with newly diagnosed Philadelphia chromosome-positive (Ph+) chronic myeloid leukemia in chronic phase (CML-CP): design of CA180-363, a phase II, open-label randomized trial [abstract]. J Clin Oncol. 2012, 30: sTPS6634-
Alves R, Ferreira M, Guarino P, Domingues C, Leite J, Gonçalves A, Sarmento-Ribeiro A: Vismodegib—an hedgehog pathway inhibitor induces cell death in an ALL cell line [abstract]. Haematologica. 2013, 98: sB1775-
Peacock CD, Wang Q, Gesell GS, Corcoran-Schwartz IM, Jones E, Kim J, Devereux WL, Rhodes JT, Huff CA, Beachy PA, Watkins DN, Matsui W: Hedgehog signaling maintains a tumor stem cell compartment in multiple myeloma. Proc Natl Acad Sci USA. 2007, 104: 4048-4053. 10.1073/pnas.0611682104.
Teglund S, Toftgard R: Hedgehog beyond medulloblastoma and basal cell carcinoma. Biochim Biophys Acta. 1805, 2010: 181-208.
Scales SJ, de Sauvage FJ: Mechanisms of hedgehog pathway activation in cancer and implications for therapy. Trends Pharmacol Sci. 2009, 30: 303-312. 10.1016/j.tips.2009.03.007.
Gailani MR, Stahle-Backdahl M, Leffell DJ, Glynn M, Zaphiropoulos PG, Pressman C, Unden AB, Dean M, Brash DE, Bale AE, Toftgard R: The role of the human homologue of Drosophila patched in sporadic basal cell carcinomas. Nat Genet. 1996, 14: 78-81. 10.1038/ng0996-78.
Xie J, Murone M, Luoh SM, Ryan A, Gu Q, Zhang C, Bonifas JM, Lam CW, Hynes M, Goddard A, Rosenthal A, Epstein EH, de Sauvage FJ: Activating Smoothened mutations in sporadic basal-cell carcinoma. Nature. 1998, 391: 90-92. 10.1038/34201.
Slade I, Murray A, Hanks S, Kumar A, Walker L, Hargrave D, Douglas J, Stiller C, Izatt L, Rahman N: Heterogeneity of familial medulloblastoma and contribution of germline PTCH1 and SUFU mutations to sporadic medulloblastoma. Fam Cancer. 2011, 10: 337-342. 10.1007/s10689-010-9411-0.
Tostar U, Malm CJ, Meis-Kindblom JM, Kindblom LG, Toftgard R, Unden AB: Deregulation of the hedgehog signalling pathway: a possible role for the PTCH and SUFU genes in human rhabdomyoma and rhabdomyosarcoma development. J Pathol. 2006, 208: 17-25. 10.1002/path.1882.
Dierks C, Grbic J, Zirlik K, Beigi R, Englund NP, Guo GR, Veelken H, Engelhardt M, Mertelsmann R, Kelleher JF, Schultz P, Warmuth M: Essential role of stromally induced hedgehog signaling in B-cell malignancies. Nat Med. 2007, 13: 944-951. 10.1038/nm1614.
Blotta S, Jakubikova J, Calimeri T, Roccaro AM, Amodio N, Azab AK, Foresta U, Mitsiades CS, Rossi M, Todoerti K, Molica S, Morabito F, Neri A, Tagliaferri P, Tassone P, Anderson KC, Munshi NC: Canonical and noncanonical hedgehog pathway in the pathogenesis of multiple myeloma. Blood. 2012, 120: 5002-5013. 10.1182/blood-2011-07-368142.
Jagani Z, Dorsch M, Warmuth M: Hedgehog pathway activation in chronic myeloid leukemia. Cell Cycle. 2010, 9: 3449-3456. 10.4161/cc.9.17.12945.
Sekulic A, Migden MR, Oro AE, Dirix L, Lewis KD, Hainsworth JD, Solomon JA, Yoo S, Arron ST, Friedlander PA, Marmur E, Rudin CM, Chang AL, Low JA, Mackey HM, Yauch RL, Graham RA, Reddy JC, Hauschild A: Efficacy and safety of vismodegib in advanced basal-cell carcinoma. N Engl J Med. 2012, 366: 2171-2179. 10.1056/NEJMoa1113713.
Gajjar A, Stewart CF, Ellison DW, Kaste S, Kun LE, Packer RJ, Goldman S, Chintagumpala M, Wallace D, Takebe N, Boyett JM, Gilbertson RJ, Curran T: Phase I study of vismodegib in children with recurrent or refractory medulloblastoma: a pediatric brain tumor consortium study. Clin Cancer Res. 2013, 19: 6305-6312. 10.1158/1078-0432.CCR-13-1425.
Jimeno A, Weiss GJ, Miller WH, Gettinger S, Eigl BJ, Chang AL, Dunbar J, Devens S, Faia K, Skliris G, Kutok J, Lewis KD, Tibes R, Sharfman WH, Ross RW, Rudin CM: Phase I study of the hedgehog pathway inhibitor IPI-926 in adult patients with solid tumors. Clin Cancer Res. 2013, 19: 2766-2774. 10.1158/1078-0432.CCR-12-3654.
Rodon J, Tawbi HA, Thomas AL, Stoller R, Turtschi CP, Baselga J, Sarantopoulos J, Mahalingam D, Shou Y, Moles MA, Yang L, Granvil C, Hurh E, Rose KL, Amakye DD, Dummer R, Mita AC: A phase 1, multicenter, open-Label, first-in-human, dose-escalation study of the oral hedgehog inhibitor sonidegib (LDE225) in patients with advanced solid tumors. Clin Cancer Res. 2014, Epub ahead of print
Kieran M, Geoerger B, Casanova M, Chisholm J, Aerts I, Bouffet E, Brandes AA, Leary SES, Sullivan M, Bailey S, Cohen K, Mason W, Kalambakas S, Deshpande P, Tai F, Hurh E, MacDonald TJ: A phase 1/2 safety and preliminary efficacy study of sonidegib (LDE225), a hedgehog pathway inhibitor, in pediatric and adult patients with relapsed or refractory medulloblastoma and other solid tumors [abstract]. Neuro Oncol. 2013, 15: s068-
Erivedge (vismodegib) Prescribing Information. 2012,http://www.cancer.gov/cancertopics/druginfo/fda-vismodegib,
Amakye D, Jagani Z, Dorsch M: Unraveling the therapeutic potential of the Hedgehog pathway in cancer. Nat Med. 2013, 19: 1410-1422. 10.1038/nm.3389.
Infinity stops phase 2 trials of saridegib in chondrosarcoma and myelofibrosis.http://www.reuters.com/article/2012/06/18/idUS87102+18-Jun-2012+BW20120618,
Jamieson C, Cortes JE, Oehler V, Baccarani M, Kantarjian HM, Papayannidis C, Rice KN, Zhang X, Shaik N, Courtney R, Levin WJ, Martinelli G: Phase 1 dose-escalation study of PF-04449913, an oral hedgehog (Hh) inhibitor, in patients with select hematologic malignancies [abstract]. Blood. 2011, 118: s424-
Huff CA, Padmanabhan S, Kelly KR, Somlo G, Camacho L, Zonder J, Fischer B, Lang L, Zhang S, Gestone T, Bennett KL: A phase I study of an oral hedgehog pathway antagonist, BMS-833923, in patients with relapsed or refractory multiple myeloma [abstract]. Blood. 2011, 118: s3993-
ClinicalTrials.gov:http://www.clinicaltrials.gov,
Lorusso PM, Rudin CM, Reddy JC, Tibes R, Weiss GJ, Borad MJ, Hann CL, Brahmer JR, Chang I, Darbonne WC, Graham RA, Zerivitz KL, Low J, Von Hoff DD: Phase I trial of hedgehog pathway inhibitor GDC-0449 in patients with refractory, locally-advanced or metastatic solid tumors. Clin Cancer Res. 2011, 17: 2502-2511. 10.1158/1078-0432.CCR-10-2745.
Guadagnuolo V, Papayannidis C, Iacobucci I, Durante S, Terragna C, Ottaviani E, Abbenante MC, Cattina F, Soverini S, Lama B, Toni L, Levin W, Courtney R, Baldazzi C, Curti A, Baccarani M, Jamieson C, Cortes J, Oehler V, McLachlan K, VanArsdale T, Martinelli G: Gas1 and Kif27 genes are strongly up-regulated biomarkers of hedgehog inhibition (PF-04449913) on leukemia stem cells in phase I acute myeloid leukemia and chronic myeloid leukemia treated patients [abstract]. Cancer Res. 2012, 72: s906-10.1158/1538-7445.AM2012-906.
Acknowledgments
The authors thank Jillian Brechbiel, PhD, and Karen Miller-Moslin, PhD, for medical editorial assistance with this manuscript. Financial support for editorial assistance was provided by Novartis Pharmaceuticals Corporation.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
RT: Research support for clinical trials from Astex, Merck, Celgene, Novartis, Tetralogic, Epizyme and Seattle Genetics. Novartis funding involves support for a clinical trial of the SMO inhibitor (LDE225) with 5-Azacitidine. RAM: Research support from Incyte, Genentech, Sanofi, Gilead, NS Pharma, Lilly, and Promedior.
Authors’ contributions
RT and RAM contributed to the literature analysis/interpretation and manuscript writing, edited/revised all drafts, and approved the final version of the manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Tibes, R., Mesa, R.A. Targeting hedgehog signaling in myelofibrosis and other hematologic malignancies. J Hematol Oncol 7, 18 (2014). https://doi.org/10.1186/1756-8722-7-18
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1756-8722-7-18