
Abstract
Recent years have witnessed much progress in both basic research and clinical trials regarding cancer immunotherapy with chimeric antigen receptor (CAR)-engineered T cells. The unique structure of CAR endows T cell tumor specific cytotoxicity and resistance to immunosuppressive microenvironment in cancers, which helps patients to better tackle the issue of immunological tolerance. Adoptive immunotherapy (AIT) using this supernatural T cell have gained momentum after decades of intense debates because of the promising results obtained from preclinical models and clinical trials. However, it is very important for us to evaluate thoroughly the challenges/obstacles before widespread clinical application, which clearly warrants more studies to improve our understanding of the mechanism underlying AIT. In this review, we focus on the critical issues related to the clinical outcomes of CAR-based adoptive immunotherapy and discuss the rationales to refine this new cancer therapeutic modality.
Similar content being viewed by others
Introduction
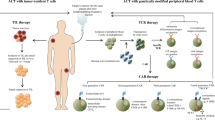
Adoptive immunotherapy (AIT) using chimeric antigen receptor-engineered T cells is a promising approach in cancer treatment. Significant progresses made in the past decades have contributed to the development of more efficient antitumor immunotherapy. Incorporation of a single chain variable fragment (scFv) of a tumor antigen specific antibody and signaling domains of T cell receptor (TCR) renders chimeric antigen receptor (CAR) the specificity of antibody as well as the cytotoxicity of cytotoxic T lymphocytes (CTLs) [1]. CAR endows T cells antigen specific recognition, activation and proliferation in an MHC independent manner. In addition, CAR bypasses many mechanisms through which cancer cells escape immunorecognition. These mechanisms include down-regulation of the MHC, reduced expression of costimulatory molecules, induction of suppressive cytokines and recruitment of regulatory T cells. Besides these beneficial effects, the technical feasibility of CAR makes it even more attractive in the development of cancer adoptive immunotherapy. The observations from preclinical and clinical studies have revealed a very encouraging therapeutic efficacy of the CAR-mediated immunotherapy in a variety of cancers including lymphoma, chronic lymphocytic leukemia, melanoma, and neuroblastoma [2]. Although there are many obstacles needed to be overcome before its widespread clinical use, it is still highly likely for the CAR-based immunotherapy to become a mainstay in clinical oncology field due to its improved tumoricidal activity. In this review, we will briefly discuss the critical issues related to the antitumor efficacy including the structure of CAR, the potential of T cells subsets, the gene modification of lymphocytes, the preconditioning regimens, etc.
Promising results of clinical trials
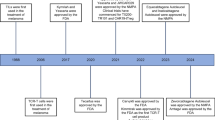
The promising observations made from in vitro studies and animal models have greatly encouraged physicians/scientists to conduct clinical studies. The efficacy and safety of adoptive CAR-engineered T cells have been evaluated in multiple oncological settings. Currently, about 30 ongoing clinical trials are under evaluation (http://www.clinicaltrials.gov). Many advances have been reported regarding the outcomes of these early clinical studies (Phase I/II) including the effect on hematological malignancies and some solid tumors.
To date, the most encouraging clinical observations have been achieved from patients with chronic lymphocytic leukemia (CLL) and lymphoma treated by CD19-orientated CAR T cells [3]. CD19 is expressed on most types of B-cell leukemia/lymphoma but absent in non-B lineage cells. Dr. June and the colleagues have made significant contributions that have greatly improved our understanding of the CAR T cells. In one of their reports, 2 out of 3 refractory CLL patients receiving CD19 CAR T cells therapy achieved complete response (CR), and another patient demonstrated partial response (PR) [4]. This proof of concept clinical investigations have demonstrated an excellent cell engraftment (up to 3 log expansion) and tumor cell lysis, eventually leading to complete remission. This pioneer work inspired numerous clinical studies focusing on CD19 for evaluating CAR technology. CD19 CAR studies account for one third of ongoing clinical trials with constantly increasing enrollments. Recent reports from seven academic institutes showed 25% overall CR rate (for review see [3, 5, 6]). In addition to CD19, other molecules such as CD20, CD22, CD30 and CD33 have also been tested for their potential as therapeutic targets for hematological malignancies [7, 8].
In parallel with the clinical trials in hematological malignancies, CAR-based therapy has also been conducted in solid tumors. CAR T cells targeting diasialoganglioside (GD2) antigen and L1-cell adhesion molecule (L1CAM) were generated for treatment of neuroblastoma [9]. Dr. Louis and the colleagues generated the first-generation CAR and modified the EBV-specific T cells. Enhanced in vivo persistence and survival of the adoptively transferred CAR-modified T cells resulted in a durable and complete response in 3 out of 11 patients [10]. Dr. Lamers’ team generated the CAR targeting carboxyl anhydrase IX (CAIX) to treat metastatic renal cell carcinoma and there was no objective clinical responses observed in any of the treated patients [11]. In addition, CARs have been generated against a number of surface molecules on solid tumors, including HER2 for colorectal cancer [12], folate receptor-α for ovarian cancer [13], carcinoembryonic antigen (CEA) for colorectal and breast cancer [14], epidermal growth factor receptor variant III (EGFRvIII) for neuroblastoma [15], and prostate-specific membrane antigen (PMSA) for prostate cancer [16]. Targeting solid tumors is more challenging compared to the treatment of hematological malignancies because of rare target antigens, poor T cells trafficking to tumor site, less cytotoxicity in local tumor immunosuppressive microenvironment.
Despite small sizes and different protocols, these phase I/II studies did demonstrate the feasibility and safety of CAR-based immunotherapy for the treatment of some types of cancer. While adverse effects are generally tolerable, the clinical efficacy of the first generation CAR is often insufficient. The CARs of the second and third generations have demonstrated improved efficacy and durable response, which motivated scientists for dynamic and innovative research in this field.
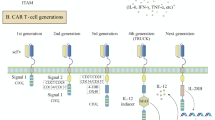
Evolving architecture of CAR
CARs have evolved considerably over the past decades. Basically, CAR is comprised of an extracellular antigen-binding moiety, a hinge and transmembrane (TM) domain, and an intracellular signaling element. The initial CAR of the first generation provides a proof of concept of the targeting and activation of T cells. The CAR of the second and third generations have been developed by addition of dual or triple costimulatory signaling domains in order to increase their cytotoxicity [17]. The most important function of multiple signaling receptors is to enhance signaling strength and persistence, subsequently increasing in their overall potency.
Targeting, the first step for the redirected T lymphocytes to function, is initiated at the single chain fragment variable (scFv) region of extracellular domain. The scFv determines the specificity of CAR and binds to target molecules through a similar mechanism as antigen-antibody interaction. Many factors including the affinity of scFv, the spatial topologic structure of epitopes, and antigen expression levels on tumor cells, have to be considered to optimize the CAR-mediated T-cell response [18]. Besides widely used scFv in many CARs, ligand or receptor can also be used as targeting moiety because the corresponding receptor or ligand are often overexpressed on tumor cells, such as peptide against vascular endothelial growth factor receptor (VEGFR) [19], and receptor for NKG2D [20]. The potential immunogenicity of murine-derived scFv limits the use of CAR in clinical setting. Utilizing humanized or fully human antibodies to construct CAR may lower the immunogenicity and avoid immune-mediated recognition leading to elimination of the genetically modified T cells.
A flexible hinge region between the targeting moiety and the TM domain seems to be important for accessibility to the epitope of the CAR-grafted T cells [21]. A study performed by Dr. Guest demonstrated that a flexible hinge region is needed for targeting membrane-proximal epitope, instead of membrane-distal epitopes [22]. Considering the spatial structure of the antigen, efficient antigen recognition of CAR might be modulated by different choices of target epitopes. Also, the TM domain is a pivotal region for CAR function, more specifically, the oligomeric status of CARs. Dr. Bridgeman and colleagues have demonstrated that CAR containing a CD3ζ TM domain form homodimers and are incorporated into the endogenous T cell receptor (TCR) complex [23]. Various transmembrane regions have been employed in CAR including those derived from CD28, CD3z, CD8, CD4, FcRγ, etc.
Intracellular signaling domain is of vital importance for CAR T cells to fulfill their antitumor function. Therefore, new generation of CAR contain a second signaling domain by adding the cytoplasmic domain of costimulatory receptors such as CD28, CD137 (4-1BB), CD134 (OX40), or inducible costimulator (ICOS). Both in vitro and in vivo studies have demonstrated that, in response to the target antigen, the modified T cells with the second or third generation CARs usually demonstrate sustained proliferation, enhanced production of cytokines and tumor lytic activity, and reduced activation-induced cell death. In a study of mouse model for comparison of the first and second generation CARs, Dr. Savoldo demonstrated the greater persistence of T cells expressing a CD28/CD3ζ-based CAR compared with T cells transduced by CAR with CD3ζ signaling only [24]. The third generation CAR usually shows better attributes of T cells than second generation due to a synergistic effect of triple signaling domains [25]. Nowadays, most researchers prefer to use the second generation CAR because of the concern about possible signaling leakage of the third generation CAR due to reduced activation threshold. As for costimulatory molecules per se, we cannot reach a clear conclusion about which one is better than the other. Previous observations indicate that inclusion of CD28 significantly increases IL-2 production compared to the inclusion of other molecules. The inclusion of CD137 may improve survival, and the use of the ICOS signaling seems the most efficient in target cell lysis [26–28].
Collectively, optimal designs of the CAR and a careful choice of the tumor associate antigen (TAA) are indispensable for the CAR-mediated therapy to attain a significant response. Although CARs in many clinical trials greatly differ in their configurations, comprehensive comparisons are still lacking. With the clinical results in mind, we need to attain a better understanding of optimal CAR signaling to promote sustained T-cell function and survival, to avoid undue proliferation, and to prevent premature death and rapid exhaustion. It is also desirable to add more elements such as homing and suicide genes to improve their functionality. It is imperative to develop a more competent and safer architecture of CAR, so-called the fourth generation CAR. Optimized design of the new generation CAR may be achieved by more extensive basic studies investigating the spatiotemporal dynamics of CAR-mediated molecular events and unraveling the molecular basis of T-cell activation by CARs.
Therapeutic potential of T cell subsets
In the past decades, researchers have tried to generate sufficient quantity of antigen-specific T cells based on the notion that the absolute number of transferred T cells is correlated with tumor responses. Most clinical trials take use of mixed populations of CD3+ T cells containing antigen-specific CD8+ and CD4+ T cells, and the phenotypic and functional attributes of the transferred T cells are subject to natural variation. As the heterogeneity and potential variable of polyclonal unselected cells could affect the efficacy and safety of cancer immunotherapy, precise detection and careful selection of the potentially most potent T cell subsets would increase their adoptive transfer efficiency.
It becomes increasingly clear that adoptive transfer of the less-differentiated T cell subsets is associated with superior T cell engraftment, persistence, and antitumor immunity, thus highly correlating with objective clinical responses [29]. An in vivo study demonstrated that adoptive transfer of central memory T cells (TCM) induced complete responses at the tested cell dose, whereas the mice in group of receiving effector memory T cells (TEM) suffered uncontrolled tumor growth [30]. This difference can be deciphered by the fact that TCM cells can self-renew and differentiate into effector T cells in vivo, while TEM cells have lost this plasticity. Subsequently, a subset of stem cell memory T cell (TSCM) has been demonstrated to have superior ability to mediate cancer regression compared to the TCM and TEM populations [31]. Therefore, ex vivo sorting/selection for superior T cell subsets like TCM or TSCM cells prior to adoptive transfer and/or gene transfer could improve in vivo persistence and therapeutic efficacy. Clinical trials have been conducting to evaluate the potential of the T cell subsets from the peripheral blood [32].
Many strategies have been utilized to obtain sufficient numbers of defined therapeutically clinical-grade CTL effectors. Current protocol includes isolation and activation using agonistic antibodies CD3 or γ-irradiated allogeneic peripheral blood mononuclear cell (PBMC) vs EBV-transformed B lymphoblastoid cell line (LCL) feeders, and a subsequent rapid expansion of cells in the presence of IL-2. Since ex vivo expansion inevitably drives T cell differentiation and results in the loss of in vivo antitumor efficacy, new strategies for handling cultured T cells have been developing recently. Traditional method for cell processing has been replaced by the automated manufacturing system. Activation of T cells can be induced by various kinds of antigen-non-specific artificial APCs (aAPCs) such as magnetic CD3/CD28 beads and K32 cell line (CD32-transfected precursor K562 chronic erythroleukemic cell line) [33].
IL-2 has been used for long time for T cell expansion. Recent evidence suggests that IL-2 may have some negative effects by depleting memory T cells and increasing the number of tumor protecting regulatory T cells (Tregs) [34]. In contrast, IL-15 can increase the persistence of CD8+ memory T lymphocytes, prevent Fas induced apoptosis and overcome tolerance of tumor specific CD8+ T cells [35]. The expansion of naive CD8+ T cells in the presence of IL-15 generates cells with the phenotypic and functional properties of naturally occurring TCM with superior antitumor capability. Therapeutic potentials of small molecular modulators in critical metabolic and developmental pathways involved in T cell differentiation have also been evaluated including those in the PI3K-AKT-mTOR and Wnt-β-catenin pathways [36].
Our understanding of the fundamental properties of heterogeneous subsets of T cells keep improving and mounting evidence indicates that selection of defined populations for gene modification is necessary to maximize the outcome of adoptive immunotherapy for cancer. The phenotypic and functional diversity of the genetically modified T cells may provide opportunities to enhance cancer immunotherapy. Because of severe adverse events reported in CAR clinical trial, it may become requisite to choose the defined T cell subsets in context of clinical setting. With recent advent of cell bioprocessing and genetic engineering technology, it is now possible to generate tumor-reactive CD8+ T cells of a defined subset. The in-depth understanding of phenotypes and functions of T cell subsets will have profound impact on future CAR T cell therapy.
Improved methods of T cell gene modification
CAR-based immunotherapy is dependent on the efficient genetic modifications of the infused T cells. Since extensive ex vivo T cell expansion is prerequisite for adoptive cell transfer, genetic modifications must be stable and require vector integration. For this reason, transduction using chromosome-integrating vectors like retroviruses or lentiviruses is superior to transfection with transient gene expression of DNA in genetically modified T cell therapy.
To date, most studies of CAR-related therapy utilize retroviruses or lentiviruses for gene delivery. Data from several sources indicate that retroviral vectors do not elicit clinically significant genotoxicity when used to deliver therapeutic genes to T cells [37]. Dr. Scholler reported long-term results from three clinical trials to evaluate retroviral vector– engineered T cells. In a clinical analysis of HIV and malignancy, which included over 200 patients with the follow-ups for as long as 11 years, transduced T cells were detected in 98% of patients with no evidence of integration-induced immortalization [38]. Lentivirus vectors have some advantages over retroviral vectors as they can transduce non-dividing cells. In addition, lentiviral vectors have higher cargo capacity and reduced susceptibility to gene silencing. Because of its safety profile, it is expected that the future use of lentiviral vectors will keep increasing [39]. Besides, Ad5/F35 adenoviral vector transduction were reported to mediate gene transfer in up to 10% of resting T cells and 30–45% of T cells after activation with phyto-haemagglutinin. Ad5/F35 vectors hold promise for adoptive transfer of engineered lymphocytes in clinical situations where expression of a transgene for less than a week is required [40].
Along with the advance of viral vectors, non-viral gene transduction systems have been developed for clinical trials as well. The lower costs and better safety profile of non-viral vectors have attracted much attention. Transposon-based system is one of the promising methods because of much higher efficiency of the transgene integration. Sleeping Beauty was the first transposon system used for modification of human T lymphocytes and demonstrated stable expression of CAR with killing of targeted cancer cells in vitro as well as in animal models [41]. PiggyBac transposons mediate stable gene expression in about 20% of primary T cells before selection, and the rate was increased to 40% after selection. The expression can sustain for over 9 weeks in culture through multiple logs of expansion [42]. Thus far, both Sleeping Beauty and PiggyBac have been used for gene modification of human T cells with CAR for HER2, CD19 and so on. RNA-based electroporation of lymphocytes is also an attractive approach because of the almost 100% transduction efficiencies, less genotoxic and transient expression [43]. The clinical trial was planned to treat the hematological malignancies.
While genetic-modification strategies provide us with the opportunity to harness the immune system against malignant disease, several issues must be taken into consideration when selecting the technology used to engineer cellular immunity. These factors include the size, number, and expression level of the transgenes, the requirement for permanent or transient genetic modification, and different delivery approaches for either constitutive or inducible transgene expression.
Optimizing regimen of host preconditioning
Numerous animal experiments as well as clinical studies in humans strongly suggest that the effectiveness of adoptively transferred T cells can be reinforced when combined with conventional cytotoxic agents such as cyclophosphamide and fludarabine, and occasionally concomitantly used with a low-dose irradiation. It was observed in adoptive transfer of tumor infiltrated lymphocytes (TILs) that preconditioning with systemic non-myeloablative chemotherapy can induce clear and reproducible responses in a substantial percentage (~50%) of patients [44]. Therefore, most recent clinical trials using CAR-engineered T cells incorporate a lymphodepletion step, which has demonstrated an increased antitumor response [45, 46]. Clearly, preconditioning chemotherapy plays a critical role in the efficacy of targeted T-cell therapy.
Several mechanisms may contribute to the increased efficacy of T cell-based immunotherapy in the lymphopaenic environment. Deletion of the suppressive CD4+CD25+ regulatory T (TReg) cells in preconditioning recipient will help the infused T cells to function normally [47]. Competition for the limited amount of the cytokines (so-called cytokine sink) between transferred and endogenous T cells interferes CD8+ T cell homeostasis. Preconditioning will eliminate cytokine sinks and creates space for the expansion of infused cells [48]. Transient eradication of endogenous lymphocytes might cause necrosis or apoptosis of tumor cells, which results in antigen presenting cells uptake of tumor antigens and the subsequent cross-presentation of these antigens to the adoptively transferred tumor-reactive CD8+ T cells [49].
The improved effectiveness of immunotherapy following a non-myeloablative lymphodepleting regimen encourages the researchers for more elegant conditioning regimens in selective or intensive ways. TReg cells might be selectively depleted with directed immunotoxins or suppressed by administering a cytokine such as tumor necrosis factor (TNF). Exogenous replenishment of certain cytokines may alleviate the sink effect coming from the endogenous T cells. Attempt of using cytokines such as IL-7, IL-15 and IL-21 might avoid suppressive function of IL-2 toward TReg cells. Intensive myeloablative preconditioning regimen with chemotherapy, total body irradiation and human stem cell transplantation (HSCT) is also under planned in human AIT protocol.
Conclusions and perspectives
CAR-modified T cells constitute a very appealing approach to cancer immunotherapy. Despite the promising results obtained from numerous clinical trials following the infusion of CAR-modified T cells, some severe adverse reactions were reported [12, 50, 51]. Recent reports have highlighted some key issues and future directions to avoid these adverse events. Care selection of candidate target antigens is essential for improved efficacy and safety of the CAR-based therapy. Additional factors needed to be considered include the density of target molecules, accessibility of the epitope, affinity of the scFv, flexibility of hinge, property of the signaling, etc. New elements are being evaluated including a controllable suicide gene like caspase 9 or HSV-TK as a safety switch [52], and a chemokine receptor for T cells to migrate to tumor site. Furthermore, the best sources of the selective T cell populations are getting attention for CAR T cell therapy. Many factors may contribute to the improved quality of the cells such as closed cell processing system, new activation/expansion methods, selection of T cell subsets with therapeutic potentials, improved culture condition, etc. Current protocols for cell bioengineering need to be simplified to facilitate widespread application of such treatment strategy. Finally, because it is technically challenging, labor intensive and very costly, most of the studies have been conducted in academic centers. Future work needs collective effort from regulatory organizations, biological companies, medical centers and academic institutions. We believe these collaborations, some of which are already in progress, would lead to the development of the comprehensive approaches to maximize the efficacy of the cancer immunotherapy.
References
Eshhar Z, Waks T, Gross G, Schindler DG: Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc Natl Acad Sci USA. 1993, 90 (2): 720-724. 10.1073/pnas.90.2.720.
Cheadle EJ, Sheard V, Hombach AA, Chmielewski M, Riet T, Berrevoets C, Schooten E, Lamers C, Abken H, Debets R: Chimeric antigen receptors for T-cell based therapy. Methods Mol Biol. 2012, 907: 645-666.
Kochenderfer JN, Rosenberg SA: Treating B-cell cancer with T cells expressing anti-CD19 chimeric antigen receptors. Nat Rev Clin Oncol. 2013, 10 (5): 267-276. 10.1038/nrclinonc.2013.46.
Porter DL, Levine BL, Kalos M, Bagg A, June CH: Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011, 365 (8): 725-733. 10.1056/NEJMoa1103849.
Brentjens RJ, Davila ML, Riviere I, Park J, Wang X, Cowell LG, Bartido S, Stefanski J, Taylor C, Olszewska M: CD19-Targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med. 2013, 5 (177): 177ra38-10.1126/scitranslmed.3005930.
Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, Teachey DT, Chew A, Hauck B, Wright JF: Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013, 368 (16): 1509-1518. 10.1056/NEJMoa1215134.
Xu XJ, Zhao HZ, Tang YM: Efficacy and safety of adoptive immunotherapy using anti-CD19 chimeric antigen receptor transduced T-cells: a systematic review of phase I clinical trials. Leuk Lymphoma. 2013, 54 (2): 255-260. 10.3109/10428194.2012.715350.
Haso W, Lee DW, Shah NN, Stetler-Stevenson M, Yuan CM, Pastan IH, Dimitrov DS, Morgan RA, FitzGerald DJ, Barrett DM: Anti-CD22-chimeric antigen receptors targeting B-cell precursor acute lymphoblastic leukemia. Blood. 2013, 121 (7): 1165-1174. 10.1182/blood-2012-06-438002.
Park JR, Digiusto DL, Slovak M, Wright C, Naranjo A, Wagner J, Meechoovet HB, Bautista C, Chang WC, Ostberg JR: Adoptive transfer of chimeric antigen receptor re-directed cytolytic T lymphocyte clones in patients with neuroblastoma. Mol Ther. 2007, 15 (4): 825-833.
Louis CU, Savoldo B, Dotti G, Pule M, Yvon E, Myers GD, Rossig C, Russell HV, Diouf O, Liu E: Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood. 2011, 118 (23): 6050-6056. 10.1182/blood-2011-05-354449.
Lamers CH, Sleijfer S, van Steenbergen S, van Elzakker P, van Krimpen B, Groot C, Vulto A, den Bakker M, Oosterwijk E, Debets R: Treatment of metastatic renal cell carcinoma with CAIX CAR-engineered T cells: clinical evaluation and management of on-target toxicity. Mol Ther. 2013, 21 (4): 904-912. 10.1038/mt.2013.17.
Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA: Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. 2010, 18 (4): 843-851. 10.1038/mt.2010.24.
Kandalaft LE, Powell DJ, Coukos G: A phase I clinical trial of adoptive transfer of folate receptor-alpha redirected autologous T cells for recurrent ovarian cancer. J Transl Med. 2012, 10: 157-10.1186/1479-5876-10-157.
Schlimper C, Hombach AA, Abken H, Schmidt-Wolf IG: Improved activation toward primary colorectal cancer cells by antigen-specific targeting autologous cytokine-induced killer cells. Clin Dev Immunol. 2012, 201 (2): 238924-
Morgan RA, Johnson LA, Davis JL, Zheng Z, Woolard KD, Reap EA, Feldman SA, Chinnasamy N, Kuan CT, Song H: Recognition of glioma stem cells by genetically modified T cells targeting EGFRvIII and development of adoptive cell therapy for glioma. Hum Gene Ther. 2012, 23 (10): 1043-1053. 10.1089/hum.2012.041.
Kloss CC, Condomines M, Cartellieri M, Bachmann M, Sadelain M: Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat Biotechnol. 2013, 31 (1): 71-75.
Shirasu N, Kuroki M: Functional design of chimeric T-cell antigen receptors for adoptive immunotherapy of cancer: architecture and outcomes. Anticancer Res. 2012, 32 (6): 2377-2383.
Hudecek M, Lupo-Stanghellini MT, Kosasih PL, Sommermeyer D, Jensen MC, Rader C, Riddell SR: Receptor affinity and extracellular domain modifications affect tumor recognition by ROR1-specific chimeric antigen receptor T cells. Clin Cancer Res. 2013, 19 (12): 3153-3164. 10.1158/1078-0432.CCR-13-0330.
Chinnasamy D, Tran E, Yu Z, Morgan RA, Restifo NP, Rosenberg SA: Simultaneous targeting of tumor antigens and the tumor vasculature using T lymphocyte transfer synergize to induce regression of established tumors in mice. Cancer Res. 2013, 73 (11): 3371-3380. 10.1158/0008-5472.CAN-12-3913.
Zhang T, Barber A, Sentman CL: Chimeric NKG2D modified T cells inhibit systemic T-cell lymphoma growth in a manner involving multiple cytokines and cytotoxic pathways. Cancer Res. 2007, 67 (22): 11029-11036. 10.1158/0008-5472.CAN-07-2251.
Hombach A, Hombach AA, Abken H: Adoptive immunotherapy with genetically engineered T cells: modification of the IgG1 Fc spacer domain in the extracellular moiety of chimeric antigen receptors avoids off-target activation and unintended initiation of an innate immune response. Gene Ther. 2010, 17 (10): 1206-1213. 10.1038/gt.2010.91.
Guest RD, Hawkins RE, Kirillova N, Cheadle EJ, Arnold J, O’Neill A, Irlam J, Chester KA, Kemshead JT, Shaw DM: The role of extracellular spacer regions in the optimal design of chimeric immune receptors: evaluation of four different scFvs and antigens. J Immunother. 2005, 28 (3): 203-211. 10.1097/01.cji.0000161397.96582.59.
Bridgeman JS, Hawkins RE, Bagley S, Blaylock M, Holland M, Gilham DE: The optimal antigen response of chimeric antigen receptors harboring the CD3zeta transmembrane domain is dependent upon incorporation of the receptor into the endogenous TCR/CD3 complex. J Immunol. 2010, 184 (12): 6938-6949. 10.4049/jimmunol.0901766.
Savoldo B, Ramos CA, Liu E, Mims MP, Keating MJ, Carrum G, Kamble RT, Bollard CM, Gee AP, Mei Z: CD28 Costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. J Clin Invest. 2011, 121 (5): 1822-1826. 10.1172/JCI46110.
Tammana S, Huang X, Wong M, Milone MC, Ma L, Levine BL, June CH, Wagner JE, Blazar BR, Zhou X: 4-1BB And CD28 signaling plays a synergistic role in redirecting umbilical cord blood T cells against B-cell malignancies. Hum Gene Ther. 2010, 21 (1): 75-86. 10.1089/hum.2009.122.
Finney HM, Akbar AN, Lawson AD: Activation of resting human primary T cells with chimeric receptors: costimulation from CD28, inducible costimulator, CD134, and CD137 in series with signals from the TCR zeta chain. J Immunol. 2004, 172 (1): 104-113.
Milone MC, Fish JD, Carpenito C, Carroll RG, Binder GK, Teachey D, Samanta M, Lakhal M, Gloss B, Danet-Desnoyers G: Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol Ther. 2009, 17 (8): 1453-1464. 10.1038/mt.2009.83.
Shen CJ, Yang YX, Han EQ, Cao N, Wang YF, Wang Y, Zhao YY, Zhao LM, Cui J, Gupta P: Chimeric antigen receptor containing ICOS signaling domain mediates specific and efficient antitumor effect of T cells against EGFRvIII expressing glioma. J Hematol Oncol. 2013, 6: 33-10.1186/1756-8722-6-33.
Klebanoff CA, Gattinoni L, Restifo NP: Sorting through subsets: which T-cell populations mediate highly effective adoptive immunotherapy?. J Immunother. 2012, 35 (9): 651-660. 10.1097/CJI.0b013e31827806e6.
Berger C, Jensen MC, Lansdorp PM, Gough M, Elliott C, Riddell SR: Adoptive transfer of effector CD8+ T cells derived from central memory cells establishes persistent T cell memory in primates. J Clin Invest. 2008, 118 (1): 294-305. 10.1172/JCI32103.
Gattinoni L, Lugli E, Ji Y, Pos Z, Paulos CM, Quigley MF, Almeida JR, Gostick E, Yu Z, Carpenito C: A human memory T cell subset with stem cell-like properties. Nat Med. 2011, 17 (10): 1290-1297. 10.1038/nm.2446.
Wang X, Naranjo A, Brown CE, Bautista C, Wong CW, Chang WC, Aguilar B, Ostberg JR, Riddell SR, Forman SJ: Phenotypic and functional attributes of lentivirus-modified CD19-specific human CD8+ central memory T cells manufactured at clinical scale. J Immunother. 2012, 35 (9): 689-701. 10.1097/CJI.0b013e318270dec7.
Turtle CJ, Riddell SR: Artificial antigen-presenting cells for use in adoptive immunotherapy. Cancer J. 2010, 16 (4): 374-381. 10.1097/PPO.0b013e3181eb33a6.
Antony PA, Paulos CM, Ahmadzadeh M, Akpinarli A, Palmer DC, Sato N, Kaiser A, Hinrichs CS, Klebanoff CA, Tagaya Y: Interleukin-2-dependent mechanisms of tolerance and immunity in vivo. J Immunol. 2006, 176 (9): 5255-5266.
Mueller YM, Makar V, Bojczuk PM, Witek J, Katsikis PD: IL-15 enhances the function and inhibits CD95/Fas-induced apoptosis of human CD4+ and CD8+ effector-memory T cells. Int Immunol. 2003, 15 (1): 49-58. 10.1093/intimm/dxg013.
Rao RR, Li Q, Odunsi K, Shrikant PA: The mTOR kinase determines effector versus memory CD8+ T cell fate by regulating the expression of transcription factors T-bet and eomesodermin. Immunity. 2010, 32 (1): 67-78. 10.1016/j.immuni.2009.10.010.
Colovos C, Villena-Vargas J, Adusumilli PS: Safety and stability of retrovirally transduced chimeric antigen receptor T cells. Immunotherapy. 2012, 4 (9): 899-902. 10.2217/imt.12.91.
Scholler J, Brady TL, Binder-Scholl G, Hwang WT, Plesa G, Hege KM, Vogel AN, Kalos M, Riley JL, Deeks SG: Decade-long safety and function of retroviral-modified chimeric antigen receptor T cells. Sci Transl Med. 2012, 4 (132): 132ra53-10.1126/scitranslmed.3003761.
McGarrity GJ, Hoyah G, Winemiller A, Andre K, Stein D, Blick G, Greenberg RN, Kinder C, Zolopa A, Binder-Scholl G: Patient monitoring and follow-up in lentiviral clinical trials. J Gene Med. 2013, 15 (2): 78-82. 10.1002/jgm.2691.
Schroers R, Hildebrandt Y, Hasenkamp J, Glass B, Lieber A, Wulf G, Piesche M: Gene transfer into human T lymphocytes and natural killer cells by Ad5/F35 chimeric adenoviral vectors. Exp Hematol. 2004, 32 (6): 536-546. 10.1016/j.exphem.2004.03.010.
Maiti SN, Huls H, Singh H, Dawson M, Figliola M, Olivares S, Rao P, Zhao YJ, Multani A, Yang G: Sleeping beauty system to redirect T-cell specificity for human applications. J Immunother. 2013, 36 (2): 112-123. 10.1097/CJI.0b013e3182811ce9.
Nakazawa Y, Huye LE, Salsman VS, Leen AM, Ahmed N, Rollins L, Dotti G, Gottschalk SM, Wilson MH, Rooney CM: PiggyBac-mediated cancer immunotherapy using EBV-specific cytotoxic T-cells expressing HER2-specific chimeric antigen receptor. Mol Ther. 2011, 19 (12): 2133-2143. 10.1038/mt.2011.131.
Barrett DM, Zhao Y, Liu X, Jiang S, Carpenito C, Kalos M, Carroll RG, June CH, Grupp SA: Treatment of advanced leukemia in mice with mRNA engineered T cells. Hum Gene Ther. 2011, 22 (12): 1575-1586. 10.1089/hum.2011.070.
Dudley ME, Yang JC, Sherry R, Hughes MS, Royal R, Kammula U, Robbins PF, Huang J, Citrin DE, Leitman SF: Adoptive cell therapy for patients with metastatic melanoma: evaluation of intensive myeloablative chemoradiation preparative regimens. J Clin Oncol. 2008, 26 (32): 5233-5239. 10.1200/JCO.2008.16.5449.
Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ, Citrin DE, Restifo NP, Robbins PF, Wunderlich JR: Durable complete responses in heavily pretreted patients with metastatic melanoma using T cell transfer immunotherapy. Clin Cancer Res. 2011, 17 (13): 4550-4557. 10.1158/1078-0432.CCR-11-0116.
Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, June CH: T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. 2011, 3 (95): 95ra73-10.1126/scitranslmed.3002842.
Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, Evdemon-Hogan M, Conejo-Garcia JR, Zhang L, Burow M: Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nature Med. 2004, 10 (9): 942-949. 10.1038/nm1093.
Gattinoni L, Finkelstein SE, Klebanoff CA, Antony PA, Palmer DC, Spiess PJ, Hwang LN, Yu Z, Wrzesinski C, Heimann DM: Removal of homeostatic cytokine sinks by lymphodepletion enhances the efficacy of adoptively transferred tumour-specific CD8+ T cells. J Exp Med. 2005, 202 (7): 907-912. 10.1084/jem.20050732.
Russo V, Tanzarella S, Dalerba P, Rigatti D, Rovere P, Villa A, Bordignon C, Traversari C: Dendritic cells acquire the MAGE-3 human tumour antigen from apoptotic cells and induce a class I-restricted T cell response. Proc Natl Acad Sci USA. 2000, 97 (5): 2185-2190. 10.1073/pnas.040540197.
Brentjens R, Yeh R, Bernal Y, Riviere I, Sadelain M: Treatment of chronic lymphocytic leukemia with genetically targeted autologous T cells: case report of an unforeseen adverse event in a phase I clinical trial. Mol Ther. 2010, 18 (4): 666-668. 10.1038/mt.2010.31.
Kochenderfer JN, Dudley ME, Feldman SA, Wilson WH, Spaner DE, Maric I, Stetler-Stevenson M, Phan GQ, Hughes MS, Sherry RM: B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric- antigen -receptor-transduced T cells. Blood. 2012, 119 (12): 2709-2720. 10.1182/blood-2011-10-384388.
Di Stasi A, Tey SK, Dotti G, Fujita Y, Kennedy-Nasser A, Martinez C, Straathof K, Liu E, Durett AG, Grilley B: Inducible apoptosis as a safety switch for adoptive cell therapy. N Engl J Med. 2011, 365 (18): 1673-1683. 10.1056/NEJMoa1106152.
Acknowledgments
This work was supported by National Natural Science Foundation of China (No. 81172415, 81241077).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
All authors have contributed to write and revise the manuscripts. All authors read and approved the final manuscript.
Ethan Q Han, Xiu-ling Li contributed equally to this work.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Han, E.Q., Li, Xl., Wang, Cr. et al. Chimeric antigen receptor-engineered T cells for cancer immunotherapy: progress and challenges. J Hematol Oncol 6, 47 (2013). https://doi.org/10.1186/1756-8722-6-47
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1756-8722-6-47