
Abstract
The diverse pathways and molecules involved in angiogenesis, the formation of new blood vessels, have been targeted for the treatment of colorectal and other cancers. Vascular endothelial growth factor (VEGF)-A binding to VEGF receptor (VEGFR)-2 is believed to be the key signaling pathway mediating angiogenesis. Other VEGF pathways involved in angiogenesis include VEGF-A, VEGF-B, and placental growth factor binding to VEGFR-1, and VEGF-C and VEGF-D binding to VEGFR-2 and VEGFR-3. VEGF signaling also intersects with other pathways, including angiopoietin/Tie, Notch, hypoxia-inducible factor, and integrin pathways. The roles of these pathways in tumor angiogenesis and in various human cancers will be explored in this article. In addition, preclinical and clinical data on bevacizumab, aflibercept (known as ziv-aflibercept in the US), and investigational antiangiogenic agents in development for the treatment of colorectal and other cancers will be reviewed.
Similar content being viewed by others

Introduction
Angiogenesis refers to the formation of new blood vessels [1]. As blood vessels are needed to supply nutrients and oxygen to tissues [2], angiogenesis plays an essential role in normal growth and development as well as in the development of cancer [1, 3, 4]. In normal development, angiogenesis is needed for embryonic development, bone formation, and the function of ovaries and other endocrine glands [4]. In cancer, angiogenesis is required for tumor growth and metastasis [1, 3].
Angiogenesis is a highly regulated, complex process orchestrated by a number of intersecting pathways, including vascular endothelial growth factor (VEGF), angiopoietins, Notch, and integrins. In normal tissues, there is a balance between proangiogenic and antiangiogenic factors [2]. During physiologic angiogenesis in adults, there is a temporary shift to proangiogenic factors; this is held in check by inhibitory mechanisms [1, 2]. In contrast to normal angiogenesis, tumor angiogenesis is outside of the control of normal physiologic inhibition, and there is an imbalance of proangiogenic and antiangiogenic factors [1, 2]. The diversity of pathways and molecules involved in angiogenesis may offer avenues for therapeutic intervention. This article aims to provide an overview of tumor angiogenesis, focusing on the role of VEGF. It will also review preclinical and clinical evidence for the use of antiangiogenic agents in the treatment of colorectal and other cancers.
Tumor angiogenesis and the role of VEGF
Tumor angiogenesis typically involves the formation of new blood vessels from pre-existing vessels in a process known as sprouting angiogenesis [1, 5]. Tumors may also co-opt pre-existing vessels [6]. Compared with the normal vasculature, the tumor vasculature is abnormal in a number of different ways. There is loss of the normal vascular hierarchy of arterioles, capillaries, and venules [7, 8]. The endothelial layer is irregular and contains spaces that contribute to the leakiness of tumor vessels [9], proliferating tumor cells cause compression of vessels [10], and interstitial pressure is increased [11]. The ability of tumor vessels to supply oxygen and remove waste products is thus compromised, resulting in hypoxia and acidosis in the tumor microenvironment [12, 13]. Abnormalities are also observed in other tumor vasculature components such as pericytes and the basement membrane. Pericytes have an abnormally weak attachment with endothelial cells and have cytoplasmic processes that extend away from the vessel wall [8]. The basement membrane is variable in thickness, has a loose association with endothelial cells and pericytes, and has extensions away from the vessel wall [14]. Endothelial cells in tumor vessels also show altered gene expression [15, 16] and cytogenetic abnormalities such as aneuploidy [17].
The VEGF signaling pathway
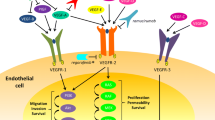
The mammalian VEGF signaling pathway includes 5 glycoproteins from the VEGF family (VEGF-A, VEGF-B, VEGF-C, VEGF-D, and placental growth factor [PlGF]), 3 receptors (VEGF receptor [VEGFR]-1 [FLT-1], VEGFR-2 [FLK-1/KDR], and VEGFR-3 [FLT-4]), and 2 co-receptors (neuropilin [NRP]-1 and NRP-2) (Figure 1) [5, 18–26]. In addition, there are multiple isoforms of VEGF-A (VEGF121, VEGF165, VEGF189, and VEGF206) [5]. The receptors for the various VEGF ligands are tyrosine kinases and are found primarily on vascular endothelial cells [18, 26].

The VEGF family. Positive and negative modulation of angiogenesis by VEGFR1 ligands [26].
VEGF-A binding to VEGFR-2 is believed to be the key signaling pathway mediating angiogenesis [18, 27]. VEGF-A enhances endothelial cell proliferation and survival, promotes endothelial cell migration, increases vascular permeability, and alters gene expression [5, 18]. Levels of VEGF-A have been shown to be increased in many cancers, including colorectal, prostate, and breast [28]. Downstream signaling activated by VEGF-A binding to VEGFR-2 includes the PLC-gamma/PKC/Ras/Raf/MEK/MAPK signaling pathway [29, 30]. Other VEGF family members, along with other signaling mediators, affect and overlap with the function of VEGF-A [26, 31, 32]. For instance, PlGF, fibroblast growth factor (FGF), and platelet-derived growth factor (PDGF) can modulate the effects of VEGF-A on angiogenesis, and VEGF-A can form heterodimers with PlGF or VEGF-B [26]. Because of alternative splice variants, an enormous diversity of heterodimerization with different functional properties may be formed, which presents a challenge to assessing the functional consequences. In addition, VEGF-A has overlapping function with VEGF-C and VEGF-D [26]. VEGF-A-mediated signaling also intersects with angiopoietin (Ang)/Tie, Notch/delta-like ligand (Dll)4, hypoxia-inducible factor (HIF)-1α and HIF-2α, and integrin pathways [26, 31, 32] (Figure 2).

Selected crosstalk between VEGF and other signaling pathways. VEGF signaling intersects with a number of other signaling pathways, including Ang/Tie, Notch/Dll4, integrins, and hypoxia to orchestrate tumor angiogenesis [26, 27, 31–35]. Ang = angiopoietin; Dll4 = delta-like ligand 4; HIF = hypoxia-inducible factor; PlGF = placenta growth factor; VEGF = vascular endothelial growth factor; VEGFR = vascular endothelial growth factor receptor. Dashed arrow indicates Ang-2 can serve as a partial Tie-2 agonist under certain conditions.
Like VEGFR-2, VEGFR-1 is expressed on vascular endothelial cells, but it is also expressed on other cell types, including monocytes and macrophages [26, 36]. The affinity of VEGFR-1 for VEGF-A is greater than that of VEGFR-2, although it has weak kinase activity, and it is thought to be a negative regulator of angiogenesis [37]. In cancer, VEGFR-1 appears to play a role in the epithelial-to-mesenchymal transition [38, 39]. VEGF-B binding to VEGFR-1 was shown to be associated with increased microvascular density in oral squamous cell carcinoma [40], but it was not associated with tumor vascularity in breast cancer [41] and it inhibited tumor growth in a mouse pancreatic neuroendocrine tumor model [42]. In a non-tumor model of pathological angiogenesis, VEGF-B promoted survival of endothelial cells, pericytes, and smooth muscle cells and upregulated the expression of prosurvival genes [43]. VEGF-B shows increased expression in multiple cancers, including ovarian, colorectal, renal cell, and prostate; its expression is associated with disease stage and expression of its receptor, VEGFR-1, predicts poor prognosis [36].
PlGF, another ligand for VEGFR-1, is thought to play a role in the angiogenic switch in pathological conditions [44]. PlGF binding to VEGFR-1 increases VEGF-A expression [45] and has a synergistic effect on VEGF-A signaling in cancer and other pathological forms of angiogenesis [44]. PlGF appears to promote the growth of tumor cells in an autocrine/paracrine manner [46]. PlGF has been shown to be upregulated in and prognostic for cancers, including gastric, colorectal, lung, breast, renal cell, hepatocellular, and brain [36].
VEGF-C and VEGF-D bind to the receptors VEGFR-2 and VEGFR-3 [26]. VEGF-C expression is associated with advanced metastatic disease for colorectal cancer [47] and it plays a role in lymphangiogenesis and/or metastasis to lymph nodes in multiple cancers, including colorectal [48] and breast [49–51]. VEGF-D is also involved in lymphangiogenesis and lymphatic metastasis [52, 53].
Crosstalk between the VEGF signaling pathway and other angiogenic signaling pathways
The angiopoietins Ang-1, Ang-2, and Ang-4 bind to the receptor tyrosine kinases Tie1 and Tie2 on vascular endothelial cells and are involved in the angiogenic switch, metastasis, and lymphangiogenesis [6, 31]. Ang-1 is expressed by mural cells, fibroblasts, and non-vascular normal and tumor cells, whereas Ang-2 is expressed primarily by endothelial cells and behaves in an autocrine manner [31]. Overexpression of Ang-2 has been shown to be associated with poor prognosis for a number of different cancers [31]. The Ang/Tie signaling pathway interacts with VEGF-A-mediated signaling in tumor angiogenesis [6, 31]. Ang-2 is upregulated by proangiogenic factors, including VEGF-A, PDGFB, and insulin-like growth factor (IGF)1 [31] (Figure 2). Ang-2 causes pericytes to dissociate from endothelial cells in pre-existing vessels and, in the presence of VEGF-A, this leads to angiogenesis [6, 31]. Ang-1 expressed by pericytes appears to help to maintain the integrity of blood vessels [31].
The Notch receptors Notch 1 and Notch 4 and the ligands jagged 1, Dll1, and Dll4 are expressed by vascular endothelial cells [27] and are involved in sprouting angiogenesis [31]. There are multiple modes of crosstalk between Notch/Dll4 and VEGF signaling (Figure 2). Dll4 expression and Notch signaling are induced by VEGF-A and VEGF-C [27, 33]. VEGFR-3 regulates Notch signaling and the conversion of tip cells to stalk cells in sprouting angiogenesis [34]. Notch upregulates VEGFR-3 and allows VEGF-A/VEGFR-2-independent angiogenesis [35]. In addition, Dll4/Notch signaling may mediate resistance to anti-VEGF therapy through multiple different mechanisms, e.g. decreased levels of hypoxia-induced VEGF and increased levels of the VEGF receptor VEGFR1 in the tumor stroma, decreased levels of VEGFR2 in large blood vessels, and reduced levels of VEGFR3 overall [54].
Integrins and hypoxia may also have an impact on VEGF and other signaling components in tumor angiogenesis (Figure 2). αv integrins, which are expressed on multiple cell types, contribute to angiogenesis [32]. These and other integrins interact with the VEGF/VEGFR and Ang/Tie signaling pathways [32]. Hypoxia, through HIF-1α and HIF-2α, leads to increased expression of VEGF-A; hypoxia may also regulate PlGF expression, which may be more complicated. Basically, tissue hypoxia may not only elevate the abundance of VEGF-A, but may also increase other angiogenic regulatory factors, therefore leading to angiogenic activity alteration [26].
VEGF-A, often referred to as VEGF without a suffix [4], is the sole target of bevacizumab, a humanized monoclonal antibody approved for the treatment of colorectal and other cancers [55] and aflibercept (VEGF Trap, known in the US as ziv-aflibercept), is a recombinant fusion protein with receptor components of VEGFR-1 and VEGFR-2 that binds multiple ligands in the angiogenesis network (VEGF-A, VEGF-B, and PlGF) [56]. Aflibercept was recently approved for use by the US FDA with the US name of ziv-aflibercept, in combination with 5-fluorouracil, leucovorin, irinotecan (FOLFIRI) for patients with metastatic colorectal cancer (mCRC) that is resistant to or has progressed following an oxaliplatin-containing regimen. In addition to agents that target the various VEGFs and VEGFRs, agents that target other angiogenic mediators, including Ang-1 and −2, Notch signaling, HIF-1α, and integrins, and that are in clinical development are listed in the NCT registry (for example NCT01210222, NCT01193868, NCT01120288, and NCT00915278).
Benefits of antiangiogenic agents in the treatment of metastatic colorectal cancer: preclinical and clinical evidence
As mentioned above, bevacizumab and aflibercept are antiangiogenic agents approved for the treatment of mCRC [55, 56]. Other antiangiogenic therapies currently in late-stage clinical trials for mCRC include the anti-VEGFR-2 monoclonal antibody ramucirumab, and the tyrosine kinase inhibitor (TKI) regorafenib; additional TKIs (brivanib alaninate, cediranib, sunitinib, and vatalanib) had negative results in phase 3 trials (Table 1). Preclinical studies with these agents supported their further development for the treatment of colorectal cancer and other malignancies.
Preclinical data
In preclinical models, bevacizumab demonstrated antitumor activity both as a single agent and in combination with chemotherapy or radiotherapy [57]. In tumor xenograft models, bevacizumab was shown to decrease tumor weight and size, tumor growth, vascular permeability, ascites formation, and the diameter and density of tumor vessels [57]. One of the proposed mechanisms by which bevacizumab is believed to enhance cytotoxic treatments is via “normalization” of the tumor vasculature [57], and the use of bevacizumab in combination with chemotherapy or radiotherapy has been shown to have at least additive activity in certain tumor models [57].
Aflibercept (VEGF Trap) is a recombinant fusion protein that contains domains from VEGFR-1 and VEGFR-2 and targets VEGF-A as well as VEGF-B and PlGF [58, 59]. In certain tumor xenograft models, aflibercept inhibited the growth of new and established tumors and tumor angiogenesis; reduced vessel density, patency, and blood flow; and inhibited metastases and ascites formation [60]. Aflibercept also increased tumor hypoxia and decreased expression of tumor vascular genes and decreased activation of vascular endothelial signaling pathways [60]. In tumor xenografts, aflibercept in combination with other agents (radiotherapy, chemotherapy, or trastuzumab) showed greater inhibition of tumor growth and tumor vasculature than was observed with the individual agents alone [60].
In a recent study, the binding characteristics of bevacizumab and aflibercept were compared using several preclinical assessments [61]. Aflibercept showed tight binding to VEGF 165; dissociation constant (KD) was significantly lower with aflibercept compared with dimerized VEGFR1 or VEGFR2. Additionally, the KD of aflibercept (0.490 pM) was approximately 100-fold lower compared with that of bevacizumab (58 pM), suggesting a 100-fold tighter binding to VEGF 165 [61].
Differences in biologic activity were also demonstrated preclinically. In a study of VEGF-A–induced activation of VEGFR1, aflibercept demonstrated 92-fold greater potency than bevacizumab in an assay in which VEGFR1 activation was induced by VEGF-A 165 or VEGF-A 121 [61]. Aflibercept also inhibited VEGFR1 activation by PlGF2, [61]. Aflibercept also inhibited activation of VEGFR2 activation induced by VEGF-A 165 or VEGF-A 121, which may suggest some clinical benefits since binding kinetics and affinity are important determinants of the biological activity of antibody-like drugs [61].
Ramucirumab, a fully human monoclonal antibody against VEGFR-2, was designed to bind to a VEGFR-2 epitope involved in ligand binding [62]. It has shown anticancer activity alone and in combination with other agents in preclinical models of leukemia, solid tumors, and metastases [63–65].
Various antiangiogenic TKIs have also demonstrated preclinical activity in cancer models. Cediranib, a VEGFR-2 TKI, inhibited tumor growth and reduced microvessel density in tumor xenograft models [66]. Regorafenib, a multikinase inhibitor whose targets include VEGFR-1-3 and Tie2, induced tumor growth inhibition or shrinkage and reduced extravasation in tumor xenograft models [67, 68]. Brivanib, a dual VEGFR-2/FGFR-1 kinase inhibitor, reduced tumor proliferation, vascular density, and microcirculation; inhibited VEGF- and FGF-driven angiogenesis; and induced apoptosis in tumor xenograft models [69, 70]. Sunitinib, a multikinase inhibitor that targets VEGFR-2, PDGFR, KIT, and FLT3, inhibited VEGFR-2 phosphorylation and VEGF-induced vascular permeability and caused tumor regression, growth arrest, or growth inhibition in tumor xenografts [71]. Vatalanib, an inhibitor of VEGFRs and other kinases, decreased tumor growth, metastasis, microvascular density, and blood flow in tumor xenograft models and induced tumor cell apoptosis [72–74].
Based on these preclinical data demonstrating anticancer activity, these agents moved forward into clinical studies for mCRC and other cancers.
Clinical data
Bevacizumab
The clinical benefit of antiantiogenesis agents in treatment of mCRC has been established. Based on encouraging data of a 3-arm phase 2 study [75], several pivotal phase 3 trials demonstrated that bevacizumab improved overall survival as first- or second-line therapy in combination with fluoropyrimidine-containing regimens (either combined with oxaliplatin or irinotecan) in patients with mCRC [76, 77]. In the initial phase 3 pivotal trial, median overall survival (mOS) was increased from 15.6 months in patients who received IFL (bolus 5-FU, leucovorin, and irinotecan), but only to 20.3 months in patients who received IFL + bevacizumab (P<0.001) as first line therapy in mCRC [76]. The E3200 study showed the benefit of bevacizumab as second line therapy when combined with FOLFOX (infusional 5-FU and oxaliplatin) with mOS of 10.8 vs. 12.9 months (P=0.0018) [77]. Together, these trials confirmed a survival benefit with bevacizumab in both the first- and second-line settings for mCRC. Bevacizumab-associated toxicities identified in early trials of bevacizumab included hemorrhage, thromboembolism, proteinuria, and hypertension [76]. In phase 3 trials of bevacizumab plus chemotherapy in patients with mCRC, the incidence of grade ≥3 bleeding/hemorrhage was 2% to 3.4% with bevacizumab versus <1% to 2.5% with comparator; the incidence of grade ≥3 thromboembolism (reported in 2 trials) was 3.4% to 10% with bevacizumab versus 2.5% to 6% with comparator; the incidence of grade ≥3 venous thromboembolic events (VTEs, reported in 1 trial) was 8% with bevacizumab versus 5% with comparator; the incidence of grade ≥3 proteinuria was <1% in either arm; and the incidence of grade ≥3 hypertension was 4% to 11% with bevacizumab versus 1% to 2.3% with comparator [76–78]. Gastrointestinal perforation was also reported in phase 3 trials of bevacizumab in patients with mCRC: <1% to 1.5% with bevacizumab versus 0% to <1% with comparator [76–78].
Since antiangiogenic agents are not traditional cytotoxic chemotherapy agents, the question remains whether their antitumor efficacy may be maintained after patients’ disease has progressed after a bevacizumab-containing chemotherapy regimen. A registry study which suggested a significant survival benefit was controversial because of the registry study design [79]. There were no randomized study data until recently. In the TML (ML 18147) study, all patients were previously treated with bevacizumab in combination with either FOLFOX or FOLFIRI. After disease progression, they were then randomized to chemotherapy regimens they were not exposed to (either FOLFIRI or FOLFOX) with continuous bevacizumab. The overall survival was significantly improved with continuous bevacizumab plus chemotherapy versus chemotherapy alone as second line in mCRC patients who had progressed on first-line bevacizumab-containing regimens (11.2 versus 9.8 months, P=0.0211) [80]. This study validates the importance of continued antiangiogenic therapy in mCRC patients following progression.
Regorafenib
Another randomized phase 3 study (CORRECT trial) with a total 760 patients who were treated with standard therapies including bevacizumab-containing regimens demonstrated the efficacy of regorafenib in both overall survival (6.4 versus 5.0 months, HR=0.773, P=0.0051) and progression-free survival (1.9 versus 1.7 months, HR=0.493, P<0.000001) compared with placebo; the most common grade ≥3 adverse events were hand-foot reaction (17%), fatigue (15%), diarrhea (8%), hyperbilirubinemia (8%), and hypertension (7%) [81]. This study was the first to demonstrate the efficacy of an oral TKI in prolonging survival in patients with mCRC.
Aflibercept
Data from a phase 3 trial (VELOUR) (N=1226, with 28.3% of patients having had previous bevacizumab exposure) demonstrated that aflibercept plus FOLFIRI in patients with mCRC who had progressed following an oxaliplatin containing regimen significantly improved overall survival (13.5 months versus 12.06 months, HR=0.817, P=0.0032) and progression-free survival (6.90 months versus 4.67 months, HR=0.758, P=0.00007) compared with placebo plus FOLFIRI in mCRC patients previously treated with FOLFOX [82, 83]. These data led to the FDA approval of aflibercept (ziv-aflibercept) for the treatment of mCRC following oxaliplatin-based chemotherapy. While the progression-free survival benefit remained in the prior bevacizumab-treated patients (6.7 versus 3.9 months, HR=0.661 [95% CI 0.512-0.852]) based on the prespecified subgroup analysis, the study was not powered to show a treatment difference between arms; therefore, no definitive conclusions may be drawn concerning the benefit of aflibercept in the prior bevacizumab-treated subgroup. The most common grade 3–4 adverse events with more than 2% higher incidence with aflibercept were diarrhea, asthenia/fatigue, stomatitis/ulceration, infections, hypertension, gastrointestinal/abdominal pains, neutropenia/neutropenic complications, and proteinuria.
Cediranib
In the HORIZON II trial, cediranib plus chemotherapy (FOLFOX or XELOX) significantly improved progression-free survival (HR=0.84) but not overall survival (HR=0.94) compared with chemotherapy plus placebo; treating to progression with cediranib plus chemotherapy appeared to have a beneficial effect [84].
Brivanib
In the NCIC Clinical Trials Group and AGITG CO.20 trial, brivanib alaninate plus cetuximab significantly improved progression-free survival (5.0 versus 3.4 months, HR=0.72, P<0.0001) but not overall survival (8.8 versus 8.1 months, HR=0.88, P=0.12) compared with cetuximab plus placebo; the most frequent grade ≥3 adverse events with brivanib alaninate were fatigue (25%), hypertension (11%), and rash (5%) [85].
Conclusions
The importance of angiogenesis in cancer, and an increased understanding of the network of signaling pathways that mediate it, have led to the development of a number of antiangiogenic agents for the treatment of cancer. Bevacizumab is approved for the first- or second-line treatment of mCRC when added to intravenous 5-fluorouracil-based regimens, and aflibercept was approved by the FDA when added to FOLFIRI in patients with mCRC previously treated with an oxaliplatin-based regimen. Other antiangiogenic agents are in late-stage clinical development. The addition of bevacizumab or aflibercept to chemotherapy in patients with mCRC has demonstrated improved overall survival compared with chemotherapy alone, and regorafenib added to best supportive care has demonstrated improved survival compared with placebo. Insight into how angiogenic signaling pathways intersect may aid in the design of agents with improved efficacy and safety profiles and a reduced risk of resistance. Additional research is needed regarding how to sequence and combine approved and investigational antiangiogenic agents for the treatment of colorectal and other cancers.
References
Folkman J, Klagsbrun M: Angiogenic factors. Science. 1987, 235: 442-447. 10.1126/science.2432664.
Jain RK: Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science. 2005, 307: 58-62. 10.1126/science.1104819.
Folkman J: Tumor angiogenesis: therapeutic implications. N Engl J Med. 1971, 285: 1182-1186. 10.1056/NEJM197111182852108.
Ferrara N: Vascular endothelial growth factor: basic science and clinical progress. Endocr Rev. 2004, 25: 581-611. 10.1210/er.2003-0027.
Ellis LM, Hicklin DJ: VEGF-targeted therapy: mechanisms of anti-tumour activity. Nat Rev Cancer. 2008, 8: 579-591. 10.1038/nrc2403.
Holash J, Maisonpierre PC, Compton D, Boland P, Alexander CR, Zagzag D, Yancopoulos GD, Wiegand SJ: Vessel cooption, regression, and growth in tumors mediated by angiopoietins and VEGF. Science. 1999, 284: 1994-1998. 10.1126/science.284.5422.1994.
Konerding MA, Malkusch W, Klapthor B, van Ackern C, Fait E, Hill SA, Parkins C, Chaplin DJ, Presta M, Denekamp J: Evidence for characteristic vascular patterns in solid tumours: quantitative studies using corrosion casts. Br J Cancer. 1999, 80: 724-732. 10.1038/sj.bjc.6690416.
Morikawa S, Baluk P, Kaidoh T, Haskell A, Jain RK, McDonald DM: Abnormalities in pericytes on blood vessels and endothelial sprouts in tumors. Am J Pathol. 2002, 160: 985-1000. 10.1016/S0002-9440(10)64920-6.
Hashizume H, Baluk P, Morikawa S, McLean JW, Thurston G, Roberge S, Jain RK, McDonald DM: Openings between defective endothelial cells explain tumor vessel leakiness. Am J Pathol. 2000, 156: 1363-1380. 10.1016/S0002-9440(10)65006-7.
Padera TP, Stoll BR, Tooredman JB, Capen D, di Tomaso E, Jain RK: Pathology: cancer cells compress intratumour vessels. Nature. 2004, 427: 695-10.1038/427695a.
Boucher Y, Baxter LT, Jain RK: Interstitial pressure gradients in tissue-isolated and subcutaneous tumors: implications for therapy. Cancer Res. 1990, 50: 4478-4484.
Fukumura D, Duda DG, Munn LL, Jain RK: Tumor microvasculature and microenvironment: novel insights through intravital imaging in pre-clinical models. Microcirculation. 2010, 17: 206-225. 10.1111/j.1549-8719.2010.00029.x.
Helmlinger G, Yuan F, Dellian M, Jain RK: Interstitial pH and pO2 gradients in solid tumors in vivo: high-resolution measurements reveal a lack of correlation. Nat Med. 1997, 3: 177-182. 10.1038/nm0297-177.
Baluk P, Morikawa S, Haskell A, Mancuso M, McDonald DM: Abnormalities of basement membrane on blood vessels and endothelial sprouts in tumors. Am J Pathol. 2003, 163: 1801-1815. 10.1016/S0002-9440(10)63540-7.
Seaman S, Stevens J, Yang MY, Logsdon D, Graff-Cherry C, St Croix B: Genes that distinguish physiological and pathological angiogenesis. Cancer Cell. 2007, 11: 539-554. 10.1016/j.ccr.2007.04.017.
St Croix B, Rago C, Velculescu V, Traverso G, Romans KE, Montgomery E, Lal A, Riggins GJ, Lengauer C, Vogelstein B, Kinzler KW: Genes expressed in human tumor endothelium. Science. 2000, 289: 1197-1202. 10.1126/science.289.5482.1197.
Hida K, Hida Y, Amin DN, Flint AF, Panigrahy D, Morton CC, Klagsbrun M: Tumor-associated endothelial cells with cytogenetic abnormalities. Cancer Res. 2004, 64: 8249-8255. 10.1158/0008-5472.CAN-04-1567.
Dvorak HF: Vascular permeability factor/vascular endothelial growth factor: a critical cytokine in tumor angiogenesis and a potential target for diagnosis and therapy. J Clin Oncol. 2002, 20: 4368-4380. 10.1200/JCO.2002.10.088.
Orlandini M, Marconcini L, Ferruzzi R, Oliviero S: Identification of a c-fos-induced gene that is related to the platelet-derived growth factor/vascular endothelial growth factor family. Erratum in: Proc Natl Acad Sci U S A. 1997, 94: 1603-Proc Natl Acad Sci U S A 1996, 93:11675–11680.
Maglione D, Guerriero V, Viglietto G, Delli-Bovi P, Persico MG: Isolation of a human placenta cDNA coding for a protein related to the vascular permeability factor. Proc Natl Acad Sci U S A. 1991, 88: 9267-9271. 10.1073/pnas.88.20.9267.
Olofsson B, Pajusola K, Kaipainen A, von Euler G, Joukov V, Saksela O, Orpana A, Pettersson RF, Alitalo K, Eriksson U: Vascular endothelial growth factor B, a novel growth factor for endothelial cells. Proc Natl Acad Sci U S A. 1996, 93: 2576-2581. 10.1073/pnas.93.6.2576.
Joukov V, Pajusola K, Kaipainen A, Chilov D, Lahtinen I, Kukk E, Saksela O, Kalkkinen N, Alitalo K: A novel vascular endothelial growth factor, VEGF-C, is a ligand for the Flt4 (VEGFR-3) and KDR (VEGFR-2) receptor tyrosine kinases. Erratum in: EMBO J. 1996, 15: 1751-EMBO J 1996, 15:290–298.
Soker S, Takashima S, Miao HQ, Neufeld G, Klagsbrun M: Neuropilin-1 is expressed by endothelial and tumor cells as an isoform-specific receptor for vascular endothelial growth factor. Cell. 1998, 92: 735-745. 10.1016/S0092-8674(00)81402-6.
Migdal M, Huppertz B, Tessler S, Comforti A, Shibuya M, Reich R, Baumann H, Neufeld G: Neuropilin-1 is a placenta growth factor-2 receptor. J Biol Chem. 1998, 273: 22272-22278. 10.1074/jbc.273.35.22272.
Neufeld G, Kessler O, Herzog Y: The interaction of Neuropilin-1 and Neuropilin-2 with tyrosine-kinase receptors for VEGF. Adv Exp Med Biol. 2002, 515: 81-90. 10.1007/978-1-4615-0119-0_7.
Cao Y: Positive and negative modulation of angiogenesis by VEGFR1 ligands. Sci Signal. 2009, 2: re1-10.1126/scisignal.259re1.
Kerbel RS: Tumor angiogenesis. N Engl J Med. 2008, 358: 2039-2049. 10.1056/NEJMra0706596.
Kut C, Mac Gabhann F, Popel AS: Where is VEGF in the body? A meta-analysis of VEGF distribution in cancer. Br J Cancer. 2007, 97: 978-985. 10.1038/sj.bjc.6603923.
Takahashi T, Ueno H, Shibuya M: VEGF activates protein kinase C-dependent, but Ras-independent Raf-MEK-MAP kinase pathway for DNA synthesis in primary endothelial cells. Oncogene. 1999, 18: 2221-2230. 10.1038/sj.onc.1202527.
Meadows KN, Bryant P, Pumiglia K: Vascular endothelial growth factor induction of the angiogenic phenotype requires Ras activation. J Biol Chem. 2001, 276: 49289-49298. 10.1074/jbc.M108069200.
Huang H, Bhat A, Woodnutt G, Lappe R: Targeting the ANGPT-TIE2 pathway in malignancy. Nat Rev Cancer. 2010, 10: 575-585. 10.1038/nrc2894.
Weis SM, Cheresh DA: αv Integrins in angiogenesis and cancer. Cold Spring Harb Perspect Med. 2011, 1: a006478-
Zheng W, Tammela T, Yamamoto M, Anisimov A, Holopainen T, Kaijalainen S, Karpanen T, Lehti K, Ylä-Herttuala S, Alitalo K: Notch restricts lymphatic vessel sprouting induced by vascular endothelial growth factor. Blood. 2011, 118: 1154-1162. 10.1182/blood-2010-11-317800.
Tammela T, Zarkada G, Nurmi H, Jakobsson L, Heinolainen K, Tvorogov D, Zheng W, Franco CA, Murtomäki A, Aranda E, Miura N, Ylä-Herttuala S, Fruttiger M, Mäkinen T, Eichmann A, Pollard JW, Gerhardt H, Alitalo K: VEGFR-3 controls tip to stalk conversion at vessel fusion sites by reinforcing Notch signalling. Nat Cell Biol. 2011, 13: 1202-1213. 10.1038/ncb2331.
Benedito R, Rocha SF, Woeste M, Zamykal M, Radtke F, Casanovas O, Duarte A, Pytowski B, Adams RH: Notch-dependent VEGFR3 upregulation allows angiogenesis without VEGF-VEGFR2 signalling. Nature. 2012, 484: 110-114. 10.1038/nature10908.
Fischer C, Mazzone M, Jonckx B, Carmeliet P: FLT1 and its ligands VEGFB and PlGF: drug targets for anti-angiogenic therapy?. Nat Rev Cancer. 2008, 8: 942-956. 10.1038/nrc2524.
Hiratsuka S, Minowa O, Kuno J, Noda T, Shibuya M: Flt-1 lacking the tyrosine kinase domain is sufficient for normal development and angiogenesis in mice. Proc Natl Acad Sci U S A. 1998, 95: 9349-9354. 10.1073/pnas.95.16.9349.
Bates RC, Goldsmith JD, Bachelder RE, Brown C, Shibuya M, Oettgen P, Mercurio AM: Flt-1-dependent survival characterizes the epithelial-mesenchymal transition of colonic organoids. Curr Biol. 2003, 13: 1721-1727. 10.1016/j.cub.2003.09.002.
Yi ZY, Feng LJ, Xiang Z, Yao H: Vascular endothelial growth factor receptor-1 activation mediates epithelial to mesenchymal transition in hepatocellular carcinoma cells. J Invest Surg. 2011, 24: 67-76. 10.3109/08941939.2010.542272.
Shintani S, Li C, Ishikawa T, Mihara M, Nakashiro K, Hamakawa H: Expression of vascular endothelial growth factor A, B, C, and D in oral squamous cell carcinoma. Oral Oncol. 2004, 40: 13-20.
Gunningham SP, Currie MJ, Han C, Robinson BA, Scott PA, Harris AL, Fox SB: VEGF-B expression in human primary breast cancers is associated with lymph node metastasis but not angiogenesis. J Pathol. 2001, 193: 325-332. 10.1002/path.814.
Albrecht I, Kopfstein L, Strittmatter K, Schomber T, Falkevall A, Hagberg CE, Lorentz P, Jeltsch M, Alitalo K, Eriksson U, Christofori G, Pietras K: Suppressive effects of vascular endothelial growth factor-B on tumor growth in a mouse model of pancreatic neuroendocrine tumorigenesis. PLoS One. 2010, 5: e14109-10.1371/journal.pone.0014109.
Zhang F, Tang Z, Hou X, Lennartsson J, Li Y, Koch AW, Scotney P, Lee C, Arjunan P, Dong L, Kumar A, Rissanen TT, Wang B, Nagai N, Fons P, Fariss R, Zhang Y, Wawrousek E, Tansey G, Raber J, Fong GH, Ding H, Greenberg DA, Becker KG, Herbert JM, Nash A, Yla-Herttuala S, Cao Y, Watts RJ, Li X: VEGF-B is dispensable for blood vessel growth but critical for their survival, and VEGF-B targeting inhibits pathological angiogenesis. Proc Natl Acad Sci U S A. 2009, 106: 6152-6157. 10.1073/pnas.0813061106.
Carmeliet P, Moons L, Luttun A, Vincenti V, Compernolle V, De Mol M, Wu Y, Bono F, Devy L, Beck H, Scholz D, Acker T, DiPalma T, Dewerchin M, Noel A, Stalmans I, Barra A, Blacher S, Vandendriessche T, Ponten A, Eriksson U, Plate KH, Foidart JM, Schaper W, Charnock-Jones DS, Hicklin DJ, Herbert JM, Collen D, Persico MG: Synergism between vascular endothelial growth factor and placental growth factor contributes to angiogenesis and plasma extravasation in pathological conditions. Nat Med. 2001, 7: 575-583. 10.1038/87904.
Roy H, Bhardwaj S, Babu M, Jauhiainen S, Herzig KH, Bellu AR, Haisma HJ, Carmeliet P, Alitalo K, Ylä-Herttuala S: Adenovirus-mediated gene transfer of placental growth factor to perivascular tissue induces angiogenesis via upregulation of the expression of endogenous vascular endothelial growth factor-A. Hum Gene Ther. 2005, 16: 1422-1428. 10.1089/hum.2005.16.1422.
Yao J, Wu X, Zhuang G, Kasman IM, Vogt T, Phan V, Shibuya M, Ferrara N, Bais C: Expression of a functional VEGFR-1 in tumor cells is a major determinant of anti-PlGF antibodies efficacy. Proc Natl Acad Sci U S A. 2011, 108: 11590-11595. 10.1073/pnas.1109029108.
Hanrahan V, Currie MJ, Gunningham SP, Morrin HR, Scott PA, Robinson BA, Fox SB: The angiogenic switch for vascular endothelial growth factor (VEGF)-A, VEGF-B, VEGF-C, and VEGF-D in the adenoma-carcinoma sequence during colorectal cancer progression. J Pathol. 2003, 200: 183-194. 10.1002/path.1339.
Wang TB, Chen ZG, Wei XQ, Wei B, Dong WG: Serum vascular endothelial growth factor-C and lymphoangiogenesis are associated with the lymph node metastasis and prognosis of patients with colorectal cancer. ANZ J Surg. 2011, 81: 694-699. 10.1111/j.1445-2197.2010.05539.x.
Skobe M, Hawighorst T, Jackson DG, Prevo R, Janes L, Velasco P, Riccardi L, Alitalo K, Claffey K, Detmar M: Induction of tumor lymphangiogenesis by VEGF-C promotes breast cancer metastasis. Nat Med. 2001, 7: 192-198. 10.1038/84643.
Wu QW, She HQ, Liang J, Huang YF, Yang QM, Yang QL, Zhang ZM: Expression and clinical significance of extracellular matrix protein 1 and vascular endothelial growth factor-C in lymphatic metastasis of human breast cancer. BMC Cancer. 2012, 12: 47-10.1186/1471-2407-12-47.
Mandriota SJ, Jussila L, Jeltsch M, Compagni A, Baetens D, Prevo R, Banerji S, Huarte J, Montesano R, Jackson DG, Orci L, Alitalo K, Christofori G, Pepper MS: Vascular endothelial growth factor-C-mediated lymphangiogenesis promotes tumour metastasis. EMBO J. 2001, 20: 672-682. 10.1093/emboj/20.4.672.
Karnezis T, Shayan R, Caesar C, Roufail S, Harris NC, Ardipradja K, Zhang YF, Williams SP, Farnsworth RH, Chai MG, Rupasinghe TW, Tull DL, Baldwin ME, Sloan EK, Fox SB, Achen MG, Stacker SA: VEGF-D promotes tumor metastasis by regulating prostaglandins produced by the collecting lymphatic endothelium. Cancer Cell. 2012, 21: 181-195. 10.1016/j.ccr.2011.12.026.
Stacker SA, Caesar C, Baldwin ME, Thornton GE, Williams RA, Prevo R, Jackson DG, Nishikawa S, Kubo H, Achen MG: VEGF-D promotes the metastatic spread of tumor cells via the lymphatics. Nat Med. 2001, 7: 186-191. 10.1038/84635.
Li JL, Sainson RC, Oon CE, Turley H, Leek R, Sheldon H, Bridges E, Shi W, Snell C, Bowden ET, Wu H, Chowdhury PS, Russell AJ, Montgomery CP, Poulsom R, Harris AL: Dll4-Notch signaling mediates tumor resistance to anti-VEGF therapy in vivo. Cancer Res. 2011, 71: 6073-6083. 10.1158/0008-5472.CAN-11-1704.
Avastin [bevacizumab) prescribing information. 2012, South San Francisco, CA: Genentech, Inc
Zaltrap (ziv-aflibercept) prescribing information. 2012, Bridgewater, NJ: Regeneron Pharmaceuticals, Inc./sanofi-aventis U.S. LLC
Gerber HP, Ferrara N: Pharmacology and pharmacodynamics of bevacizumab as monotherapy or in combination with cytotoxic therapy in preclinical studies. Cancer Res. 2005, 65: 671-680.
Tew WP, Gordon M, Murren J, Dupont J, Pezzulli S, Aghajanian C, Sabbatini P, Mendelson D, Schwartz L, Gettinger S, Psyrri A, Cedarbaum JM, Spriggs DR: Phase 1 study of aflibercept administered subcutaneously to patients with advanced solid tumors. Clin Cancer Res. 2010, 16: 358-366. 10.1158/1078-0432.CCR-09-2103.
Holash J, Davis S, Papadopoulos N, Croll SD, Ho L, Russell M, Boland P, Leidich R, Hylton D, Burova E, Ioffe E, Huang T, Radziejewski C, Bailey K, Fandl JP, Daly T, Wiegand SJ, Yancopoulos GD, Rudge JS: VEGF-Trap: a VEGF blocker with potent antitumor effects. Proc Natl Acad Sci U S A. 2002, 99: 11393-11398. 10.1073/pnas.172398299.
Gaya A, Tse V: A preclinical and clinical review of aflibercept for the management of cancer. Cancer Treat Rev. 2012, 38: 484-493. 10.1016/j.ctrv.2011.12.008.
Papadopoulos N, Martin J, Ruan Q, Rafique A, Rosconi MP, Shi E, Pyles EA, Yancopoulos GD, Stahl N, Wiegand SJ: Binding and neutralization of vascular endothelial growth factor (VEGF) and related ligands by VEGF Trap, ranibizumab and bevacizumab. Angiogenesis. 2012, 15: 171-185. 10.1007/s10456-011-9249-6.
Spratlin JL, Cohen RB, Eadens M, Gore L, Camidge DR, Diab S, Leong S, O'Bryant C, Chow LQ, Serkova NJ, Meropol NJ, Lewis NL, Chiorean EG, Fox F, Youssoufian H, Rowinsky EK, Eckhardt SG: Phase I pharmacologic and biologic study of ramucirumab (IMC-1121B), a fully human immunoglobulin G1 monoclonal antibody targeting the vascular endothelial growth factor receptor-2. J Clin Oncol. 2010, 28: 780-787. 10.1200/JCO.2009.23.7537.
Zhu Z, Hattori K, Zhang H, Jimenez X, Ludwig DL, Dias S, Kussie P, Koo H, Kim HJ, Lu D, Liu M, Tejada R, Friedrich M, Bohlen P, Witte L, Rafii S: Inhibition of human leukemia in an animal model with human antibodies directed against vascular endothelial growth factor receptor 2. Correlation between antibody affinity and biological activity. Leukemia. 2003, 17: 604-611. 10.1038/sj.leu.2402831.
Prewett M, Huber J, Li Y, Santiago A, O'Connor W, King K, Overholser J, Hooper A, Pytowski B, Witte L, Bohlen P, Hicklin DJ: Antivascular endothelial growth factor receptor (fetal liver kinase 1) monoclonal antibody inhibits tumor angiogenesis and growth of several mouse and human tumors. Cancer Res. 1999, 59: 5209-5218.
Bruns CJ, Liu W, Davis DW, Shaheen RM, McConkey DJ, Wilson MR, Bucana CD, Hicklin DJ, Ellis LM: Vascular endothelial growth factor is an in vivo survival factor for tumor endothelium in a murine model of colorectal carcinoma liver metastases. Cancer. 2000, 89: 488-499. 10.1002/1097-0142(20000801)89:3<488::AID-CNCR3>3.0.CO;2-X.
Wedge SR, Kendrew J, Hennequin LF, Valentine PJ, Barry ST, Brave SR, Smith NR, James NH, Dukes M, Curwen JO, Chester R, Jackson JA, Boffey SJ, Kilburn LL, Barnett S, Richmond GH, Wadsworth PF, Walker M, Bigley AL, Taylor ST, Cooper L, Beck S, Jürgensmeier JM, Ogilvie DJ: AZD2171: a highly potent, orally bioavailable, vascular endothelial growth factor receptor-2 tyrosine kinase inhibitor for the treatment of cancer. Cancer Res. 2005, 65: 4389-4400. 10.1158/0008-5472.CAN-04-4409.
Mross K, Frost A, Steinbild S, Hedbom S, Büchert M, Fasol U, Unger C, Kratzschmar J, Heinig R, Boix O, Christensen O: A phase I dose-escalation study of regorafenib (BAY 73–4506), an inhibitor of oncogenic, angiogenic and stromal kinases, in patients with advanced solid tumors. Clin Cancer Res. 2012, 18: 2658-2667. 10.1158/1078-0432.CCR-11-1900.
Wilhelm SM, Dumas J, Adnane L, Lynch M, Carter CA, Schütz G, Thierauch KH, Zopf D: Regorafenib (BAY 73–4506): a new oral multikinase inhibitor of angiogenic, stromal and oncogenic receptor tyrosine kinases with potent preclinical antitumor activity. Int J Cancer. 2011, 129: 245-255. 10.1002/ijc.25864.
Huynh H, Ngo VC, Fargnoli J, Ayers M, Soo KC, Koong HN, Thng CH, Ong HS, Chung A, Chow P, Pollock P, Byron S, Tran E: Brivanib alaninate, a dual inhibitor of vascular endothelial growth factor receptor and fibroblast growth factor receptor tyrosine kinases, induces growth inhibition in mouse models of human hepatocellular carcinoma. Clin Cancer Res. 2008, 14: 6146-6153. 10.1158/1078-0432.CCR-08-0509.
Bhide RS, Lombardo LJ, Hunt JT, Cai ZW, Barrish JC, Galbraith S, Jeyaseelan R, Mortillo S, Wautlet BS, Krishnan B, Kukral D, Malone H, Lewin AC, Henley BJ, Fargnoli J: The antiangiogenic activity in xenograft models of brivanib, a dual inhibitor of vascular endothelial growth factor receptor-2 and fibroblast growth factor receptor-1 kinases. Mol Cancer Ther. 2010, 9: 369-378. 10.1158/1535-7163.MCT-09-0472.
Mendel DB, Laird AD, Xin X, Louie SG, Christensen JG, Li G, Schreck RE, Abrams TJ, Ngai TJ, Lee LB, Murray LJ, Carver J, Chan E, Moss KG, Haznedar JO, Sukbuntherng J, Blake RA, Sun L, Tang C, Miller T, Shirazian S, McMahon G, Cherrington JM: In vivo antitumor activity of SU11248, a novel tyrosine kinase inhibitor targeting vascular endothelial growth factor and platelet-derived growth factor receptors: determination of a pharmacokinetic/pharmacodynamic relationship. Clin Cancer Res. 2003, 9: 327-337.
Wood JM, Bold G, Buchdunger E, Cozens R, Ferrari S, Frei J, Hofmann F, Mestan J, Mett H, O'Reilly T, Persohn E, Rösel J, Schnell C, Stover D, Theuer A, Towbin H, Wenger F, Woods-Cook K, Menrad A, Siemeister G, Schirner M, Thierauch KH, Schneider MR, Drevs J, Martiny-Baron G, Totzke F: PTK787/ZK 222584, a novel and potent inhibitor of vascular endothelial growth factor receptor tyrosine kinases, impairs vascular endothelial growth factor-induced responses and tumor growth after oral administration. Cancer Res. 2000, 60: 2178-2189.
Drevs J, Hofmann I, Hugenschmidt H, Wittig C, Madjar H, Müller M, Wood J, Martiny-Baron G, Unger C, Marmé D: Effects of PTK787/ZK 222584, a specific inhibitor of vascular endothelial growth factor receptor tyrosine kinases, on primary tumor, metastasis, vessel density, and blood flow in a murine renal cell carcinoma model. Cancer Res. 2000, 60: 4819-4824.
Liu Y, Poon RT, Li Q, Kok TW, Lau C, Fan ST: Both antiangiogenesis- and angiogenesis-independent effects are responsible for hepatocellular carcinoma growth arrest by tyrosine kinase inhibitor PTK787/ZK222584. Cancer Res. 2005, 65: 3691-3699. 10.1158/0008-5472.CAN-04-3462.
Kabbinavar F, Hurwitz HI, Fehrenbacher L, Meropol NJ, Novotny WF, Lieberman G, Griffing S, Bergsland E: Phase II, randomized trial comparing bevacizumab plus fluorouracil (FU)/leucovorin (LV) with FU/LV alone in patients with metastatic colorectal cancer. J Clin Oncol. 2003, 21: 60-65. 10.1200/JCO.2003.10.066.
Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, Heim W, Berlin J, Baron A, Griffing S, Holmgren E, Ferrara N, Fyfe G, Rogers B, Ross R, Kabbinavar F: Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004, 350: 2335-2342. 10.1056/NEJMoa032691.
Giantonio BJ, Catalano PJ, Meropol NJ, O'Dwyer PJ, Mitchell EP, Alberts SR, Schwartz MA, Benson AB: ROM: Bevacizumab in combination with oxaliplatin, fluorouracil, and leucovorin (FOLFOX4) for previously treated metastatic colorectal cancer:results from the Eastern Cooperative Oncology Group Study E3200. J Clin Oncol. 2007, 25: 1539-1544. 10.1200/JCO.2006.09.6305.
Saltz LB, Clarke S, Díaz-Rubio E, Scheithauer W, Figer A, Wong R, Koski S, Lichinitser M, Yang TS, Rivera F, Couture F, Sirzén F, Cassidy J: Bevacizumab in combination with oxaliplatin-based chemotherapy as first-line therapy in metastatic colorectal cancer: a randomized phase III study. J Clin Oncol. 2008, 26: 2013-2019. 10.1200/JCO.2007.14.9930. Erratum in: J Clin Oncol 2008, 26:3110. J Clin Oncol 2009, 27: 653.
Grothey A, Sugrue MM, Purdie DM, Dong W, Sargent D, Hedrick E, Kozloff M: Bevacizumab beyond first progression is associated with prolonged overall survival in metastatic colorectal cancer: results from a large observational cohort study (BRiTE). J Clin Oncol. 2008, 26: 5326-5334. 10.1200/JCO.2008.16.3212.
Arnold D, Andre T, Bennouna J, Sastre J, Osterlund PJ, Greil R, Van Cutsem E, Von Moos R, Reyes-Rivera I, Bendahmane B, Kubicka S: Bevacizumab (BEV) plus chemotherapy (CT) continued beyond first progression in patients with metastatic colorectal cancer (mCRC) previously treated with BEV plus CT: Results of a randomized phase III intergroup study (TML study). J Clin Oncol. 2012, 30 (suppl): CRA3503-
Van Cutsem E, Sobrero AF, Siena S, Falcone A, Ychou M, Humblet Y, Bouche O, Mineur L, Barone C, Adenis A, Tabernero J, Yoshino T, Lenz H-J, Goldberg RM, Sargent DJ, Cihon F, Wagner A, Laurent D, Grothey A, on behalf of CORRECT Investigators: Phase III CORRECT trial of regorafenib in metastatic colorectal cancer (mCRC). J Clin Oncol. 2012, 30 (suppl): 3502-
Van Cutsem E, Tabernero J, Lakomy R, Prenen H, Prausová J, Macarulla T, Ruff P, Van Hazel GA, Moiseyenko V, Ferry D, McKendrick J, Polikoff J, Tellier A, Castan R, Allegra C: Addition of aflibercept to FOLFIRI improves survival in a phase III randomized trial in patients with metastatic colorectal cancer previously treated with an oxaliplatin-based regimen. J Clin Oncol. 2012, 10.1200/JCO.2012.42.8201.
Allegra CJ, Lakomy R, Tabernero J, Prausová J, Ruff P, Van Hazel G, Moiseyenko VM, Ferry DR, McKendrick JJ, Van Cutsem E: Effects of prior bevacizumab (B) use on outcomes from the VELOUR study: A phase III study of aflibercept (Afl) and FOLFIRI in patients (pts) with metastatic colorectal cancer (mCRC) after failure of an oxaliplatin regimen. J Clin Oncol. 2012, 30 (suppl): 3505-
Fielding A, Hoff PM, Pike L, Wilson D, Robertson JD: Exploration of the effect of treatment to progression with the VEGF signaling inhibitor cediranib (CED) plus chemotherapy (CT) on the results of the HORIZON (HZ) II trial in first-line metastatic colorectal cancer. J Clin Oncol. 2011, 29 (suppl): e14052-
Siu LL, Shapiro JD, Jonker DJ, Karapetis CS, Zalcberg JR, Simes J, Couture F, Moore MJ, Price TJ, Siddiqui J, Nott LM, Charpentier D, Liauw WS, Sawyer MB, Jefford M, Magoski NM, Haydon AM, Walters IB, Tu D, O'Callaghan CJ, NCIC Clinical Trials Group and AGITG: Phase III randomized trial of cetuximab (CET) plus either brivanib alaninate (BRIV) or placebo in patients (pts) with metastatic (MET) chemotherapy refractory K-RAS wild-type (WT) colorectal carcinoma (CRC): The NCIC Clinical Trials Group and AGITG CO.20 trial. J Clin Oncol. 2012, 30 (suppl 4): 3504-
Acknowledgements
Medical editorial writing assistance was provided by Susan DePetris, PhD, of Phase Five Communications Inc., and supported by sanofi-aventis US LLC, in collaboration with Regeneron Pharmaceuticals. The author retained full editorial control over the content of the manuscript and received no compensation from any party for this work.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interest
I have participated in the advisors meetings of Genentech and Sanofi in the past 1-2 years.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Sun, W. Angiogenesis in metastatic colorectal cancer and the benefits of targeted therapy. J Hematol Oncol 5, 63 (2012). https://doi.org/10.1186/1756-8722-5-63
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1756-8722-5-63