
Abstract
In the 9th ETA-CRN Meeting (September 2009, Lisbon) some recommendations from the American Thyroid Association (ATA) guidelines for the management of medullary thyroid cancer (MTC) were discussed by an European Panel of Experts (EPE). Among the 12 ATA recommendations related to hereditary MTC and to the optimal range of RET mutations to be tested (recommendations 1-5 and 9-15), 7 were shared and 5 were not shared by the EPE. In the present paper, the related comments and suggestions will be reported and discussed.
Similar content being viewed by others


Introduction
In the 9th ETA-CRN Meeting (September 2009, Lisbon) some recommendations from the American Thyroid Association (ATA) guidelines for the management of medullary thyroid cancer (MTC) [1] were discussed by an European Panel of Experts (EPE). In the present paper, the comments risen on ATA recommendations (ATA-R) related to hereditary MTC and to the optimal range of RET mutations to be tested will be reported and discussed (Additional file 1).
European comments to ATA-R 1-5 and 9-15
ATA recommendations shared by the EPE
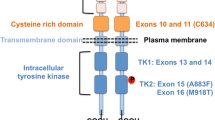
All experts fully agreed with the need to perform RET testing in subjects at risk for autosomal dominant inheritance of MEN 2 (multiple endocrine neoplasia type 2) or FMTC (Familial Medullary Thyroid Cancer) and, in particular, shortly after birth for MEN 2B and before 5 years of age for MEN 2A and FMTC (ATA-R #3). Agreement was also found about the need for a pre- and post-RET testing expert genetic counseling (ATA-R #5) and it was stated that a list of European laboratories should probably be useful for patients. Nevertheless, though at the Orphanet EuroGentest website (http://www.orpha.net/consor/cgi-bin/index.php) more than 100 European laboratories are listed, this is probably far to be the complete list, also considering that it largely changes over times. No concerns were risen about the need to offer RET mutation analysis to all first-degree relatives of known mutation carriers (ATA-R #9). The topic of ATA-R #12 relates to the indication for the sequencing of the entire coding region of RET in patients with MEN 2 and negative for mutations in exons 8, 10, 11, 13-16. It should be underlined that the remaining 13 exons (1-7, 9, 12, 17-21) have never been found to be mutated [2]. Nevertheless, though this approach was considered poorly cost effective, most experts and the 46% of the participants agreed with the ATA indications, since rare mutations comprise up to 30% of all hereditary cases, based on the literature data [3–5]. An additional recommendation would be to start the analysis of the additional regions of the gene from exon 5, which has been reported to be mutated (G321R) [6]. Strictly related to this topic, the possibility to analyze, in this category of patients, pheocrocytoma (PHAEO)- related genes was also discussed. The background was that 40-45% of apparently sporadic PHAEO or paragangliomas are hereditary and germline mutations are found in VHL (von Hippel-Lindau tumor suppressor, E3 ubiquitin protein ligase), SDHB/C/D (succinate dehydrogenase complex, subunit B, subunit C, subunit D) and RET (ret proto-oncogene) [7]. Nevertheless, the prevalence of mutated cases is extremely variable among different Countries, and in particular very low for the RET gene. This must be considered when the decision is taken to analyze PHAEO-related genes in RET negative cases.
In the clinical setting of MEN 2B, since virtually all patients harbor mutations in exons 15 and 16 (A883F and M918T, respectively), the panel agreed with ATA indication to analyze both exons and to test the entire RET coding region if those 2 exons are negative for mutations (ATA-R #13 and #14). Finally, total agreement was also expressed regarding the ATA-R #15 which indicates to treat MEN 2B patients harboring codon 804 mutation associated to another mutation similarly to those with the more typical MEN 2B RET causing mutations. Indeed, data from the literature report that the phenotype associated to V804M mutation alone is mostly FMTC [8, 9], though also MEN 2A cases have been described [9–11]. However, when this mutation is associated to another on the same allele (Y806C, S904C, E805K, V778I) the phenotype resembles that of MEN 2B [12–16].
ATA recommendations not shared by the EPE
The ATA-R #1, related to the indication to offer the germline RET testing to all patients with a personal medical history of primary C cell hyperplasia (CCH), MTC, or MEN2, was extensively discussed. In particular, while no concerns were risen about the need to perform RET analysis in a patient with MEN, it was observed that RET mutations are extremely rare in patients with CCH (0.03%), as reported by Scheuba et al. in 2009 [17]. Thus, the genetic analysis was not considered cost-effective for CCH cases by about half of the EPE, though the indication to recommend it was shared by other experts and by the majority of the participants. Similarly, there were some pros and cons also regarding apparently sporadic MTC. On one hand, the presentation of sporadic and familial-non MEN2A /2B- forms is almost identical and they cannot be differentiated on a clinical basis. Moreover, it has been reported that 6.5-10% of apparently sporadic cases are indeed hereditary forms [6, 18]. On the other hand, it was outlined that the extensive and non-selective use of RET testing could lead to a stop in the reimbursement of the genetic screening (at least of the less involved exons) by the National Health Care Systems in patients with MTC, citing a lack of evidence to suggest hereditary disease [19]. The final recommendation was thus to modulate RET testing in apparently sporadic MTC according to clinical data and the local General Health System.
The ATA-R #11 is related to the identification of the optimal range of RET mutations to be tested. Most of the experts and about half of the participants suggested to extend the analysis to exon 8, at least in all regions where mutations in that exon have been described, always in non MEN 2A/2B hereditary MTCs (Brazil, Greece, France, Italy) [20–23].
ATA-R #10 (grade A) suggests to perform the testing of exon 10 in individuals with Hirschprung disease (HSCR), in order to detect families with activating mutations 618 o 620 in whom HSCR and MEN 2A are associated. Nevertheless, it should be highlighted that this association is frequent for MEN 2A (25-40% of families), but it is rare (2%) [24] in a very common disease such as HSCR, which has been related to mutations in at least 10 different genes. RET testing was thus considered to be not cost-effective and most experts indicated to downgrade this recommendation, and proposed to test HSCR patients for basal Ct.
Another important topic was related to the lichen planus amyloidosis, in the presence of which ATA guidelines recommend (#4, grade C) to perform the RET genetic testing for 634 mutation. The EPE and the large majority of participants proposed to upgrade this recommendation based on the data of the literature showing that this early cutaneous manifestation is more frequent than previously suspected, being present in up to 36% of MEN 2A cases [25]. In addition, lichen planus has been associated not only to codon 634, but also to codon 804 mutations [26]. Thus, the presence of lichen planus amyloidosis or pruritis in the central upper back should prompt Ct measurement in adults and genetic testing in children, possibly after 4 mm punch biopsy and staining with thioflavin T in order to identify amyloid deposits.
ATA-R #2 (grade B) is related to the indication to perform RET testing in cases of intestinal ganglioneuromatosis. The background refers to data reporting that in MEN 2B, de novo mutations are extremely more frequent (50-90%) than for the other familial MTCs (2-9%) [27]. For this reason, the early diagnosis of MEN 2B in several cases cannot be based on familial RET screening. Moreover, extrathyroidal MEN 2B symptoms develop age dependently, and, though the majority of the “classic” symptoms is not expressed before 4-6 years of age, ganglioneuromatosis is always present and it is one of the earliest signs [28]. In keeping with the general agreement that in MEN 2B the clinical diagnosis of the syndrome is crucial, EPE was concordant in the indication to perform the RET screening in cases presenting with one or more signs related to the syndromes, such as ganglioneuromatosis, but also bumpy lips with mucosal neuroma, and corneal nerves thickening (with a prevalence of 69% in MEN 2B and 29% in MEN 2A) [29, 30]. Another crucial sign, not reported in the ATA guidelines, is the tearless crying which can be recognized in more than 85% of the MEN 2A/B infants and which seems to be one of the most promising signs [27], though more data are likely needed to include this manifestation as indication for RET testing in infants without any other MEN 2 symptoms and with negative family history.
Finally, the EPE discussed the opportunity to include some recommendation about RET polymorphism, not reported in the ATA guidelines. Conflicting results have been published in recent years about the possible role of RET polymorphisms as genetic modifiers either in sporadic or hereditary MTC. In particular, and only considering familial MTCs, an association between G691S and S904S with an earlier age at onset in MEN2A [31] was observed, in patients with the V804M mutation, the association between L769L and earlier age of onset was reported [32], whereas in patients with G533C mutation, the variant IVS1-126G > T was found to be associated with age at diagnosis and IVS8 +82A > G; 85-86 insC with node metastases at diagnosis [33]. More recently, the RET S836S variant has been found to be associated with early onset and increased risk for metastatic disease [34]. Nevertheless, the data available to date have not been significantly replicated and discordant data have been obtained in different series [35]. Thus, the EPE and most of the participants supported the idea to draw a recommendation highlighting the lack of clinical significance of polymorphic variants.
Abbreviations
- CCH:
-
C cell hyperplasia
- FMTC:
-
Familial Medullary Thyroid Cancer
- HSCR:
-
Hirschprung disease
- MEN 2:
-
multiple endocrine neoplasia type 2
- MTC:
-
Medullary Thyroid Cancer
- PHAEO:
-
pheocrocytoma
References
American Thyroid Association Guidelines Task Force, Kloos RT, Eng C, Evans DB, Francis GL, Gagel RF, Gharib H, Moley JF, Pacini F, Ringel MD, Schlumberger M, Wells SA Jr: Medullary thyroid cancer: management guidelines of the American Thyroid Association. Thyroid 2009, 19: 565–612. 10.1089/thy.2008.0403
Stenson PD, Ball EV, Howells K, Phillips AD, Mort M, Cooper DN: The Human Gene Mutation Database: providing a comprehensive central mutation database for molecular diagnostics and personalized genomics. Hum Genomics 2009, 4: 69–72. 10.1186/1479-7364-4-2-69
Romei C, Mariotti S, Fugazzola L, Taccaliti A, Pacini F, Opocher G, Mian C, Castellano M, degli Uberti E, Ceccherini I, Cremonini N, Seregni E, Orlandi F, Ferolla P, Puxeddu E, Giorgino F, Colao A, Loli P, Bondi F, Cosci B, Bottici V, Cappai A, Pinna G, Persani L, Verga U, Boscaro M, Castagna MG, Cappelli C, Zatelli MC, Faggiano A, Francia G, Brandi ML, Falchetti A, Pinchera A, Elisei R, ItaMEN network: Multiple endocrine neoplasia type 2 syndromes (MEN 2): results from the ItaMEN network analysis on the prevalence of different genotypes and phenotypes. Eur J Endocrinol 2010, 163: 301–308. 10.1530/EJE-10-0333
Cordella D, Muzza M, Alberti L, Colombo P, Travaglini P, Beck-Peccoz P, Fugazzola L, Persani L: An in-frame complex germline mutation in the juxtamembrane intracellular domain causing RET activation in familial medullary thyroid carcinoma. Endocr Relat Cancer 2006, 13: 945–953. 10.1677/erc.1.01144
Lindsey SC, Kunii IS, Germano-Neto F, Sittoni MY, Camacho CP, Valente FO, Yang JH, Signorini PS, Delcelo R, Cerutti JM, Maciel RM, Dias-da-Silva MR: Extended RET gene analysis in patients with apparently sporadic medullary thyroid cancer: clinical benefits and cost. Horm Cancer 2012, 3: 181–186. 10.1007/s12672-012-0109-7
Romei C, Cosci B, Renzini G, Bottici V, Molinaro E, Agate L, Passannanti P, Viola D, Biagini A, Basolo F, Ugolini C, Materazzi G, Pinchera A, Vitti P, Elisei R: RET genetic screening of sporadic medullary thyroid cancer (MTC) allows the preclinical diagnosis of unsuspected gene carriers and the identification of a relevant percentage of hidden familial MTC (FMTC). Clin Endocrinol (Oxf) 2011, 74: 241–247. 10.1111/j.1365-2265.2010.03900.x
Opocher G, Schiavi F: Genetics of pheochromocytomas and paragangliomas. Best Pract Res Clin Endocrinol Metab 2010, 24: 943–956. 10.1016/j.beem.2010.05.001
Feldman GL, Edmonds MW, Ainsworth PJ, Schuffenecker I, Lenoir GM, Saxe AW, Talpos GB, Roberson J, Petrucelli N, Jackson CE: Variable expressivity of familial medullary thyroid carcinoma (FMTC) due to a RET V804M (GTG-->ATG) mutation. Surgery 2000, 128: 93–98. 10.1067/msy.2000.107103
Pinna G, Orgiana G, Riola A, Ghiani M, Lai ML, Carcassi C, Mariotti S: RET proto-oncogene in Sardinia: V804M is the most frequent mutation and may be associated with FMTC/MEN-2A phenotype. Thyroid 2007, 17: 101–104. 10.1089/thy.2006.0198
Nilsson O, Tisell LE, Jansson S, Ahlman H, Gimm O, Eng C: Adrenal and extra-adrenal pheochromocytomas in a family with germline RET V804L mutation. JAMA 1999, 281: 1587–1588. 10.1001/jama.281.17.1587
Gibelin H, Bezieau S, Misso C, Bouin-Pineau MH, Maréchaud R, Kraimps JL: Germline RET V804M mutation associated with multiple endocrine neoplasia type 2A. Br J Surg 2004, 91: 1458–1459. 10.1002/bjs.4718
Miyauchi A, Futami H, Hai N, Yokozawa T, Kuma K, Aoki N, Kosugi S, Sugano K, Yamaguchi K: Two germline missense mutations at codons 804 and 806 of the RET proto-oncogene in the same allele in a patient with multiple endocrine neoplasia type 2B without codon 918 mutation. Jpn J Cancer Res 1999, 90: 1–5. 10.1111/j.1349-7006.1999.tb00658.x
Menko FH, van der Luijt RB, de Valk IA, Toorians AW, Sepers JM, van Diest PJ, Lips CJ: Atypical MEN type 2B associated with two germline RET mutations on the same allele not involving codon 918. J Clin Endocrinol Metab 2002, 87: 393–397. 10.1210/jcem.87.1.8136
Cranston AN, Carniti C, Oakhill K, Radzio-Andzelm E, Stone EA, McCallion AS, Hodgson S, Clarke S, Mondellini P, Leyland J, Pierotti MA, Whittaker J, Taylor SS, Bongarzone I, Ponder BA: RET is constitutively activated by novel tandem mutations that alter the active site resulting in multiple endocrine neoplasia type 2B. Cancer Res 2006, 66: 10179–10187. 10.1158/0008-5472.CAN-06-0884
Kasprzak L, Nolet S, Gaboury L, Pavia C, Villabona C, Rivera-Fillat F, Oriola J, Foulkes WD: Familial medullary thyroid carcinoma and prominent corneal nerves associated with the germline V804M and V778I mutations on the same allele of RET. J Med Genet 2001, 38: 784–787. 10.1136/jmg.38.11.784
Mukherjee S, Zakalik D: RET codon 804 mutations in multiple endocrine neoplasia 2: genotype-phenotype correlations and implications in clinical management. Clin Genet 2011, 79: 1–16. 10.1111/j.1399-0004.2010.01453.x
Scheuba C, Kaserer K, Moritz A, Drosten R, Vierhapper H, Bieglmayer C, Haas OA, Niederle B: Sporadic hypercalcitoninemia: clinical and therapeutic consequences. Endocr Relat Cancer 2009, 16: 243–253.
Wiench M, Wygoda Z, Gubala E, Wloch J, Lisowska K, Krassowski J, Scieglinska D, Fiszer-Kierzkowska A, Lange D, Kula D, Zeman M, Roskosz J, Kukulska A, Krawczyk Z, Jarzab B: Estimation of risk of inherited medullary thyroid carcinoma in apparent sporadic patients. J Clin Oncol 2001, 19: 1374–80.
Machens A, Dralle H: Prophylactic thyroidectomy in RET carriers at risk for hereditary medullary thyroid cancer. Thyroid 2009, 19: 551–554. 10.1089/thy.2009.0144
Da Silva AM, Maciel RM, Da Silva MR, Toledo SR, De Carvalho MB, Cerutti JM: A novel germ-line point mutation in RET exon 8 (Gly(533)Cys) in a large kindred with familial medullary thyroid carcinoma. J Clin Endocrinol Metab 2003, 88: 5438–5443. 10.1210/jc.2003-030997
Fazioli F, Piccinini G, Appolloni G, Bacchiocchi R, Palmonella G, Recchioni R, Pierpaoli E, Silvetti F, Scarpelli M, Bruglia M, Melillo RM, Santoro M, Boscaro M, Taccaliti A: A new germline point mutation in Ret exon 8 (cys515ser) in a family with medullary thyroid carcinoma. Thyroid 2008, 18: 775–782. 10.1089/thy.2007.0365
Muzza M, Cordella D, Bombled J, Bressac-de Paillerets B, Guizzardi F, Francis Z, Beck-Peccoz P, Schlumberger M, Persani L, Fugazzola L: Four novel RET germline variants in exons 8 and 11 display an oncogenic potential in vitro. Eur J Endocrinol 2010, 162: 771–777. 10.1530/EJE-09-0929
Sarika HL, Papathoma A, Garofalaki M, Vasileiou V, Vlassopoulou B, Anastasiou E, Alevizaki M: High prevalence of exon 8 g533c mutation in apparently sporadic medullary thyroid carcinoma (mtc) in Greece. Clin Endocrinol (Oxf) 2012, 7. doi: 10.1111/j.1365–2265.2012.04462.x. [Epub ahead of print]
Amiel J, Sproat-Emison E, Garcia-Barcelo M, Lantieri F, Burzynski G, Borrego S, Pelet A, Arnold S, Miao X, Griseri P, Brooks AS, Antinolo G, de Pontual L, Clement-Ziza M, Munnich A, Kashuk C, West K, Wong KK, Lyonnet S, Chakravarti A, Tam PK, Ceccherini I, Hofstra RM, Fernandez R, Hirschsprung Disease Consortium: Hirschsprung disease, associated syndromes and genetics: a review. J Med Gene 2008, 45: 1–14. 10.1136/jmg.2007.055129
Verga U, Fugazzola L, Cambiaghi S, Pritelli C, Alessi E, Cortelazzi D, Gangi E, Beck-Peccoz P: Frequent association between MEN 2A and cutaneous lichen amyloidosis. Clin Endocrinol (Oxf) 2003, 59: 156–161. 10.1046/j.1365-2265.2003.01782.x
Rothberg AE, Raymond VM, Gruber SB, Sisson J: Familial medullary thyroid carcinoma associated with cutaneous lichen amyloidosis. Thyroid 2009, 19: 651–655. 10.1089/thy.2009.0021
Brauckhoff M, Gimm O: Extrathyroidal manifestations of multiple endocrine neoplasia type 2. Thyroid 2009, 19: 555–557. 10.1089/thy.2009.0151
Camacho CP, Hoff AO, Lindsey SC, Signorini PS, Valente FO, Oliveira MN, Kunii IS, Biscolla RP, Cerutti JM, Maciel RM: Early diagnosis of multiple endocrine neoplasia type 2B: a challenge for physicians. Arq Bras Endocrinol Metabol 2008, 52: 1393–1398. 10.1590/S0004-27302008000800031
Vasen HF, van der Feltz M, Raue F, Kruseman AN, Koppeschaar HP, Pieters G, Seif FJ, Blum WF, Lips CJ: The natural course of multiple endocrine neoplasia type IIb. A study of 18 cases. Arch Intern Med 1992, 152: 1250–1252. 10.1001/archinte.1992.00400180104016
Kinoshita S, Tanaka F, Ohashi Y, Ikeda M, Takai S: Incidence of prominent corneal nerves in multiple endocrine neoplasia type 2A. Am J Ophthalmol 1991, 111: 307–311.
Robledo M, Gil L, Pollán M, Cebrián A, Ruíz S, Azañedo M, Benitez J, Menárguez J, Rojas JM: Polymorphisms G691S/S904S of RET as genetic modifiers of MEN 2A. Cancer Res 2003, 63: 1814–1817.
Magalhães PK, de Castro M, Elias LL, Soares EG, Maciel LM: Polymorphisms in the RET proto-oncogene and the phenotypic presentation of familial medullary thyroid carcinoma. Thyroid 2004, 14: 848–852. 10.1089/thy.2004.14.848
Tamanaha R, Camacho CP, Pereira AC, da Silva AM, Maciel RM, Cerutti JM: Evaluation of RET polymorphisms in a six-generation family with G533C RET mutation: specific RET variants may modulate age at onset and clinical presentation. Clin Endocrinol (Oxf) 2009, 71: 56–64. 10.1111/j.1365-2265.2008.03491.x
Siqueira DR, Romitti M, da Rocha AP, Ceolin L, Meotti C, Estivalet A, Puñales MK, Maia AL: The RET polymorphic allele S836S is associated with early metastatic disease in patients with hereditary or sporadic medullary thyroid carcinoma. Endocr Relat Cancer 2010, 17: 953–963. 10.1677/ERC-09-0312
Ceolin L, Siqueira DR, Romitti M, Ferreira CV, Maia AL: Molecular Basis of Medullary Thyroid Carcinoma: The Role of RET Polymorphisms. Int J Mol Sci 2012, 13: 221–239.
Declarations
This article has been published as part of Thyroid Research Volume 6 Supplement 1, 2013: European comments on Medullary Thyroid Cancer Management Guidelines of the American Thyroid Association. The full contents of the supplement are available online at http://www.thyroidresearchjournal.com/supplements/6/S1. Publication of this supplement has been supported by the European Thyroid Association-Cancer Research Network.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
No competing interests exist for me and my co-authors.
Electronic supplementary material
13044_2013_93_MOESM1_ESM.docx
Additional file 1: Table 1. American Thyroid Association recommendations (ATA R) and the corresponding recommendation resulting from the European Panel of Experts (EPE R rev). (DOCX 14 KB)
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Fugazzola, L., De Leo, S. & Perrino, M. The optimal range of RET mutations to be tested: European comments to the guidelines of the American Thyroid Association. Thyroid Res 6 (Suppl 1), S8 (2013). https://doi.org/10.1186/1756-6614-6-S1-S8
Published:
DOI: https://doi.org/10.1186/1756-6614-6-S1-S8