
Abstract
Loss of the translational repressor FMRP causes Fragile X syndrome. In healthy neurons, FMRP modulates the local translation of numerous synaptic proteins. Synthesis of these proteins is required for the maintenance and regulation of long-lasting changes in synaptic strength. In this role as a translational inhibitor, FMRP exerts profound effects on synaptic plasticity.
Similar content being viewed by others


Background
The long-term maintenance of many forms of synaptic plasticity requires the synthesis of new proteins. While the role of experience-dependent somatic gene transcription in long-term memory has been well studied [1], many mRNAs are trafficked to dendrites suggesting an additional role for local synaptic control of protein synthesis [2]. Indeed, activity-dependent translation of pre-existing dendritic mRNA at the synapse is necessary for the expression of multiple forms of synaptic plasticity [3–5]. Fragile X mental retardation protein (FMRP) influences this synaptic plasticity by functioning as a key regulator of mRNA translation [6–10].
FMRP was first characterized in the context of Fragile X syndrome. The FMR1 gene is silenced in Fragile X (FX), and the consequent loss of FMRP leads to the symptoms of the disorder, often including intellectual disability and autism. In the Fmr1 KO mouse model [11], loss of FMRP results in increased levels of protein synthesis [9, 12]. The downstream consequences of this increase are believed to at the core of FX pathophysiology [13–15]. Rapid progress has been made characterizing how loss of FMRP influences synaptic function and plasticity, and this knowledge has led to several strategies to correct the disorder that have been validated in animals and are now being tested in humans [16–19].
Here we briefly review the evidence, mostly from the Fmr1 KO mouse, suggesting a role for FMRP in synaptic plasticity. Although the distinction is not always clear-cut, it is conceptually important to separate disruptions of synaptic plasticity that are consequences of altered brain development from those disruptions of synaptic plasticity that cause altered brain function in the Fmr1 KO. While both are important for understanding disease pathophysiology, only the latter is relevant to the question of how FMRP contributes to synaptic plasticity in the wild-type brain.
FMRP regulates translation
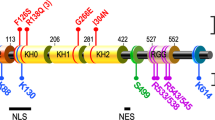
FMRP is an RNA-binding protein and a repressor of translation which is well-conserved from mouse to human. FMRP associates with mRNAs through one of three RNA-binding domains [20, 21], in some cases in conjunction with adaptor proteins [22, 23]. There is evidence that FMRP can repress translation both by blocking initiation and elongation [15, 24, 25]. A point mutation in one FMRP/mRNA binding domain is sufficient to recapitulate plasticity phenotypes seen in the Fmr1 KO mouse [26] and in at least one case FX in a human patient [27]. Thus it is likely that FMRP regulates plasticity mainly in its role as a repressor of translation.
FMRP is regulated by posttranslational modifications. Phosphorylated FMRP stalls ribosomal translocation and inhibits translation, whereas dephosphorylation of FMRP upregulates translation [28–30]. Bidirectional regulation of FMRP phosphorylation by the S6 kinase and protein phosphatase 2A (PP2A) in response to activity provide a potential link between synaptic stimulation and local translation [24].
FMRP is well-positioned to regulate synaptic plasticity
FMRP is well-positioned to be a key regulator of synaptic plasticity for three main reasons. First, the protein is found in dendritic spines [31–34], important postsynaptic sites of plasticity induction and maintenance. Secondly, FMRP regulates dendritic mRNA translation [16, 17], which is required for multiple forms of plasticity [35]. Finally, FMRP itself is dynamically regulated by activity: experience and synaptic activation can trigger its local translation and rapid degradation, in addition to the posttranslational regulation mentioned above. Multiple experimental manipulations associated with synaptic plasticity have been shown to increase FMRP levels, including exposure to an enriched environment, a complex learning task, and pharmacological activation of group 1 metabotropic glutatmate receptors (mGluRs) [31, 36–38]. Importantly, FMRP is synthesized rapidly, on the same time scale (10–30 minutes) as induction of stable synaptic plasticity [37]. In hippocampal cultures, activity- and mGluR-dependent increases in dendritic FMRP may result from increased trafficking of existing FMRP, rather than de novo FMRP synthesis [33, 39, 40]. Either way, FMRP is an ideal candidate to be involved in regulating synaptic plasticity because of its rapid, transient rise in dendrites following well-characterized plasticity induction paradigms, as well as its role as an inhibitor of translation.
FMRP regulates mGluR-LTD via protein synthesis
Long-term potentiation (LTP) and long-term depression (LTD) are well-characterized forms of synaptic plasticity associated with learning and memory. These persistent changes in synaptic strength can be induced by a variety of manipulations and their expression mechanisms are diverse. Different induction protocols rely on different mechanisms for maintenance, including the requirement for protein synthesis. A particularly compelling example of a form of plasticity requiring local translation is metabotropic glutamate receptor-dependent LTD (mGluR-LTD) in the CA1 region of the hippocampus. Activation of group 1 mGluRs (mGluR1 and 5), either with paired-pulse low-frequency synaptic stimulation (PP-LFS) [4] or with the selective agonist (S)-3,5-dihydroxyphenylglycine (DHPG) [41–43], results in a persistent decrease in synaptic strength that is mechanistically distinct from classical NMDA receptor (NMDAR)-dependent LTD [41, 44]. It is important to note that there are several mechanisms downstream of mGluR activation that can depress synaptic transmission, and these can be differentially expressed depending on the induction protocol, age, rearing history, and species (e.g., [44–48]). However, under appropriate experimental conditions the maintenance of mGluR-LTD requires rapid protein synthesis within minutes of induction [4, 49]. This protein synthesis is likely to be synaptic, as mGluR-LTD can still be induced if the dendritic layer is physically severed from the cell body layer [4]. mGluR-LTD is expressed, in part, by the removal of AMPA receptors from synapses, which also requires rapid de novo translation [50]. The new protein synthesis may be instructive rather than merely permissive for synaptic plasticity since activation of group 1 mGluRs rapidly stimulates protein synthesis in hippocampal slices [12], dendrites and synaptoneurosomes [51, 52].
Fmr1 knockout mice show enhanced hippocampal mGluR-LTD [8, 14, 49, 53] (Table 1). A subsequent study found a similar enhancement in cerebellar mGluR-LTD, which shares many of the same expression mechanisms [54]. Consistent with the electophysiological data, loss of FMRP leads to excessive mGluR-mediated AMPAR internalization [55]. In addition, mGluR-LTD no longer requires new protein synthesis in the Fmr1 KO mice [49, 56]. These results, combined with what is known about FMRP function, suggest that FMRP acts to inhibit the synthesis of proteins required for mGluR-LTD. In the absence of FMRP, these “LTD proteins” are already available or over-expressed in dendrites resulting in enhanced magnitude and protein synthesis-independent persistence of this form of plasticity (Figure 1A) [13]. Conversely, postnatal overexpression of FMRP reduces the magnitude of mGluR-LTD in both wildtype and Fmr1 KO neurons [49] and restores its protein synthesis dependence [57]. Moreover, reducing mGluR5 signaling in Fmr1 KO mice restores both protein synthesis rates and LTD magnitude in the hippocampus to wildtype levels [53, 58], suggesting that mGluR5 and FMRP act in functional opposition to maintain an optimal level of synaptic protein synthesis throughout development and into adulthood (Figure 1A).

The role of FMRP in translation-dependent synaptic plasticity. (A) FMRP and mGluR5 impose opposite regulation on the local mRNA translation required for mGluR-LTD expression. In the absence of FMRP, there is excessive protein synthesis and exaggerated LTD. (B) While FMRP is known to regulate the translation required for LTD, evidence suggests it is not involved in the expression of L-LTP. There may be different pools of mRNA available at the synapse that are differentially required for LTD versus LTP, and FMRP may specifically regulate the pool required for LTD. (C) FMRP is explicitly involved in the regulation of dendritically localized translation and may not regulate somatic translation. Consequently, FMRP may only impact forms of plasticity that require local translation, such as mGluR-LTD. (D) In addition to mGluR-LTD, FMRP regulates the protein synthesis involved in mGluR-dependent facilitation of LTP. This finding suggests that the proteins whose translation is controlled by FMRP may be involved in bi-directional maintenance of plasticity rather than being specific to LTD.
L-LTP appears normal in Fmr1 KO mice
While the effects of protein synthesis inhibition on mGluR-LTD can be seen within minutes, most forms of synaptic plasticity do not require de novo synthesis until several hours after induction. This is best characterized by late phase LTP (L-LTP), a persistent form of potentiation lasting at least 4 hours. The late maintenance phase of L-LTP requires protein synthesis but initial induction does not [59, 60]. Due to FMRP’s conjectured role in translation regulation, L-LTP was one of the first forms of plasticity studied in the Fmr1 KO mouse [61]. Interestingly, no difference has been found in the magnitude of L-LTP in the Fmr1 KO [61, 62]. The fact that removal of FMRP affects protein synthesis-dependent LTD but not LTP suggests that FMRP may specifically regulate the translation of proteins required for the expression of LTD (Figure 1B). However, while the magnitude of L-LTP is unchanged, it is possible that L-LTP is qualitatively different in its requirement for new protein synthesis when FMRP is absent, as is the case for mGluR-LTD (and LTP priming, see below). Therefore, it will be important to test the protein synthesis-dependency of L-LTP in Fmr1 KO mice to show that FMRP truly does not play a role in regulating the persistence of LTP.
Alternatively, FMRP may be required for the regulation of local but not somatic translation in the context of L-LTP (Figure 1C). L-LTP is traditionally induced by multiple trains of high frequency tetanus or theta burst stimulation, protocols that rely on cell-wide transcription and translation [63–65]. L-LTP was characterized in the Fmr1 KO mouse using these classical paradigms [61, 62]. However, using a less intense induction protocol results in L-LTP that is maintained specifically by local dendritic translation [5, 66]. This form of L-LTP, similar to mGluR-LTD, is sensitive to inhibitors of translation but not transcription, and can be maintained in isolated dendrites. It will be interesting to determine if this locally expressed form of L-LTP is regulated by FMRP.
FMRP regulates LTP priming
While the role of FMRP in L-LTP is unclear, FMRP is known to be involved in LTP in other contexts. In particular, FMRP is involved in regulation of an mGluR-dependent form of metaplasticity that sets the threshold for LTP. Originally described in rats [67], weak activation of group 1 mGluRs, in itself insufficient for LTD induction, facilitates the subsequent induction of LTP (“LTP priming”). As with mGluR-LTD, this facilitation requires translation but not transcription [68]. This prompted the examination of the role of FMRP in LTP priming [69]. mGluR-dependent priming of LTP is of comparable magnitude in WT and Fmr1 KO mice; however, while LTP priming requires acute stimulation of protein synthesis in WT mice, it is no longer protein synthesis-dependent in the Fmr1 KO. Thus, while mGluR-LTD and LTP priming are qualitatively different functional consequences of Gp1 mGluR-stimulated protein synthesis in the hippocampus, both processes are altered by the removal of FMRP (Figure 1D). These results suggest that the mRNA under translational control of FMRP may code for proteins required for bidirectional changes in synaptic strength. Thus, the proteins regulated by FMRP should be conceptualized as plasticity gatekeepers rather than solely “LTD proteins.”
The induction threshold for LTP and STD-LTP is raised in Fmr1 KO mice
In Fmr1 KO hippocampal slices, LTP induction is deficient with a weak 5 theta burst protocol but is normal with a strong 10 theta burst protocol (Figure 2A) [70]. In addition, FMRP modulates the induction threshold for spike-timing dependent long-term potentiation (STD-LTP). This form of Hebbian plasticity is induced by temporally staggered presynaptic and postsynaptic activity within a very short window [71, 72]. In somatosensory and prefrontal cortices, STD-LTP is deficient in Fmr1 KO neurons [73, 74]. However, if the postsynaptic stimulus strength is increased from a single spike to a burst of five spikes, STD-LTP does occur in KO neurons (Figure 2A) [74]. Therefore FMRP is not required for expression of STD-LTP, but the threshold is raised in its absence. A possible mechanism for ongoing regulation of LTP thresholds by FMRP is discussed later in this review.

FMRP and Kv4.2 regulate the threshold for inducing synaptic potentiation. (A) FMRP sets the threshold for LTP and STD-LTP. Fmr1 KO mice have deficient hippocampal LTP and cortical STD-LTP only with a “weak” induction protocol. (B) Kv4.2 is a dendritic A-type K+ channel that attenuates action potential backpropagation (bAP) and dendritic excitability. (C) Inhibition of Kv4.2 restores LTP following a weak induction protocol in Fmr1 KO mice.
FMRP and other translation-dependent forms of plasticity
In addition to its role in translation-dependent forms of Hebbian plasticity, FMRP can also modulate some forms of homeostatic plasticity. Synaptic scaling is a form of homeostatic plasticity that acts to keep the strength of synapses within a functional range in response to extreme changes in activity. Broadly, a decrease in activity leads to a subsequent cell-wide increase in synaptic strength (“scaling up”) and an increase in activity leads to a decrement in synaptic strength (“scaling down”) [75]. Two types of scaling up have been described in hippocampal slice culture: one that requires transcription [76] and one that requires local translation [77]. Interestingly, only the translation-dependent form of synaptic scaling is deficient in neurons lacking FMRP. Postsynaptic viral expression of FMRP corrects deficient translation-dependent scaling up in Fmr1 KO neurons [78]. Scaling down of synapses in response to high levels of activity (following prolonged blockade of inhibition) has also been observed [79] and requires mGluR5 activation [80, 81]. However, the role of FMRP and local protein synthesis in scaling down has not been directly examined.
While the role of FMRP has been best characterized in mGluR-dependent forms of plasticity, it is not specific to these receptors. Removal of FMRP occludes TrkB-mediated increases in protein synthesis [12] and alters other forms of G protein-coupled receptor (GPCR)-dependent LTD and LTP [82, 83]. The common thread between these processes is their reliance on local dendritic translation. Indeed, evidence suggests that FMRP may specifically be important for the regulation of local rather than somatic translation (Figure 1C), as removal of FMRP affects translation but not transcription-dependent forms of Hebbian and homeostatic plasticity.
FMRP and translation-independent plasticity
While many forms of translation-dependent synaptic plasticity are abnormal in Fmr1 KO mice, other forms of hippocampal plasticity, including NMDAR-dependent LTD and early-phase LTP, are normal [8, 61, 69, 84, 85]. These observations suggest that FMRP regulates plasticity mainly in its role as a regulator of translation. However, removal of FMRP has also been shown to affect some forms of synaptic plasticity that do not require de novo translation, such as early-phase LTP in other brain areas, including the cortex and amygdala [61, 85–89]. Some of these effects could be explained by FMRP modulation of protein synthesis-dependent plasticity thresholds; however it seems likely that many represent end-stage consequences of altered synaptic development in the Fmr1 KO.
A case in point is altered LTP in the amygdala. A substantial deficit in basal transmission was reported at the same synapses that showed impaired LTP [88]. Reduced synaptic connectivity might have caused the defective LTP, and might have arisen as a consequence of increased FMRP-dependent protein synthesis during the development of amygdala circuitry.
Candidate plasticity gating proteins regulated by FMRP
In order to determine how FMRP regulates synaptic plasticity, we must identify the synaptic proteins whose translation is regulated by FMRP. FMRP has a wide variety of targets - it has been shown to selectively bind approximately 4% of the mRNA in the mammalian brain [90]. Recently, over 800 mRNA binding targets of FMRP were identified using a novel high throughput cross-linking immunoprecipitation (HITS-CLIP) assay [10]. These targets include genes coding for pre- and post-synaptically expressed proteins: 27% of pre-synaptic protein mRNAs (90 genes) and 23% of postsynaptic protein mRNAs (257 genes) are FMRP targets [10]. More specifically, the HITS-CLIP study found that 31% of mRNAs coding for proteins in the NMDAR complex (58 genes), 62% in the mGluR5 complex (32 genes), and 33% in the AMPAR complex (3 genes) are FMRP targets. These three receptor complexes are important for the induction and maintenance of synaptic plasticity, suggesting that FMRP likely acts broadly as a translational regulator rather than solely regulating one or two “plasticity proteins.”
The finding that many FMRP targets encode presynaptic proteins is interesting and illuminating. In the mature nervous system the evidence for local protein synthesis in axons or axon terminals is still sparse; however during early axon development and synapse formation local protein synthesis is believed to play an important role in pathway and target selection [91, 92]. Thus, the absence of FMRP regulation of protein synthesis during early development very likely alters synaptic connectivity well before the onset of experience-dependent postnatal plasticity. In addition, outside the CNS, local control of translation in sensory afferent terminals plays a role in nociceptive sensitization and neuropathic pain [93]. FMRP is localized to these terminals and Fmr1 KO mice show altered nociceptive sensitization [94]. These results suggest that in the spinal cord, presynaptic FMRP may inhibit local translation and can regulate pain plasticity even into adulthood.
We have discussed two major categories of plasticity defects in Fmr1 KO mice: (1) forms of plasticity requiring FMRP/local translation for their maintenance (mGluR-LTD) and (2) forms of plasticity where FMRP regulates their induction threshold (STD-LTP). We will discuss a few proteins in both categories that are likely involved given their regulation by FMRP and their known roles in plasticity maintenance and threshold-setting in wild-type synapses. These “candidate proteins” are meant to serve as exemplars of how FMRP might regulate synaptic plasticity.
Plasticity maintenance proteins: MAP1B, Arc, and STEP
Recent work has identified proteins whose translation is regulated by FMRP and are involved in mGluR-LTD, including microtubule-associated protein 1B (MAP1B) and activity-regulated cytoskeleton-associated protein (Arc) [17, 18]. MAP1B is required for mGluR-depdendent AMPA receptor endocytosis [95], the mechanism by which mGluR-LTD is expressed. FMRP associates with MAP1B mRNA and represses its translation [90, 96–98], and Fmr1 KO mice show increased hippocampal MAP1B expression [49]. However, there may be mouse strain and region-specific variations in how FMRP regulates MAP1B translation. For example, in the cerebellum and hippocampus of FVB mice, FMRP may positively regulate MAP1B expression [99].
Arc is involved in AMPAR endocytosis [100] and is upregulated in dendrites following mGluR activation [101, 102] and behavior [103]. Arc is required for hippocampal mGluR-LTD and L-LTP, which are both protein synthesis-dependent, and Arc−/− mice have multiple learning deficits [101, 102, 104]. FMRP binds Arc mRNA and suppresses its translation. As a result, Arc expression is increased in Fmr1 KO dendrites [98, 105, 106]. Since (a) mGluR-LTD is increased in Fmr1 KO mice, (b) Arc is increased in Fmr1 KO dendrites, and (c) Arc is required for mGluR-LTD, it seems likely that FMRP regulates mGluR-LTD via Arc. This hypothesis was tested directly using Fmr1/Arc double knockout mice which show deficient (rather than exaggerated) mGluR-LTD [8, 102]. This finding suggests that increased Arc expression may partially account for the enhanced mGluR-LTD seen in Fmr1 KO mice.
Mechanistically, dephosphorylation of FMRP by the phosphatase PP2A is required for rapid mGluR-mediated increases in Arc protein. However in Fmr1 KO neurons, Arc levels are basally increased, occluding a further effect of DHPG treatment. Acute viral reintroduction of FMRP into Fmr1 KO neurons normalizes dendritic Arc levels and restores rapid mGluR-mediated Arc synthesis. This provides further evidence that the acute loss of FMRP, rather than developmental abnormality, underlies synaptic plasticity phenotypes in the Fmr1 knockout mouse. eregulation of translation.
In addition to MAP1B and Arc, numerous other candidate LTD proteins have been identified in the Fmr1 KO mouse. One interesting example is striatal-enriched protein tyrosine phosphatase (STEP). Translation of STEP is increased during mGluR-LTD [107, 108], and STEP mRNA binds to FMRP [10]. Genetic reduction of STEP corrects behavioral phenotypes in the Fmr1 KO mouse; but it is not known whether corresponding LTD phenotypes are affected [109]. Additional candidate proteins include APP [110, 111], OPHN1 [112], CaMKIIα [49, 98, 113], PSD-95 [113–115], and PI3K [116].
Plasticity threshold-regulating proteins: Kv4.2
A recent review discussing the role of potassium channels in Fragile X provides insight into how FMRP may regulate excitability [117]. FMRP directly regulates the translation of at least three potassium channels: Kv4.2, Kv3.1b, and Slack [118–122]. FMRP’s control of Kv4.2 translation may have indirect consequences on regulating the threshold for LTP and STD-LTP induction.
Kv4.2 is an A-type potassium channel that regulates dendritic excitability and the extent of action potential backpropagation [123, 124]. A-type currents act to dampen dendritic excitability and AP backpropagation (Figure 2B). By modulating the strength of backpropagation, Kv4.2 also has been shown to regulate the threshold for LTP and STD-LTP [123, 125]. In the absence of Kv4.2, dendrites are more excitable and there is a decreased threshold for LTP induction [123, 126].
Fmr1 KO mice have an increased threshold for LTP and STD-LTP induction, as discussed earlier (Figure 2A) [73, 74]. One potential hypothesis for this phenomenon is that FMRP inhibits the translation of Kv4.2, and Fmr1 KO mice have excessive Kv4.2 protein synthesized in dendrites. Indeed, FMRP does directly associate with and negatively regulate the translation of Kv4.2 mRNA [118]. But does this account for the altered LTP/STD-LTP threshold in Fmr1 KO mice? Pharmacological inhibition of Kv4.2 in Fmr1 KO mice does correct deficient weak-stimulus hippocampal LTP while strong-stimulus LTP remains unchanged [118] (Figure 2C). This finding suggests that the increased threshold for LTP in the Fmr1 KO mouse may be accounted for by increased translation of the potassium channel Kv4.2.
Interestingly, another group has recently shown that under their conditions, FMRP positively regulates the translation of Kv4.2 [119]. This study did not address the potential consequences of decreased Kv4.2 in the Fmr1 KO on synaptic plasticity. One would expect increased dendritic excitability, which has been previously reported in other contexts [127], and a decreased LTP threshold. It will be important to determine the precise experimental and in vivo conditions under which each of these opposing patterns of regulation can occur, but it is clear that FMRP’s regulation of Kv4.2 in either direction would have important consequences for plasticity.
FMRP, synaptic plasticity and learning
Long-lasting synaptic potentiation and depression have long been considered potential neural correlates of learning and memory. In conjunction with FMRP’s role in synaptic plasticity in multiple brain areas, FMRP is also important for a wide range of behavioral learning tasks in mice. Fmr1 KO mice show deficient amygdalar trace fear memory [87], cerebellar learning [54], inhibitory avoidance learning [58], and have difficulties with a prefrontal cognitive learning task [128]. Drosophila mutants lacking FMRP also have impaired long-term memory [129]. Overall, learning and memory deficits in the Fmr1 KO mouse are a likely behavioral consequence of abnormal synaptic plasticity.
Conclusions
FMRP participates in the regulation of numerous forms of synaptic plasticity, including mGluR-LTD, LTP priming, and synaptic scaling. It seems that FMRP is particularly important for synaptic plasticity that requires dendritic translation, as these forms of plasticity all require local translation and FMRP is a well-established regulator of local translation. The current evidence suggests that FMRP plays an essential role in regulating the synaptic expression of proteins required for bidirectional changes in synaptic strength (Figure 1). It is likely that FMRP controls the expression of proteins not only acutely required for expression of synaptic plasticity, but also proteins that regulate the threshold for plasticity induction (Figure 2). Therefore FMRP’s role in synaptic plasticity is two-fold: it regulates the translation of proteins that directly participate in the induction and expression of plasticity as well as proteins that can indirectly modulate the properties of plasticity.
A key goal in the Fragile X field is to identify which proteins are regulated by FMRP and how increases or decreases in these proteins may account for phenotypes of the disorder. Determining the proteins that are regulated by FMRP (and altered in FX) will also lead to a better understanding of the neuronal processes that are essential for synaptic plasticity and learning/memory. The HITS-CLIP screen has identified hundreds of candidates and a significant number of these are putatively involved in synaptic plasticity. It is unlikely that there is one global “plasticity protein” - multiple proteins likely regulate different processes in parallel. Mapping which proteins are essential for which processes is the important next step for understanding the role of FMRP in the pathological and non-pathological brain.
The Fmr1 KO mouse provides a model for assessing the role of FMRP in synaptic plasticity - but on their own, studies in Fmr1 KO mice leave open the possibility that developmental rather than acute changes result in altered synaptic plasticity. In multiple contexts, acute manipulations of FMRP suggest that FMRP does actively regulate synaptic plasticity as a regulator of translation. There is ample evidence that FMRP can directly impact synaptic plasticity through its control of protein synthesis. Future work that allows for better temporal and spatial control of FMRP expression will help dissect the role of FMRP in development from its acute effects on synaptic plasticity.
References
Kandel ER: The molecular biology of memory storage: a dialogue between genes and synapses. Science. 2001, 294: 1030-1038. 10.1126/science.1067020.
Steward O, Levy WB: Preferential localization of polyribosomes under the base of dendritic spines in granule cells of the dentate gyrus. J Neurosci. 1982, 2: 284-291.
Kang H, Schuman EM: A requirement for local protein synthesis in neurotrophin-induced hippocampal synaptic plasticity. Science. 1996, 273: 1402-1406. 10.1126/science.273.5280.1402.
Huber KM, Kayser MS, Bear MF: Role for rapid dendritic protein synthesis in hippocampal mGluR-dependent long-term depression. Science. 2000, 288: 1254-1257. 10.1126/science.288.5469.1254.
Huang YY, Kandel ER: Theta frequency stimulation induces a local form of late phase LTP in the CA1 region of the hippocampus. Learn Mem. 2005, 12: 587-593. 10.1101/lm.98905.
Laggerbauer B, Ostareck D, Keidel EM, Ostareck-Lederer A, Fischer U: Evidence that fragile X mental retardation protein is a negative regulator of translation. Hum Mol Genet. 2001, 10: 329-338. 10.1093/hmg/10.4.329.
Li Z, Zhang Y, Ku L, Wilkinson KD, Warren ST, Feng Y: The fragile X mental retardation protein inhibits translation via interacting with mRNA. Nucleic Acids Res. 2001, 29: 2276-2283. 10.1093/nar/29.11.2276.
Huber KM, Gallagher SM, Warren ST, Bear MF: Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc Natl Acad Sci USA. 2002, 99: 7746-7750. 10.1073/pnas.122205699.
Qin M, Kang J, Burlin TV, Jiang C, Smith CB: Postadolescent changes in regional cerebral protein synthesis: an in vivo study in the FMR1 null mouse. J Neurosci. 2005, 25: 5087-5095. 10.1523/JNEUROSCI.0093-05.2005.
Darnell JC, Van Driesche SJ, Zhang C, Hung KY, Mele A, Fraser CE, Stone EF, Chen C, Fak JJ, Chi SW: FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell. 2011, 146: 247-261. 10.1016/j.cell.2011.06.013.
Consortium TD-BFX: Fmr1 knockout mice: a model to study fragile X mental retardation. Cell. 1994, 78: 23-33.
Osterweil EK, Krueger DD, Reinhold K, Bear MF: Hypersensitivity to mGluR5 and ERK1/2 leads to excessive protein synthesis in the hippocampus of a mouse model of fragile X syndrome. J Neurosci. 2010, 30: 15616-15627. 10.1523/JNEUROSCI.3888-10.2010.
Bear MF, Huber KM, Warren ST: The mGluR theory of fragile X mental retardation. Trends Neurosci. 2004, 27: 370-377. 10.1016/j.tins.2004.04.009.
Bhattacharya A, Kaphzan H, Alvarez-Dieppa AC, Murphy JP, Pierre P, Klann E: Genetic removal of p70 S6 kinase 1 corrects molecular, synaptic, and behavioral phenotypes in fragile X syndrome mice. Neuron. 2012, 76: 325-337. 10.1016/j.neuron.2012.07.022.
Bhakar AL, Dolen G, Bear MF: The pathophysiology of fragile X (and what it teaches us about synapses). Annu Rev Neurosci. 2012, 35: 417-443. 10.1146/annurev-neuro-060909-153138.
Garber K, Smith KT, Reines D, Warren ST: Transcription, translation and fragile X syndrome. Curr Opin Genet Dev. 2006, 16: 270-275. 10.1016/j.gde.2006.04.010.
Bassell GJ, Warren ST: Fragile X syndrome: loss of local mRNA regulation alters synaptic development and function. Neuron. 2008, 60: 201-214. 10.1016/j.neuron.2008.10.004.
Pfeiffer BE, Huber KM: The state of synapses in fragile X syndrome. Neuroscientist. 2009, 15: 549-567. 10.1177/1073858409333075.
Krueger DD, Bear MF: Toward fulfilling the promise of molecular medicine in fragile X syndrome. Annu Rev Med. 2011, 62: 411-429. 10.1146/annurev-med-061109-134644.
Ashley CT, Wilkinson KD, Reines D, Warren ST: FMR1 protein: conserved RNP family domains and selective RNA binding. Science. 1993, 262: 563-566. 10.1126/science.7692601.
Siomi H, Siomi MC, Nussbaum RL, Dreyfuss G: The protein product of the fragile X gene, FMR1, has characteristics of an RNA-binding protein. Cell. 1993, 74: 291-298. 10.1016/0092-8674(93)90420-U.
Napoli I, Mercaldo V, Boyl PP, Eleuteri B, Zalfa F, De Rubeis S, Di Marino D, Mohr E, Massimi M, Falconi M: The fragile X syndrome protein represses activity-dependent translation through CYFIP1, a new 4E-BP. Cell. 2008, 134: 1042-1054. 10.1016/j.cell.2008.07.031.
El Fatimy R, Tremblay S, Dury AY, Solomon S, De Koninck P, Schrader JW, Khandjian EW: Fragile X mental retardation protein interacts with the RNA-binding protein Caprin1 in neuronal RiboNucleoProtein complexes [corrected]. PLoS One. 2012, 7: e39338-10.1371/journal.pone.0039338.
Santoro MR, Bray SM, Warren ST: Molecular mechanisms of fragile X syndrome: a twenty-year perspective. Annu Rev Pathol. 2012, 7: 219-245. 10.1146/annurev-pathol-011811-132457.
Bagni C, Greenough WT: From mRNP trafficking to spine dysmorphogenesis: the roots of fragile X syndrome. Nat Rev Neurosci. 2005, 6: 376-387.
Zang JB, Nosyreva ED, Spencer CM, Volk LJ, Musunuru K, Zhong R, Stone EF, Yuva-Paylor LA, Huber KM, Paylor R: A mouse model of the human Fragile X syndrome I304N mutation. PLoS Genet. 2009, 5: e1000758-10.1371/journal.pgen.1000758.
De Boulle K, Verkerk AJ, Reyniers E, Vits L, Hendrickx J, Van Roy B, Van den Bos F, de Graaff E, Oostra BA, Willems PJ: A point mutation in the FMR-1 gene associated with fragile X mental retardation. Nat Genet. 1993, 3: 31-35. 10.1038/ng0193-31.
Coffee RL, Williamson AJ, Adkins CM, Gray MC, Page TL, Broadie K: In vivo neuronal function of the fragile X mental retardation protein is regulated by phosphorylation. Hum Mol Genet. 2012, 21: 900-915. 10.1093/hmg/ddr527.
Muddashetty RS, Nalavadi VC, Gross C, Yao X, Xing L, Laur O, Warren ST, Bassell GJ: Reversible inhibition of PSD-95 mRNA translation by miR-125a, FMRP phosphorylation, and mGluR signaling. Mol Cell. 2011, 42: 673-688. 10.1016/j.molcel.2011.05.006.
Ceman S, O'Donnell WT, Reed M, Patton S, Pohl J, Warren ST: Phosphorylation influences the translation state of FMRP-associated polyribosomes. Hum Mol Genet. 2003, 12: 3295-3305. 10.1093/hmg/ddg350.
Weiler IJ, Irwin SA, Klintsova AY, Spencer CM, Brazelton AD, Miyashiro K, Comery TA, Patel B, Eberwine J, Greenough WT: Fragile X mental retardation protein is translated near synapses in response to neurotransmitter activation. Proc Natl Acad Sci USA. 1997, 94: 5395-5400. 10.1073/pnas.94.10.5395.
Feng Y, Absher D, Eberhart DE, Brown V, Malter HE, Warren ST: FMRP associates with polyribosomes as an mRNP, and the I304N mutation of severe fragile X syndrome abolishes this association. Mol Cell. 1997, 1: 109-118. 10.1016/S1097-2765(00)80012-X.
Antar LN, Afroz R, Dictenberg JB, Carroll RC, Bassell GJ: Metabotropic glutamate receptor activation regulates fragile x mental retardation protein and FMR1 mRNA localization differentially in dendrites and at synapses. J Neurosci. 2004, 24: 2648-2655. 10.1523/JNEUROSCI.0099-04.2004.
Ferrari F, Mercaldo V, Piccoli G, Sala C, Cannata S, Achsel T, Bagni C: The fragile X mental retardation protein-RNP granules show an mGluR-dependent localization in the post-synaptic spines. Mol Cell Neurosci. 2007, 34: 343-354. 10.1016/j.mcn.2006.11.015.
Sutton MA, Schuman EM: Dendritic protein synthesis, synaptic plasticity, and memory. Cell. 2006, 127: 49-58. 10.1016/j.cell.2006.09.014.
Irwin SA, Swain RA, Christmon CA, Chakravarti A, Weiler IJ, Greenough WT: Evidence for altered Fragile-X mental retardation protein expression in response to behavioral stimulation. Neurobiol Learn Mem. 2000, 74: 87-93.
Gabel LA, Won S, Kawai H, McKinney M, Tartakoff AM, Fallon JR: Visual experience regulates transient expression and dendritic localization of fragile X mental retardation protein. J Neurosci. 2004, 24: 10579-10583. 10.1523/JNEUROSCI.2185-04.2004.
Todd PK, Malter JS, Mack KJ: Whisker stimulation-dependent translation of FMRP in the barrel cortex requires activation of type I metabotropic glutamate receptors. Brain Res Mol Brain Res. 2003, 110: 267-278. 10.1016/S0169-328X(02)00657-5.
Antar LN, Dictenberg JB, Plociniak M, Afroz R, Bassell GJ: Localization of FMRP-associated mRNA granules and requirement of microtubules for activity-dependent trafficking in hippocampal neurons. Genes Brain Behav. 2005, 4: 350-359. 10.1111/j.1601-183X.2005.00128.x.
Dictenberg JB, Swanger SA, Antar LN, Singer RH, Bassell GJ: A direct role for FMRP in activity-dependent dendritic mRNA transport links filopodial-spine morphogenesis to fragile X syndrome. Dev Cell. 2008, 14: 926-939. 10.1016/j.devcel.2008.04.003.
Palmer MJ, Irving AJ, Seabrook GR, Jane DE, Collingridge GL: The group I mGlu receptor agonist DHPG induces a novel form of LTD in the CA1 region of the hippocampus. Neuropharmacology. 1997, 36: 1517-1532. 10.1016/S0028-3908(97)00181-0.
Huber KM, Bear MF: Activation of group 1 metabotropic glutamate receptors induces long-term depression of synaptic transmission in area CA1 of rat hippocampus. Soc Neurosci Abstr. 1998, 22:
Huber KM, Roder JC, Bear MF: Chemical induction of mGluR5- and protein synthesis-dependent long-term depression in hippocampal area CA1. J Neurophysiol. 2001, 86: 321-325.
Oliet SH, Malenka RC, Nicoll RA: Two distinct forms of long-term depression coexist in CA1 hippocampal pyramidal cells. Neuron. 1997, 18: 969-982. 10.1016/S0896-6273(00)80336-0.
Zakharenko SS, Zablow L, Siegelbaum SA: Altered presynaptic vesicle release and cycling during mGluR-dependent LTD. Neuron. 2002, 35: 1099-1110. 10.1016/S0896-6273(02)00898-X.
Nosyreva ED, Huber KM: Developmental switch in synaptic mechanisms of hippocampal metabotropic glutamate receptor-dependent long-term depression. J Neurosci. 2005, 25: 2992-3001. 10.1523/JNEUROSCI.3652-04.2005.
Moult PR, Correa SA, Collingridge GL, Fitzjohn SM, Bashir ZI: Co-activation of p38 mitogen-activated protein kinase and protein tyrosine phosphatase underlies metabotropic glutamate receptor-dependent long-term depression. J Physiol. 2008, 586: 2499-2510. 10.1113/jphysiol.2008.153122.
Auerbach BD, Osterweil EK, Bear MF: Mutations causing syndromic autism define an axis of synaptic pathophysiology. Nature. 2011, 480: 63-68. 10.1038/nature10658.
Hou L, Antion MD, Hu D, Spencer CM, Paylor R, Klann E: Dynamic translational and proteasomal regulation of fragile X mental retardation protein controls mGluR-dependent long-term depression. Neuron. 2006, 51: 441-454. 10.1016/j.neuron.2006.07.005.
Snyder EM, Philpot BD, Huber KM, Dong X, Fallon JR, Bear MF: Internalization of ionotropic glutamate receptors in response to mGluR activation. Nat Neurosci. 2001, 4: 1079-1085. 10.1038/nn746.
Job C, Eberwine J: Identification of sites for exponential translation in living dendrites. Proc Natl Acad Sci USA. 2001, 98: 13037-13042. 10.1073/pnas.231485698.
Weiler IJ, Greenough WT: Metabotropic glutamate receptors trigger postsynaptic protein synthesis. Proc Natl Acad Sci USA. 1993, 90: 7168-7171. 10.1073/pnas.90.15.7168.
Michalon A, Sidorov M, Ballard TM, Ozmen L, Spooren W, Wettstein JG, Jaeschke G, Bear MF, Lindemann L: Chronic Pharmacological mGlu5 Inhibition Corrects Fragile X in Adult Mice. Neuron. 2012, 74: 49-56. 10.1016/j.neuron.2012.03.009.
Koekkoek SK, Yamaguchi K, Milojkovic BA, Dortland BR, Ruigrok TJ, Maex R, De Graaf W, Smit AE, VanderWerf F, Bakker CE: Deletion of FMR1 in Purkinje cells enhances parallel fiber LTD, enlarges spines, and attenuates cerebellar eyelid conditioning in Fragile X syndrome. Neuron. 2005, 47: 339-352. 10.1016/j.neuron.2005.07.005.
Nakamoto M, Nalavadi V, Epstein MP, Narayanan U, Bassell GJ, Warren ST: Fragile X mental retardation protein deficiency leads to excessive mGluR5-dependent internalization of AMPA receptors. Proc Natl Acad Sci USA. 2007, 104: 15537-15542. 10.1073/pnas.0707484104.
Nosyreva ED, Huber KM: Metabotropic receptor-dependent long-term depression persists in the absence of protein synthesis in the mouse model of fragile X syndrome. J Neurophysiol. 2006, 95: 3291-3295. 10.1152/jn.01316.2005.
Zeier Z, Kumar A, Bodhinathan K, Feller JA, Foster TC, Bloom DC: Fragile X mental retardation protein replacement restores hippocampal synaptic function in a mouse model of fragile X syndrome. Gene Ther. 2009, 16: 1122-1129. 10.1038/gt.2009.83.
Dolen G, Osterweil E, Rao BS, Smith GB, Auerbach BD, Chattarji S, Bear MF: Correction of fragile X syndrome in mice. Neuron. 2007, 56: 955-962. 10.1016/j.neuron.2007.12.001.
Stanton PK, Sarvey JM: Blockade of long-term potentiation in rat hippocampal CA1 region by inhibitors of protein synthesis. J Neurosci. 1984, 4: 3080-3088.
Frey U, Krug M, Reymann KG, Matthies H: Anisomycin, an inhibitor of protein synthesis, blocks late phases of LTP phenomena in the hippocampal CA1 region in vitro. Brain Res. 1988, 452: 57-65. 10.1016/0006-8993(88)90008-X.
Paradee W, Melikian HE, Rasmussen DL, Kenneson A, Conn PJ, Warren ST: Fragile X mouse: strain effects of knockout phenotype and evidence suggesting deficient amygdala function. Neuroscience. 1999, 94: 185-192. 10.1016/S0306-4522(99)00285-7.
Zhang J, Hou L, Klann E, Nelson DL: Altered hippocampal synaptic plasticity in the FMR1 gene family knockout mouse models. J Neurophysiol. 2009, 101: 2572-2580.
Abraham WC, Williams JM: Properties and mechanisms of LTP maintenance. Neuroscientist. 2003, 9: 463-474. 10.1177/1073858403259119.
Krug M, Lossner B, Ott T: Anisomycin blocks the late phase of long-term potentiation in the dentate gyrus of freely moving rats. Brain Res Bull. 1984, 13: 39-42. 10.1016/0361-9230(84)90005-4.
Nguyen PV, Abel T, Kandel ER: Requirement of a critical period of transcription for induction of a late phase of LTP. Science. 1994, 265: 1104-1107. 10.1126/science.8066450.
Kelleher RJ, Govindarajan A, Jung HY, Kang H, Tonegawa S: Translational control by MAPK signaling in long-term synaptic plasticity and memory. Cell. 2004, 116: 467-479. 10.1016/S0092-8674(04)00115-1.
Cohen AS, Abraham WC: Facilitation of long-term potentiation by prior activation of metabotropic glutamate receptors. J Neurophysiol. 1996, 76: 953-962.
Raymond CR, Thompson VL, Tate WP, Abraham WC: Metabotropic glutamate receptors trigger homosynaptic protein synthesis to prolong long-term potentiation. J Neurosci. 2000, 20: 969-976.
Auerbach BD, Bear MF: Loss of the fragile X mental retardation protein decouples metabotropic glutamate receptor dependent priming of long-term potentiation from protein synthesis. J Neurophysiol. 2010, 104: 1047-1051. 10.1152/jn.00449.2010.
Lauterborn JC, Rex CS, Kramar E, Chen LY, Pandyarajan V, Lynch G, Gall CM: Brain-derived neurotrophic factor rescues synaptic plasticity in a mouse model of fragile X syndrome. J Neurosci. 2007, 27: 10685-10694. 10.1523/JNEUROSCI.2624-07.2007.
Markram H, Lubke J, Frotscher M, Sakmann B: Regulation of synaptic efficacy by coincidence of postsynaptic APs and EPSPs. Science. 1997, 275: 213-215. 10.1126/science.275.5297.213.
Bi GQ, Poo MM: Synaptic modifications in cultured hippocampal neurons: dependence on spike timing, synaptic strength, and postsynaptic cell type. J Neurosci. 1998, 18: 10464-10472.
Desai NS, Casimiro TM, Gruber SM, Vanderklish PW: Early postnatal plasticity in neocortex of Fmr1 knockout mice. J Neurophysiol. 2006, 96: 1734-1745. 10.1152/jn.00221.2006.
Meredith RM, Holmgren CD, Weidum M, Burnashev N, Mansvelder HD: Increased threshold for spike-timing-dependent plasticity is caused by unreliable calcium signaling in mice lacking fragile X gene FMR1. Neuron. 2007, 54: 627-638. 10.1016/j.neuron.2007.04.028.
Turrigiano GG: The self-tuning neuron: synaptic scaling of excitatory synapses. Cell. 2008, 135: 422-435. 10.1016/j.cell.2008.10.008.
Ibata K, Sun Q, Turrigiano GG: Rapid synaptic scaling induced by changes in postsynaptic firing. Neuron. 2008, 57: 819-826. 10.1016/j.neuron.2008.02.031.
Sutton MA, Ito HT, Cressy P, Kempf C, Woo JC, Schuman EM: Miniature neurotransmission stabilizes synaptic function via tonic suppression of local dendritic protein synthesis. Cell. 2006, 125: 785-799. 10.1016/j.cell.2006.03.040.
Soden ME, Chen L: Fragile X protein FMRP is required for homeostatic plasticity and regulation of synaptic strength by retinoic acid. J Neurosci. 2010, 30: 16910-16921. 10.1523/JNEUROSCI.3660-10.2010.
Turrigiano GG, Leslie KR, Desai NS, Rutherford LC, Nelson SB: Activity-dependent scaling of quantal amplitude in neocortical neurons. Nature. 1998, 391: 892-896. 10.1038/36103.
Hu JH, Park JM, Park S, Xiao B, Dehoff MH, Kim S, Hayashi T, Schwarz MK, Huganir RL, Seeburg PH: Homeostatic scaling requires group I mGluR activation mediated by Homer1a. Neuron. 2010, 68: 1128-1142. 10.1016/j.neuron.2010.11.008.
Zhong X, Li H, Chang Q: MeCP2 phosphorylation is required for modulating synaptic scaling through mGluR5. J Neurosci. 2012, 32: 12841-12847. 10.1523/JNEUROSCI.2784-12.2012.
Volk LJ, Pfeiffer BE, Gibson JR, Huber KM: Multiple Gq-coupled receptors converge on a common protein synthesis-dependent long-term depression that is affected in fragile X syndrome mental retardation. J Neurosci. 2007, 27: 11624-11634. 10.1523/JNEUROSCI.2266-07.2007.
Connor SA, Hoeffer CA, Klann E, Nguyen PV: Fragile X mental retardation protein regulates heterosynaptic plasticity in the hippocampus. Learn Mem. 2011, 18: 207-220. 10.1101/lm.2043811.
Godfraind JM, Reyniers E, De Boulle K, D'Hooge R, De Deyn PP, Bakker CE, Oostra BA, Kooy RF, Willems PJ: Long-term potentiation in the hippocampus of fragile X knockout mice. Am J Med Genet. 1996, 64: 246-251. 10.1002/(SICI)1096-8628(19960809)64:2<246::AID-AJMG2>3.0.CO;2-S.
Li J, Pelletier MR, Perez Velazquez JL, Carlen PL: Reduced cortical synaptic plasticity and GluR1 expression associated with fragile X mental retardation protein deficiency. Mol Cell Neurosci. 2002, 19: 138-151. 10.1006/mcne.2001.1085.
Hayashi ML, Rao BS, Seo JS, Choi HS, Dolan BM, Choi SY, Chattarji S, Tonegawa S: Inhibition of p21-activated kinase rescues symptoms of fragile X syndrome in mice. Proc Natl Acad Sci USA. 2007, 104: 11489-11494. 10.1073/pnas.0705003104.
Zhao MG, Toyoda H, Ko SW, Ding HK, Wu LJ, Zhuo M: Deficits in trace fear memory and long-term potentiation in a mouse model for fragile X syndrome. J Neurosci. 2005, 25: 7385-7392. 10.1523/JNEUROSCI.1520-05.2005.
Suvrathan A, Hoeffer CA, Wong H, Klann E, Chattarji S: Characterization and reversal of synaptic defects in the amygdala in a mouse model of fragile X syndrome. Proc Natl Acad Sci USA. 2010, 107: 11591-11596. 10.1073/pnas.1002262107.
Wilson BM, Cox CL: Absence of metabotropic glutamate receptor-mediated plasticity in the neocortex of fragile X mice. Proc Natl Acad Sci USA. 2007, 104: 2454-2459. 10.1073/pnas.0610875104.
Brown V, Jin P, Ceman S, Darnell JC, O'Donnell WT, Tenenbaum SA, Jin X, Feng Y, Wilkinson KD, Keene JD: Microarray identification of FMRP-associated brain mRNAs and altered mRNA translational profiles in fragile X syndrome. Cell. 2001, 107: 477-487. 10.1016/S0092-8674(01)00568-2.
Jung H, Yoon BC, Holt CE: Axonal mRNA localization and local protein synthesis in nervous system assembly, maintenance and repair. Nat Rev Neurosci. 2012, 13: 308-324.
Akins MR, Berk-Rauch HE, Fallon JR: Presynaptic translation: stepping out of the postsynaptic shadow. Front Neural Circuits. 2009, 3: 17-
Price TJ, Melemedjian OK: Fragile X mental retardation protein (FMRP) and the spinal sensory system. Results Probl Cell Differ. 2012, 54: 41-59. 10.1007/978-3-642-21649-7_4.
Price TJ, Rashid MH, Millecamps M, Sanoja R, Entrena JM, Cervero F: Decreased nociceptive sensitization in mice lacking the fragile X mental retardation protein: role of mGluR1/5 and mTOR. J Neurosci. 2007, 27: 13958-13967. 10.1523/JNEUROSCI.4383-07.2007.
Davidkova G, Carroll RC: Characterization of the role of microtubule-associated protein 1B in metabotropic glutamate receptor-mediated endocytosis of AMPA receptors in hippocampus. J Neurosci. 2007, 27: 13273-13278. 10.1523/JNEUROSCI.3334-07.2007.
Lu R, Wang H, Liang Z, Ku L, O'Donnell WT, Li W, Warren ST, Feng Y: The fragile X protein controls microtubule-associated protein 1B translation and microtubule stability in brain neuron development. Proc Natl Acad Sci USA. 2004, 101: 15201-15206. 10.1073/pnas.0404995101.
Darnell JC, Jensen KB, Jin P, Brown V, Warren ST, Darnell RB: Fragile X mental retardation protein targets G quartet mRNAs important for neuronal function. Cell. 2001, 107: 489-499. 10.1016/S0092-8674(01)00566-9.
Zalfa F, Giorgi M, Primerano B, Moro A, Di Penta A, Reis S, Oostra B, Bagni C: The fragile X syndrome protein FMRP associates with BC1 RNA and regulates the translation of specific mRNAs at synapses. Cell. 2003, 112: 317-327. 10.1016/S0092-8674(03)00079-5.
Wei ZX, Yi YH, Sun WW, Wang R, Su T, Bai YJ, Liao WP: Expression changes of microtubule associated protein 1B in the brain of Fmr1 knockout mice. Neurosci Bull. 2007, 23: 203-208. 10.1007/s12264-007-0030-1.
Chowdhury S, Shepherd JD, Okuno H, Lyford G, Petralia RS, Plath N, Kuhl D, Huganir RL, Worley PF: Arc/Arg3.1 interacts with the endocytic machinery to regulate AMPA receptor trafficking. Neuron. 2006, 52: 445-459. 10.1016/j.neuron.2006.08.033.
Waung MW, Pfeiffer BE, Nosyreva ED, Ronesi JA, Huber KM: Rapid translation of Arc/Arg3.1 selectively mediates mGluR-dependent LTD through persistent increases in AMPAR endocytosis rate. Neuron. 2008, 59: 84-97. 10.1016/j.neuron.2008.05.014.
Park S, Park JM, Kim S, Kim JA, Shepherd JD, Smith-Hicks CL, Chowdhury S, Kaufmann W, Kuhl D, Ryazanov AG: Elongation factor 2 and fragile X mental retardation protein control the dynamic translation of Arc/Arg3.1 essential for mGluR-LTD. Neuron. 2008, 59: 70-83. 10.1016/j.neuron.2008.05.023.
Shepherd JD, Bear MF: New views of Arc, a master regulator of synaptic plasticity. Nat Neurosci. 2011, 14: 279-284. 10.1038/nn.2708.
Plath N, Ohana O, Dammermann B, Errington ML, Schmitz D, Gross C, Mao X, Engelsberg A, Mahlke C, Welzl H: Arc/Arg3.1 is essential for the consolidation of synaptic plasticity and memories. Neuron. 2006, 52: 437-444. 10.1016/j.neuron.2006.08.024.
Niere F, Wilkerson JR, Huber KM: Evidence for a fragile X mental retardation protein-mediated translational switch in metabotropic glutamate receptor-triggered Arc translation and long-term depression. J Neurosci. 2012, 32: 5924-5936. 10.1523/JNEUROSCI.4650-11.2012.
Iacoangeli A, Rozhdestvensky TS, Dolzhanskaya N, Tournier B, Schutt J, Brosius J, Denman RB, Khandjian EW, Kindler S, Tiedge H: On BC1 RNA and the fragile X mental retardation protein. Proc Natl Acad Sci USA. 2008, 105: 734-739. 10.1073/pnas.0710991105.
Zhang Y, Venkitaramani DV, Gladding CM, Kurup P, Molnar E, Collingridge GL, Lombroso PJ: The tyrosine phosphatase STEP mediates AMPA receptor endocytosis after metabotropic glutamate receptor stimulation. J Neurosci. 2008, 28: 10561-10566. 10.1523/JNEUROSCI.2666-08.2008.
Goebel-Goody SM, Baum M, Paspalas CD, Fernandez SM, Carty NC, Kurup P, Lombroso PJ: Therapeutic implications for striatal-enriched protein tyrosine phosphatase (STEP) in neuropsychiatric disorders. Pharmacol Rev. 2012, 64: 65-87. 10.1124/pr.110.003053.
Goebel-Goody SM, Wilson-Wallis ED, Royston S, Tagliatela SM, Naegele JR, Lombroso PJ: Genetic manipulation of STEP reverses behavioral abnormalities in a fragile X syndrome mouse model. Genes Brain Behav. 2012, 11: 586-600. 10.1111/j.1601-183X.2012.00781.x.
Westmark CJ, Malter JS: FMRP mediates mGluR5-dependent translation of amyloid precursor protein. PLoS Biol. 2007, 5: e52-10.1371/journal.pbio.0050052.
Westmark CJ, Westmark PR, O'Riordan KJ, Ray BC, Hervey CM, Salamat MS, Abozeid SH, Stein KM, Stodola LA, Tranfaglia M: Reversal of fragile X phenotypes by manipulation of AbetaPP/Abeta levels in Fmr1KO mice. PLoS One. 2011, 6: e26549-10.1371/journal.pone.0026549.
Nadif Kasri N, Nakano-Kobayashi A, Van Aelst L: Rapid synthesis of the X-linked mental retardation protein OPHN1 mediates mGluR-dependent LTD through interaction with the endocytic machinery. Neuron. 2011, 72: 300-315. 10.1016/j.neuron.2011.09.001.
Muddashetty RS, Kelic S, Gross C, Xu M, Bassell GJ: Dysregulated metabotropic glutamate receptor-dependent translation of AMPA receptor and postsynaptic density-95 mRNAs at synapses in a mouse model of fragile X syndrome. J Neurosci. 2007, 27: 5338-5348. 10.1523/JNEUROSCI.0937-07.2007.
Todd PK, Mack KJ, Malter JS: The fragile X mental retardation protein is required for type-I metabotropic glutamate receptor-dependent translation of PSD-95. Proc Natl Acad Sci USA. 2003, 100: 14374-14378. 10.1073/pnas.2336265100.
Zalfa F, Eleuteri B, Dickson KS, Mercaldo V, De Rubeis S, di Penta A, Tabolacci E, Chiurazzi P, Neri G, Grant SG, Bagni C: A new function for the fragile X mental retardation protein in regulation of PSD-95 mRNA stability. Nat Neurosci. 2007, 10: 578-587. 10.1038/nn1893.
Gross C, Nakamoto M, Yao X, Chan CB, Yim SY, Ye K, Warren ST, Bassell GJ: Excess phosphoinositide 3-kinase subunit synthesis and activity as a novel therapeutic target in fragile X syndrome. J Neurosci. 2010, 30: 10624-10638. 10.1523/JNEUROSCI.0402-10.2010.
Lee HY, Jan LY: Fragile X syndrome: mechanistic insights and therapeutic avenues regarding the role of potassium channels. Curr Opin Neurobiol. 2012, 22: 1-8. 10.1016/j.conb.2012.01.004.
Lee HY, Ge WP, Huang W, He Y, Wang GX, Rowson-Baldwin A, Smith SJ, Jan YN, Jan LY: Bidirectional regulation of dendritic voltage-gated potassium channels by the fragile X mental retardation protein. Neuron. 2011, 72: 630-642. 10.1016/j.neuron.2011.09.033.
Gross C, Yao X, Pong DL, Jeromin A, Bassell GJ: Fragile X mental retardation protein regulates protein expression and mRNA translation of the potassium channel Kv4.2. J Neurosci. 2011, 31: 5693-5698. 10.1523/JNEUROSCI.6661-10.2011.
Strumbos JG, Brown MR, Kronengold J, Polley DB, Kaczmarek LK: Fragile X mental retardation protein is required for rapid experience-dependent regulation of the potassium channel Kv3.1b. J Neurosci. 2010, 30: 10263-10271. 10.1523/JNEUROSCI.1125-10.2010.
Brown MR, Kronengold J, Gazula VR, Chen Y, Strumbos JG, Sigworth FJ, Navaratnam D, Kaczmarek LK: Fragile X mental retardation protein controls gating of the sodium-activated potassium channel Slack. Nat Neurosci. 2010, 13: 819-821. 10.1038/nn.2563.
Zhang Y, Brown MR, Hyland C, Chen Y, Kronengold J, Fleming MR, Kohn AB, Moroz LL, Kaczmarek LK: Regulation of neuronal excitability by interaction of fragile x mental retardation protein with slack potassium channels. J Neurosci. 2012, 32: 15318-15327. 10.1523/JNEUROSCI.2162-12.2012.
Chen X, Yuan LL, Zhao C, Birnbaum SG, Frick A, Jung WE, Schwarz TL, Sweatt JD, Johnston D: Deletion of Kv4.2 gene eliminates dendritic A-type K+ current and enhances induction of long-term potentiation in hippocampal CA1 pyramidal neurons. J Neurosci. 2006, 26: 12143-12151. 10.1523/JNEUROSCI.2667-06.2006.
Hoffman DA, Magee JC, Colbert CM, Johnston D: K+ channel regulation of signal propagation in dendrites of hippocampal pyramidal neurons. Nature. 1997, 387: 869-875. 10.1038/43119.
Watanabe S, Hoffman DA, Migliore M, Johnston D: Dendritic K+ channels contribute to spike-timing dependent long-term potentiation in hippocampal pyramidal neurons. Proc Natl Acad Sci USA. 2002, 99: 8366-8371. 10.1073/pnas.122210599.
Jung SC, Kim J, Hoffman DA: Rapid, bidirectional remodeling of synaptic NMDA receptor subunit composition by A-type K+ channel activity in hippocampal CA1 pyramidal neurons. Neuron. 2008, 60: 657-671. 10.1016/j.neuron.2008.08.029.
Chuang SC, Zhao W, Bauchwitz R, Yan Q, Bianchi R, Wong RK: Prolonged epileptiform discharges induced by altered group I metabotropic glutamate receptor-mediated synaptic responses in hippocampal slices of a fragile X mouse model. J Neurosci. 2005, 25: 8048-8055. 10.1523/JNEUROSCI.1777-05.2005.
Krueger DD, Osterweil EK, Chen SP, Tye LD, Bear MF: Cognitive dysfunction and prefrontal synaptic abnormalities in a mouse model of fragile X syndrome. Proc Natl Acad Sci USA. 2011, 108: 2587-2592. 10.1073/pnas.1013855108.
Bolduc FV, Bell K, Cox H, Broadie KS, Tully T: Excess protein synthesis in Drosophila fragile X mutants impairs long-term memory. Nat Neurosci. 2008, 11: 1143-1145. 10.1038/nn.2175.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
All authors drafted, read, and approved the manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Sidorov, M.S., Auerbach, B.D. & Bear, M.F. Fragile X mental retardation protein and synaptic plasticity. Mol Brain 6, 15 (2013). https://doi.org/10.1186/1756-6606-6-15
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1756-6606-6-15