
Abstract
Alzheimer’s disease (AD) is characterized by episodic memory impairment that often precedes clinical diagnosis by many years. Probing the mechanisms of such impairment may provide much needed means of diagnosis and therapeutic intervention at an early, pre-dementia, stage. Prior to the onset of significant neurodegeneration, the structural and functional integrity of synapses in mnemonic circuitry is severely compromised in the presence of amyloidosis. This review examines recent evidence evaluating the role of amyloid-ß protein (Aβ) in causing rapid disruption of synaptic plasticity and memory impairment. We evaluate the relative importance of different sizes and conformations of Aβ, including monomer, oligomer, protofibril and fibril. We pay particular attention to recent controversies over the relevance to the pathophysiology of AD of different water soluble Aβ aggregates and the importance of cellular prion protein in mediating their effects. Current data are consistent with the view that both low-n oligomers and larger soluble assemblies present in AD brain, some of them via a direct interaction with cellular prion protein, cause synaptic memory failure. At the two extremes of aggregation, monomers and fibrils appear to act in vivo both as sources and sinks of certain metastable conformations of soluble aggregates that powerfully disrupt synaptic plasticity. The same principle appears to apply to other synaptotoxic amyloidogenic proteins including tau, α-synuclein and prion protein.
Similar content being viewed by others

Introduction
Many different amyloidogenic proteins form water insoluble deposits in the brains of patients who die from neurodegenerative diseases [1–3]. The common observation of extensive synaptic loss and mixed neuropathology in many of these diseases suggests that different amyloidogenic proteins may share similar synaptic actions and effects [4–8]. The most frequent cause of neurodegenerative dementia, Alzheimer’s disease (AD), is characterized by profound episodic memory loss which usually presages cognitive decline. The discovery that the hallmark extracellular senile plaques found in the patients’ brains are largely composed of water insoluble fibrillar amyloid ß-protein (Aβ) laid the foundation of the amyloid cascade hypotheses of disease aetiology and led to the investigation of the deleterious effects of Aβ on memory and related neurophysiological processing [9–11].
In the light of the many recent reviews of the cellular mechanisms [12–16], the present review focuses on defining the roles of different Aβ assemblies [17, 18] in Aβ-mediated synaptic and memory disruption. Since cognitive status in patients with AD is much more strongly correlated with brain concentration of water soluble Aβ rather than insoluble fibrillar Aβ-containing plaque load [19, 20], most recent research has focused on soluble species of Aβ.
In order to investigate the effects of different Aβ species on memory and related synaptic mechanisms, acute treatment with Aβ provides a relatively simple but very attractive and manipulable model system, compared to transgenic amyloid precursor protein (APP) animal models [21]. The acute application approach gives the opportunity to control and characterize the biophysical state of aggregation-prone Aβ preparations prior to use. Since pioneering in vivo studies found that injection of synthetic Aβ-related peptides of undefined assembly can impair learning [22, 23] and reduce synaptic transmission in the hippocampus of the rat brain [24], this approach has been exploited in order to examine the role of different Aβ assemblies. By comparing the relative activity of different soluble preparations of Aβ in these acute models it is hoped that it will be possible to determine the nature and actions of synaptic and memory disrupting assemblies. These assemblies vary in primary sequence, size and putative generic conformation. They include monomers, low-n oligomers, larger oligomers such as Aβ derived diffusible ligands (ADDLs) [25, 26] and globulomers [27], and protofibrils which are usually shorter and thinner than insoluble amyloid fibrils [28] (Figure 1). Currently there is little agreement as to which, if any, of these assemblies is most culpable in causing synaptic plasticity and memory disruption. The present review examines recent evidence, including the actions of other amyloidogenic peptides and the possible involvement of cellular prion protein (PrPC) as a selective target of certain oligomers.

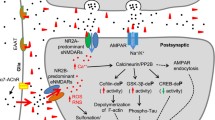
Schematic representation of Aβ processing and aggregation. (A) Primary sequence of human Aß1-42 with examples of natural or designed intra-Aß mutations (above sequence) and post-translational modifications (below sequence). (B) Amyloid precursor protein (APP) cleavage by β- and γ- secretases releases aggregation-prone Aβ peptides, particularly Aβ1-42. Intra-Aβ mutations and post-translational modifications increase Aβ ability to aggregate even more. It has been suggested that diffusible Aβ aggregates rather than monomer form or fibrils are the synaptotoxic species. These aggregates include ADDLS (Aß-derived diffusible ligands), globulomers (globule-like 12-mers), Aß*56 (56 kDa Aß-containing aggregates derived from brain), protofibrils (soluble, short fibril-shaped often “worm-like” structures) and annular protofibrils (protofibrils that can form pores in membranes). Other aggregation-prone proteins also form synaptotoxic soluble species that may share conformation recognized by antibodies.
Acute synaptic and behavioural effects of Aβ
Two of the most sensitive and robust measures of the acute synaptic disruptive effects of Aβ are inhibition of long-term potentiation (LTP) [29] and facilitation of long-term depression (LTD) of excitatory synaptic transmission [30], both of which engage plasticity mechanisms believed to underlie certain types of learning and memory [31–33]. Baseline synaptic efficacy appears more resistant to the effects of Aβ in most acute studies. Some of the most sensitive behavioural indicants of rapid impairment of cognition and memory include performance of operant tasks [34] and aversive learning [35].
Aβ amino acid sequence and post-translational modification
The cleavage of APP by the γ-secretase complex is permissive, with Aβ1-40 the dominant Aß species (Figure 1) [17]. In AD brain the concentrations of highly amyloidogenic species, especially the more potent synaptic plasticity-disrupting Aβ1-42 [29, 36], increase. Since the discovery of rare early-onset autosomal dominant familial forms of AD caused by missense mutations of the APP gene within the Aβ region, synthetic peptides bearing familial and design mutations have been used to investigate the potential importance of primary sequence in determining Aβ aggregation, toxicity and synaptic disruption [37]. Some years ago we found that Arctic synthetic mutant Aβ1-40(E22G) peptide, which has a much greater tendency than Aβ1-40 to form soluble aggregates including protofibrils, is accompanied by a greater potency to block LTP [38]. More recently Tomiyama et al. [39] reported that familial AD-associated Aβ that lacks glutamate-22 showed enhanced oligomerization in the apparent absence of fibril formation, and was a more potent inhibitor of LTP.
Beyond the primary sequence, biochemical modifications of Aβ, including post-translational processing, can lead to the generation of highly aggregation prone species in the brain [40, 41]. Aminopeptidase removal of residues 1 and 2 of Aß followed by glutaminyl cylase-mediated cyclization of the exposed glutamate to a pyroglutamate, leads to the production of N-terminally truncated pyroglutamate –modified variants of Aβ (Aβ3pE-4x) [42] (Figure 1) which have been proposed to be particularly pathogenic [43]. In agreement, Aβ3pE-42 impairs spatial working memory and retention of reference memory in mice after intracerebroventricular (i.c.v.) injection with a similar potency to Aβ1-42 [44]. In a detailed structure-activity relationship analysis, freshly prepared synthetic Aβ3pE-x inhibited LTP in vitro with the following order of potency: Aβ3pE-42 > Aβ1-42 = Aβ3pE-38 = Aβ3pE-40 >> Aβ1-40, Aβ1-38 or Aβ3-40, the latter three being inactive at the highest concentration tested [45]. The authors found that this activity correlated with the relative ability to rapidly form oligomers and short fibrillar aggregates. Clearly the N-terminus of Aβ can play a critical role in determining aggregation and hence, presumably, ability to disrupt synaptic plasticity.
Somewhat similarly, nitration of Aß at tyrosine 10 also promotes aggregation and increases the magnitude of inhibition of LTP by Aß1-42 in hippocampal slices [46]. Such nitration is likely to arise subsequent to the formation of secondary products of NO production by pro-inflammatory upregulation of inducible nitric oxide synthase and can be pharmacologically targeted [46]. It will be interesting in future studies to determine if the activity in acute synaptic plasticity and memory models of other Aβ species found in AD brain, including Aβx-43 [47], phosphorylated Aβ [48] and glycosylated Aβ [49] relates to their tendency to form specific aggregates. One of the difficulties of working with aggregation prone peptides is to ensure consistent starting material in the absence of extensive solvent pretreatment. Recently, it has been shown that Aβ1-42 aggregates can be reliably generated from a precursor isopeptide by direct dissolution in physiological buffers [50]. Aβ oligomers prepared in this manner impede spatial learning and inhibit LTP both in vitro[50] and in vivo (Klyubin et al., unpublished) (Figure 2).

Aβ1-42 oligomers prepared from a precursor isopeptide by direct dissolution in physiological buffer [50]inhibits long-term potentiation in the hippocampus in vivo . Animals were injected with either vehicle (open circles) or Aβ1-42 (2.5 μg, closed circles) by intracerbroventricular injection (asterisk) 10 min before the conditioning high frequency stimulation (HFS, arrow) in the CA1 area of the anaesthetized rat hippocampus. Values are the mean ± SEM baseline field EPSP (fEPSP) (n = 6-7 per group).
Aβ assembly size
Aβ monomers
What do we know about the synaptic and mnemonic activity of Aβ monomers? Despite the natural ability of Aβ, especially the human sequence, to form aggregates, the majority of Aβ prepared by chemical synthesis or Aβ produced naturally by cells in vitro and the brain in vivo usually contains a sizable fraction of Aβ monomers. Biophysical methods such as size exclusion chromatography (SEC) are employed to enrich them. The results obtained by our group and by others suggest that Aβ monomers probably have little or no ability to disrupt synaptic functioning. Firstly, Aβ monomers produced by cultured CHO cells overexpressing human APP, known as 7PA2 cell line, did not affect LTP in vivo[51] or learned behavior [34]. Secondly, SEC fractions of Aβ monomers from AD brain homogenates and native human cerebrospinal fluid (CSF) failed to inhibit LTP in vitro and in vivo, respectively [35, 52]. Thirdly, SEC-separated monomers of a synthetic analog of Aβ1-40, Aβ1-40(S26C), had no effect on LTP in the CA1 area in vivo[53]. Fourthly, and most recently, Aβ 1-42 monomers, used in the preparation of oligomers using photo-induced cross-linking of unmodified proteins (PICUP), failed to inhibit LTP or facilitate LTD induction in hippocampal slices [54].
Some “physiological” effects of Aß on synaptic transmission, plasticity and learning have been described [55–59]. For example, extremely low concentrations of Aβ, both exogenously applied and endogenously generated, can enhance synaptic LTP and improve performance of learning tasks [58]. Because neurotrophic and neuroprotective effects of Aβ in cultured cells [60] have been attributed to Aβ monomers [61], it might be expected that these apparently positive effects are mediated by Aβ monomers [58, 59]. However, Puzzo et al. [58] reported that whereas pre-aggregated Aβ1-42 reversed the impairment of LTP caused by an anti-rodent Aβ antibody, they did not detect any effect of a synthetic Aβ1-42 preparation enriched in monomers. It will be of interest to determine if the same applies to the more abundant Aβ1-40 or cell-derived Aβ.
Small and large aggregates
Given the findings that Aβ monomers per se don't appear to impair synaptic function, the question arises as to which soluble Aβ aggregates are disruptive. Several lines of evidence suggest that small highly diffusible Aβ aggregates may be responsible for memory impairment in AD [2, 62]. The size of these aggregates varies from Aβ dimers, containing only two Aβ molecules, to approximately 20-mers. Although several protocols for the generation of well-characterized synthetic Aβ aggregates have been established to date, the question remains as to how relevant these Aβ assemblies are to the situation in an AD brain in vivo where there is a complex mixture of potentially interacting species [17, 18, 63].
Acute administration of extremely low doses of low-n Aβ oligomer-enriched fractions of conditioned medium from cultured 7PA2 cells rapidly disrupts synaptic plasticity [51] and performance of learned behaviours [34, 64]. In contrast, medium from APP transfected HEK293 cells that contained Aβ1-x or Aβ3-x peptides as a mixture of monomers and dimers (total Aβ concentration ~700 nM) did not significantly inhibit LTP [45]. However medium containing soluble large oligomers of Aβ3pE-x peptides in addition to an equivalent amount of monomer/dimer, inhibited LTP, indicating that larger assemblies of some natural Aβ may be particularly active.
Interestingly, the impairment of avoidance learning by 7PA2 conditioned medium, that contains low-n Aβ oligomers but no detectible large soluble assemblies such as protofibrils, is associated with disruption of synaptic remodeling in the dentate gyrus [65]. Furthermore, recall of hippocampus-dependent contextual fear learning is more susceptible to impairment than recall of amygdala-dependent cued learning after i.c.v. injection of 7PA2 conditioned medium [66]. These studies indicate that low-n oligomers may have preferential interactions with synapses in key hippocampal pathways.
The group led by Selkoe, having shown the presence of various Aβ assemblies in AD brain, suggested that soluble Aβ dimers are the smallest synaptotoxic species [35]. Indeed, a combination of biochemical analysis, electrophysiological experiments and behavioral tests revealed that sodium dodecyl sulfate (SDS) stable Aβ dimers found in water soluble extracts of AD brain disrupt the performance of an aversive learning task, inhibit LTP and facilitate LTD in rodents [35, 67–69]. AD brain soluble Aβ, containing SDS-stable dimers, disrupts synaptic plasticity in a dose-dependent manner and is very potent (Klyubin et al., unpublished observations) (Figure 3). In contrast, the larger Aβ*56 oligomers extracted from APP transgenic mouse brain [70] appear to be much less potent than cell-derived low-n oligomers or human brain Aβ dimer-containing soluble extracts at causing deficits in cognitive tasks [35, 64].

Aβ in soluble extracts of Alzheimer’s disease brain inhibits LTP in vivo . Representative example of LTP impairment after intracerebroventricular (i.c.v.) injection (asterisk) of Tris-buffered saline (TBS) extract of an AD brain containing Aβ (130 pg in total) (closed circles). In vehicle-injected controls, HFS (arrow) induces stable LTP (open circles) in the CA1 area of the anaesthetized rat.
In apparent support of the proposal that dimers are key synaptotoxic species, synthetic Aβ1-40 dimers blocked LTP both in vivo and in vitro when acutely applied at a concentration approximately 50-fold lower than unmodified Aβ1-40 [35, 53]. These dimers were created using Aβ with a single conservative amino acid substitution (cysteine in place of serine 26, S26C) enabling covalent cross-linking with a disulfide bond under oxidizing conditions. However, soon after this, Walsh and colleagues found evidence that these dimers need to assemble into large protofibril-like aggregates before being able to potently inhibit LTP [71]. In fact, freshly prepared non-aggregated synthetic Aβ1-40(S26C) dimers, like monomers (see above), were found to have no significant effect on synaptic plasticity whereas protofibril-rich assemblies of these dimers strongly inhibited LTP in vitro. An explanation given by the authors for the discrepancy from the earlier findings was a lack of definition of aggregation state of materials used in previous studies. This conclusion is in agreement with the work of another group who demonstrated that tissue transglutaminase, an enzyme implicated in neurodegeneration with the catalytic capability to covalently cross-link “wild type” Aβ between lysine and glutamine residues, induced synthetic Aβ1-40 to form large assemblies including protofibrils which potently inhibited LTP in the CA1 area in vitro[72]. In contrast, a similar low concentration (100 nM) of untreated Aβ1-40 had no effect.
Some synthetic Aβ low-n and high-n oligomers are not harmful to neurons. Thus aggregation of synthetic Aß1-42 where the glycine residue at position 33 is substituted with alanine generated Aβ1-42(G33A) tetramers which failed to inhibit LTP, as was the case with Aβ1-42(G33I) which only formed high-n oligomers when aggregated [73].
Just as in the case of the disruptive effects of synthetic Aβ on synaptic plasticity, there is evidence that only certain “intermediate” synthetic Aβ assemblies, including protofibrils, can rapidly impair learning [74, 75]. However, regardless of the relationship between size of soluble Aβ aggregates and synaptic dysfunction, insoluble fibrils per se are unlikely candidates for memory impairment in AD. Rather, plaque-containing insoluble fibrils are likely to provide a major source and sink of memory disrupting soluble Aβ [35, 74].
Because of difficulties in determining the size of biologically active Aβ aggregates, especially under non-denaturing conditions, size-selective ligands such as antibodies should prove useful. Recently, O'Nuallain et al. [76] developed an antibody, 3C6, that preferentially binds soluble aggregates of covalently cross-linked dimers of Aβ1-40(S26C), and recognizes only a portion of SDS-stable dimers in aqueous extracts of AD brain [76]. Importantly, such apparent selectivity was sufficient to prevent block of LTP by the AD brain soluble extract in vivo. It is possible that 3C6-mediated abrogation of LTP inhibition triggered by AD brain soluble Aβ was due to rapid direct neutralization of aggregates of Aβ larger than single SDS-stable dimers.
In an analogous approach with synthetic Aβ, N7, an agent believed to selectively block large Aβ assemblies that form ion-permeable pores in membranes, prevented Aβ aggregate-induced depletion of presynaptic glutamatergic vesicles and consequent depression of spontaneous synaptic currents in cultured hippocampal neurons [77].
Conformation versus size
Not only size, but also the spatial conformation of synapse-disruptive soluble Aβ aggregates varies. Thus Aβ aggregates can be classified based on the ability of conformation-specific antibodies to recognize aggregates in a manner that appears relatively independent of size [78]. Such conformation-specific antibodies, for example, are used to distinguish between so-called “prefibrillar” and “fibrillar” types of aggregates regardless of their size. Thus, ADDLs and globulomers are likely to be “fibril”-type whereas Aβ*56 is probably “prefibrillar”. As a corollary to the ability of different sized aggregates to adopt similar conformations, the same sized Aβ aggregates may have different sub-populations of different conformers.
Evidence suggestive of a relatively “size-independent” role for an N-terminal ß strand conformation in the synaptic plasticity disrupting effects of synthetic Aß oligomers and protofibrils has been reported [79]. Thus, synthetic Aß1-40, containing oligomers and protofibrils in the presence of a ß-sheet breaker peptide corresponding to residues 4-10 of Aß, designed to reduce the relative amount of N-terminal ß strand conformation, failed to inhibit LTP. In contrast, synthetic Aß1-40 containing a point mutation (P4F) that promoted the formation of protofibrils, including those with an N-terminal ß-strand conformation, inhibited LTP in vitro with a similar potency to an oligomer preparation of wild type Aß1-40 with a similar ß-strand conformation.
Like Aβ, many other amyloidogenic proteins form aqueous soluble oligomers that are neurotoxic [2, 80]. Intriguingly, many of these neurotoxic oligomers adopt similar conformations to Aβ recognized by conformation-selectivez antibodies [78, 81, 82]. The conformations adopted are relatively independent of their primary amino acid sequence, as is the case for fibrils [83]. For example, the antibody A11, originally generated against Aβ oligomers, recognizes a common conformation adopted by oligomers of many peptides, including α−synuclein and an amyloidogenic fragment of PrPC, PrP106-126 [84, 85]. Whether or not conformational epitopes on Aβ and other peptide aggregates determine their ability to selectively bind to specific synaptic sites and thereby disrupt memory mechanisms has yet to be resolved but there is growing suggestive evidence consistent with the hypothesis, as outlined below.
Soluble oligomers of tau, the main protein deposited intracellularly as insoluble fibrils in AD and fronto-temporal dementia, can rapidly impair object recognition memory and reduce levels of synaptic vesicle-associated proteins when applied intrahippocampally in vivo[86]. In contrast, monomers and fibrils of tau appeared inactive under the same acute treatment protocol.
Insoluble aggregates of α−synuclein are the main constituent of intracellular inclusions, Lewy bodies, in the brains of patients with Parkinson’s disease and related dementias, but soluble oligomers are released extracellularly and are neurotoxic [87]. Intriguingly, low nanomolar concentrations of large α−synuclein oligomers can rapidly trigger a selective increase in AMPA receptor-mediated synaptic transmission in autaptic neuronal cultures [88]. In contrast, other oligomers of α−synuclein were reported to inhibit LTP without affecting baseline transmission, and to impair learning an avoidance task [89].
The prion peptide fragment PrP106-126 is used to model the neurotoxic, rather than the infective, aspects of prion-mediated transmissible spongiform encephalopathies (TSEs)[90–95]. In prion diseases synaptic mechanisms are often disrupted at a relatively early stage [96]. Intriguingly, i.c.vinjection of PrP106-126 inhibits LTP of synaptic transmission in the CA1 area of the hippocampus in vivo (Cullen et al., unpublished observations) (Figure 4).

The aggregation-prone prion protein fragment PrP106-126 inhibits LTP in vivo . Animals were injected with either vehicle (open circles) or PrP106-126 (96 ng, closed circles) by i.c.v. injection (asterisk) 10 min before the HFS (arrow) in the CA1 area of the anaesthetized rat hippocampus. Values are the mean ± SEM baseline fEPSP (n = 10 per group).
Another amyloidogenic peptide, ADan, is found deposited in the brains of patients with familial Danish dementia, a rare autosomal dominant form of cognitive impairment with AD-like neuropathology. The N-terminally truncated pyroglutamate form of ADan was found to be especially prone to aggregate into large oligomers and appeared to be more potent than unmodified ADan at inhibiting LTP in vitro[45].
In view of the shared ability of conformational antibodies to recognize these aggregates and their shared ability to inhibit LTP it is tempting to speculate that a common conformation is critical for the synaptic plasticity and hence memory disrupting actions of these very different peptides. In line with this and similar to the situation with regards the role of Aβ aggregate size, future studies should attempt to resolve which, if any, specific conformation of soluble Aβ assemblies is more disruptive to synapses and memory.
Cellular prion protein and Aβ-mediated disruption of synaptic plasticity and learning
Given the likely key pathogenic role of a partially protease-resistant misfolded form of PrPC (PrPSc), and the critical requirement for PrPC, in transmissible spongiform encephalopathies [8, 97], the relationship and commonalities between prion-mediated neurodegenerative diseases and AD have become a major focus of research [98–104].
Recently synthetic Aβ oligomer-mediated inhibition of LTP at hippocampal synapses in vitro was reported to be dependent on PrPC[105] with Aβ oligomers, but not monomers or fibrils, potently and selectively binding specific regions of PrPC, especially in the vicinity of amino acids 95-105 [105–107]. Antibodies that bind PrPC within the region of 93-109 [105] or 93-102 [107] prevented the inhibition of hippocampal LTP by synthetic Aβ1-42 oligomers in vitro. Consistent with these reports, the in vivo synaptic plasticity disrupting actions of AD brain extracts containing water soluble Aβ were dependent on PrPC[67]. Thus, the disruptive effect of Aβ was abrogated by D13, an antigen recognizing antibody fragment (Fab) that binds selectively to PrP96-104C. It is likely that these antibodies and related agents are directly obstructing the binding of Aβ oligomers to PrPC. In addition, an antibody to the alpha helix of PrPC also prevented the inhibition of LTP by AD brain Aβ oligomers both in vitro and in vivo[107] whereas a Fab directed to the C-terminus of PrPC appeared to be inactive [67]. Since the alpha helix of PrPC does not overlap with the putative binding site of Aβ oligomers, one possible explanation for these findings is that the antibody to the alpha helix is interfering with PrP:PrP contact. Interestingly, direct intra-hippocampal injection of bivalent D13 antibodies, but not monovalent D13 Fabs, can cause delayed apoptotic neurodegeneration in mice [108], but see [109], indicating that abnormal cross-linking of PrPC in the 96-106 region by oligomers may contribute to their damaging effects. Indeed, cross-linking of PrPC has been associated with synaptic damage caused by cell-derived low-n oligomers of Aβ in cultured neurons [110]. Such cross-linking Aβ oligomers may prevent PrP-dependent inactivation of N-methyl-d-aspartate (NMDA) receptor-mediated currents leading to abnormally enhanced NMDA receptor-mediated glutamatergic transmission [111]. Furthermore, cross-linking of other adjacent membrane proteins, in particular metabotropic glutamate receptor 5, may go hand-in-hand with this process in mediating Aβ oligomer-induced synaptotoxicity [112].
In apparent direct contradiction to the findings of Lauren et al. and Freir et al. [67, 105, 107], Kessels et al. [113] reported that Aβ1-42 oligomers impaired LTP in hippocampal slices from transgenic mice lacking PrPC. Moreover, in APP transgenic mice a deficit in LTP was similar in the presence or absence of PrPC[114]. Differences in the Aβ oligomer concentration/assembly are likely to explain these apparently contradictory findings [107]. In the Kessels et al. study [113], in contrast to most other reports on acute effects of Aβ oligomers on synaptic plasticity, the inhibition of LTP was accompanied by a marked rapid reduction in baseline synaptic transmission. This indicates that concentrations of certain Aβ oligomer-containing preparations sufficient to rapidly reduce baseline transmission can bypass a requirement for PrPC to disrupt synaptic function.
At the behavioural level, there is also strong evidence that Aβ-mediated memory impairment is PrPC-dependent [115, 116]. However, an apparent acute disruption of object recognition memory caused by Aβ1-42 was not prevented in mice lacking PrPC[117]. Moreover, Cissé et al [118] in a recent paper observed the same cognitive deficits in APP transgenic mice in the presence or absence of PrPC. These authors provided strong evidence, instead, that impairments of synaptic plasticity and memory were due to a direct interaction of Aβ oligomers with the Ephrin B2 receptor EphB2 [119]. It will be important to determine if the fact that different APP transgenic mice at different ages express different potentially synaptotoxic Aβ assemblies [18, 120] can help explain this controversy.
Conclusion
The commonalities and differences between amyloidogenic proteins in different neurodegenerative diseases are of great theoretical and practical interest. The ability of certain assemblies of these proteins to rapidly disrupt synaptic plasticity and memory mechanisms indicates that there may be shared mechanisms across diseases. An obvious limitation of the acute application approach is that although it is now feasible to apply relatively homogenous protein assemblies, it is not clear how these relatively labile preparations behave structurally throughout the full duration of the experiments and how this may depend on the existing milieu of endogenous amyloidogenic proteins which is known to depend on the ongoing neuronal activity amongst many factors [100, 121]. In the light of the chronic nature of these diseases this may prove a difficult but important question to address. Furthermore, the question remains as to how well exogenously applied proteins, especially synthetic aggregates, in rodents, mimic the actions and effects of endogenously generated proteins in situ in the brains of patients. To date, the evidence for the involvement of different sizes of aggregates and different cellular targets in these models is compelling. If the same conclusion applies to patients, perhaps with different assemblies playing a leading role at different stages of disease, it probably will be necessary to take this diversity into account when developing new diagnostic and therapeutic approaches. On the other hand, if common conformations of different proteins are pathophysiologically relevant, selectively neutralizing them [51, 122], or changing their aggregation kinetics such that monomers are stabilized [54] or even by accelerating their conversion to fibrillar material [123], may have utility in a wide spectrum of neurodegenerative disorders.
Consent
Each brain donor consented to have their post-mortem tissue used for research.
References
Roberts GW, Lofthouse R, Allsop D, Landon M, Kidd M, Prusiner SB, Crow TJ: CNS amyloid proteins in neurodegenerative diseases. Neurology. 1988, 38: 1534-1540. 10.1212/WNL.38.10.1534.
Haass C, Selkoe DJ: Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid beta-peptide. Nat Rev Mol Cell Biol. 2007, 8: 101-112. 10.1038/nrm2101.
Palop JJ, Chin J, Mucke L: A network dysfunction perspective on neurodegenerative diseases. Nature. 2006, 443: 768-773. 10.1038/nature05289.
Selkoe DJ: Alzheimer's disease is a synaptic failure. Science. 2002, 298: 789-791. 10.1126/science.1074069.
Schneider JA, Arvanitakis Z, Bang W, Bennett DA: Mixed brain pathologies account for most dementia cases in community-dwelling older persons. Neurology. 2007, 69: 2197-2204. 10.1212/01.wnl.0000271090.28148.24.
Gray BC, Siskova Z, Perry VH, O'Connor V: Selective presynaptic degeneration in the synaptopathy associated with ME7-induced hippocampal pathology. Neurobiol Dis. 2009, 35: 63-74. 10.1016/j.nbd.2009.04.001.
Bate C, Gentleman S, Williams A: Alpha-synuclein induced synapse damage is enhanced by amyloid-beta1-42. Mol Neurodegener. 2010, 5: 55-10.1186/1750-1326-5-55.
Moreno JA, Mallucci GR: Dysfunction and recovery of synapses in prion disease: implications for neurodegeneration. Biochem Soc Trans. 2010, 38: 482-487. 10.1042/BST0380482.
Hardy J, Allsop D: Amyloid deposition as the central event in the aetiology of Alzheimer's disease. Trends Pharmacol Sci. 1991, 12: 383-388.
Randall AD, Witton J, Booth C, Hynes-Allen A, Brown JT: The functional neurophysiology of the amyloid precursor protein (APP) processing pathway. Neuropharmacology. 2010, 59: 243-267. 10.1016/j.neuropharm.2010.02.011.
The biology of Alzheimer's disease. Edited by: Selkoe DJ, Mandelkow E, Holtzman DM. 2012, New York: Cold Spring Harbor Laboratory Press
Patel AN, Jhamandas JH: Neuronal receptors as targets for the action of amyloid-beta protein (Abeta) in the brain. Expet Rev Mol Med. 2012, 14: e2-
Hu NW, Ondrejcak T, Rowan MJ: Glutamate receptors in preclinical research on Alzheimer's disease: update on recent advances. Pharmacol Biochem Behav. 2012, 100: 855-862. 10.1016/j.pbb.2011.04.013.
Ma T, Klann E: Amyloid beta: linking synaptic plasticity failure to memory disruption in Alzheimer's disease. J Neurochem. 2012, 120 (Suppl 1): 140-148.
Marchetti C, Marie H: Hippocampal synaptic plasticity in Alzheimer's disease: what have we learned so far from transgenic models?. Rev Neurosci. 2011, 22: 373-402.
Malinow R: New developments on the role of NMDA receptors in Alzheimer's disease. Curr Opin Neurobiol. 2012, 22: 559-563. 10.1016/j.conb.2011.09.001.
Benilova I, Karran E, De Strooper B: The toxic Abeta oligomer and Alzheimer's disease: an emperor in need of clothes. Nat Neurosci. 2012, 15: 349-357. 10.1038/nn.3028.
Larson ME, Lesne SE: Soluble Abeta oligomer production and toxicity. J Neurochem. 2012, 120 (Suppl 1): 125-139.
Lue LF, Kuo YM, Roher AE, Brachova L, Shen Y, Sue L, Beach T, Kurth JH, Rydel RE, Rogers J: Soluble amyloid beta peptide concentration as a predictor of synaptic change in Alzheimer's disease. Am J Pathol. 1999, 155: 853-862. 10.1016/S0002-9440(10)65184-X.
Wang J, Dickson DW, Trojanowski JQ, Lee VM: The levels of soluble versus insoluble brain Abeta distinguish Alzheimer's disease from normal and pathologic aging. Exp Neurol. 1999, 158: 328-337. 10.1006/exnr.1999.7085.
Chambon C, Wegener N, Gravius A, Danysz W: Behavioural and cellular effects of exogenous amyloid-beta peptides in rodents. Behav Brain Res. 2011, 225: 623-641. 10.1016/j.bbr.2011.08.024.
Flood JF, Morley JE, Roberts E: Amnestic effects in mice of four synthetic peptides homologous to amyloid beta protein from patients with Alzheimer disease. Proc Natl Acad Sci U S A. 1991, 88: 3363-3366. 10.1073/pnas.88.8.3363.
McDonald MP, Dahl EE, Overmier JB, Mantyh P, Cleary J: Effects of an exogenous beta-amyloid peptide on retention for spatial learning. Behav Neural Biol. 1994, 62: 60-67. 10.1016/S0163-1047(05)80059-7.
Cullen WK, Wu J, Anwyl R, Rowan MJ: beta-Amyloid produces a delayed NMDA receptor-dependent reduction in synaptic transmission in rat hippocampus. Neuroreport. 1996, 8: 87-92. 10.1097/00001756-199612200-00018.
Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, et al: Diffusible, nonfibrillar ligands derived from A beta(1-42) are potent central nervous system neurotoxins. Proc Natl Acad Sci U S A. 1998, 95: 6448-6453. 10.1073/pnas.95.11.6448.
Lacor PN, Buniel MC, Chang L, Fernandez SJ, Gong Y, Viola KL, Lambert MP, Velasco PT, Bigio EH, Finch CE, et al: Synaptic targeting by Alzheimer's-related amyloid beta oligomers. J Neurosci. 2004, 24: 10191-10200. 10.1523/JNEUROSCI.3432-04.2004.
Nimmrich V, Grimm C, Draguhn A, Barghorn S, Lehmann A, Schoemaker H, Hillen H, Gross G, Ebert U, Bruehl C: Amyloid beta oligomers (A beta(1-42) globulomer) suppress spontaneous synaptic activity by inhibition of P/Q-type calcium currents. J Neurosci. 2008, 28: 788-797. 10.1523/JNEUROSCI.4771-07.2008.
Ross CA, Poirier MA: Opinion: what is the role of protein aggregation in neurodegeneration?. Nat Rev Mol Cell Biol. 2005, 6: 891-898. 10.1038/nrm1742.
Cullen WK, Suh YH, Anwyl R, Rowan MJ: Block of LTP in rat hippocampus in vivo by ß-amyloid precursor protein fragments. Neuroreport. 1997, 8: 3213-3217. 10.1097/00001756-199710200-00006.
Kim JH, Anwyl R, Suh YH, Djamgoz MB, Rowan MJ: Use-dependent effects of amyloidogenic fragments of (beta)-amyloid precursor protein on synaptic plasticity in rat hippocampus in vivo. J Neurosci. 2001, 21: 1327-1333.
Lynch MA: Long-term potentiation and memory. Physiol Rev. 2004, 84: 87-136. 10.1152/physrev.00014.2003.
Neves G, Cooke SF, Bliss TV: Synaptic plasticity, memory and the hippocampus: a neural network approach to causality. Nat Rev Neurosci. 2008, 9: 65-75. 10.1038/nrn2303.
Collingridge GL, Peineau S, Howland JG, Wang YT: Long-term depression in the CNS. Nat Rev Neurosci. 2010, 11: 459-473.
Cleary JP, Walsh DM, Hofmeister JJ, Shankar GM, Kuskowski MA, Selkoe DJ, Ashe KH: Natural oligomers of the amyloid-beta protein specifically disrupt cognitive function. Nat Neurosci. 2005, 8: 79-84. 10.1038/nn1372.
Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, et al: Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med. 2008, 14: 837-842. 10.1038/nm1782.
Nomura I, Takechi H, Kato N: Intraneuronally injected amyloid beta inhibits long-term potentiation in rat hippocampal slices. J Neurophysiol. 2012, 107: 2526-2531. 10.1152/jn.00589.2011.
Hard T: Protein engineering to stabilize soluble amyloid beta-protein aggregates for structural and functional studies. FEBS J. 2011, 278: 3884-3892. 10.1111/j.1742-4658.2011.08295.x.
Klyubin I, Walsh DM, Cullen WK, Fadeeva JV, Anwyl R, Selkoe DJ, Rowan MJ: Soluble arctic amyloid beta protein inhibits hippocampal long-term potentiation in vivo. Eur J Neurosci. 2004, 19: 2839-2846. 10.1111/j.1460-9568.2004.03389.x.
Tomiyama T, Nagata T, Shimada H, Teraoka R, Fukushima A, Kanemitsu H, Takuma H, Kuwano R, Imagawa M, Ataka S, et al: A new amyloid beta variant favoring oligomerization in Alzheimer's-type dementia. Ann Neurol. 2008, 63: 377-387. 10.1002/ana.21321.
Portelius E, Bogdanovic N, Gustavsson MK, Volkmann I, Brinkmalm G, Zetterberg H, Winblad B, Blennow K: Mass spectrometric characterization of brain amyloid beta isoform signatures in familial and sporadic Alzheimer's disease. Acta neuropathologica. 2010, 120: 185-193. 10.1007/s00401-010-0690-1.
Mori H, Takio K, Ogawara M, Selkoe DJ: Mass spectrometry of purified amyloid beta protein in Alzheimer's disease. J Biol Chem. 1992, 267: 17082-17086.
Schilling S, Zeitschel U, Hoffmann T, Heiser U, Francke M, Kehlen A, Holzer M, Hutter-Paier B, Prokesch M, Windisch M, et al: Glutaminyl cyclase inhibition attenuates pyroglutamate Abeta and Alzheimer's disease-like pathology. Nat Med. 2008, 14: 1106-1111. 10.1038/nm.1872.
Jawhar S, Wirths O, Bayer TA: Pyroglutamate amyloid-beta (Abeta): a hatchet man in Alzheimer disease. J Biol Chem. 2011, 286: 38825-38832. 10.1074/jbc.R111.288308.
Youssef I, Florent-Bechard S, Malaplate-Armand C, Koziel V, Bihain B, Olivier JL, Leininger-Muller B, Kriem B, Oster T, Pillot T: N-truncated amyloid-beta oligomers induce learning impairment and neuronal apoptosis. Neurobiol Aging. 2008, 29: 1319-1333. 10.1016/j.neurobiolaging.2007.03.005.
Schlenzig D, Ronicke R, Cynis H, Ludwig HH, Scheel E, Reymann K, Saido T, Hause G, Schilling S, Demuth HU: N-Terminal pyroglutamate formation of Abeta38 and Abeta40 enforces oligomer formation and potency to disrupt hippocampal long-term potentiation. J Neurochem. 2012, 121: 774-784. 10.1111/j.1471-4159.2012.07707.x.
Kummer MP, Hermes M, Delekarte A, Hammerschmidt T, Kumar S, Terwel D, Walter J, Pape HC, Konig S, Roeber S, et al: Nitration of tyrosine 10 critically enhances amyloid beta aggregation and plaque formation. Neuron. 2011, 71: 833-844. 10.1016/j.neuron.2011.07.001.
Saito T, Suemoto T, Brouwers N, Sleegers K, Funamoto S, Mihira N, Matsuba Y, Yamada K, Nilsson P, Takano J, et al: Potent amyloidogenicity and pathogenicity of Abeta43. Nat Neurosci. 2011, 14: 1023-1032. 10.1038/nn.2858.
Kumar S, Rezaei-Ghaleh N, Terwel D, Thal DR, Richard M, Hoch M, Mc Donald JM, Wullner U, Glebov K, Heneka MT, et al: Extracellular phosphorylation of the amyloid beta-peptide promotes formation of toxic aggregates during the pathogenesis of Alzheimer's disease. EMBO J. 2011, 30: 2255-2265. 10.1038/emboj.2011.138.
Halim A, Brinkmalm G, Ruetschi U, Westman-Brinkmalm A, Portelius E, Zetterberg H, Blennow K, Larson G, Nilsson J: Site-specific characterization of threonine, serine, and tyrosine glycosylations of amyloid precursor protein/amyloid beta-peptides in human cerebrospinal fluid. Proc Natl Acad Sci U S A. 2011, 108: 11848-11853. 10.1073/pnas.1102664108.
Bozso Z, Penke B, Simon D, Laczko I, Juhasz G, Szegedi V, Kasza A, Soos K, Hetenyi A, Weber E, et al: Controlled in situ preparation of A beta(1-42) oligomers from the isopeptide "iso-A beta(1-42)", physicochemical and biological characterization. Peptides. 2010, 31: 248-256. 10.1016/j.peptides.2009.12.001.
Klyubin I, Walsh DM, Lemere CA, Cullen WK, Shankar GM, Betts V, Spooner ET, Jiang LY, Anwyl R, Selkoe DJ, Rowan MJ: Amyloid beta protein immunotherapy neutralizes A beta oligomers that disrupt synaptic plasticity in vivo. Nature Medicine. 2005, 11: 556-561. 10.1038/nm1234.
Klyubin I, Betts V, Welzel AT, Blennow K, Zetterberg H, Wallin A, Lemere CA, Cullen WK, Peng Y, Wisniewski T, et al: Amyloid beta protein dimer-containing human CSF disrupts synaptic plasticity: prevention by systemic passive immunization. J Neurosci. 2008, 28: 4231-4237. 10.1523/JNEUROSCI.5161-07.2008.
Hu NW, Smith IM, Walsh DM, Rowan MJ: Soluble amyloid-beta peptides potently disrupt hippocampal synaptic plasticity in the absence of cerebrovascular dysfunction in vivo. Brain. 2008, 131: 2414-2424. 10.1093/brain/awn174.
Ono K, Li L, Takamura Y, Yoshiike Y, Zhu L, Han F, Mao X, Ikeda T, Takasaki JI, Nishijo H, et al: Phenolic compounds prevent amyloid beta-protein oligomerization and synaptic dysfunction by site specific binding. J Biol Chem. 2012, 287: 14631-14643. 10.1074/jbc.M111.325456.
Abramov E, Dolev I, Fogel H, Ciccotosto GD, Ruff E, Slutsky I: Amyloid-beta as a positive endogenous regulator of release probability at hippocampal synapses. Nat Neurosci. 2009, 12: 1567-1576. 10.1038/nn.2433.
Garcia-Osta A, Alberini CM: Amyloid beta mediates memory formation. Learn Mem. 2009, 16: 267-272. 10.1101/lm.1310209.
Morley JE, Farr SA, Banks WA, Johnson SN, Yamada KA, Xu L: A physiological role for amyloid-beta protein:enhancement of learning and memory. J Alzheimers Dis. 2010, 19: 441-449.
Puzzo D, Privitera L, Fa M, Staniszewski A, Hashimoto G, Aziz F, Sakurai M, Ribe EM, Troy CM, Mercken M, et al: Endogenous amyloid-beta is necessary for hippocampal synaptic plasticity and memory. Ann Neurol. 2011, 69: 819-830. 10.1002/ana.22313.
Puzzo D, Privitera L, Leznik E, Fa M, Staniszewski A, Palmeri A, Arancio O: Picomolar amyloid-beta positively modulates synaptic plasticity and memory in hippocampus. J Neurosci. 2008, 28: 14537-14545. 10.1523/JNEUROSCI.2692-08.2008.
Pearson HA, Peers C: Physiological roles for amyloid beta peptides. J Physiol. 2006, 575: 5-10. 10.1113/jphysiol.2006.111203.
Giuffrida ML, Caraci F, Pignataro B, Cataldo S, De Bona P, Bruno V, Molinaro G, Pappalardo G, Messina A, Palmigiano A, et al: Beta-amyloid monomers are neuroprotective. J Neurosci. 2009, 29: 10582-10587. 10.1523/JNEUROSCI.1736-09.2009.
Krafft GA, Klein WL: ADDLs and the signaling web that leads to Alzheimer's disease. Neuropharmacology. 2010, 59: 230-242. 10.1016/j.neuropharm.2010.07.012.
Mc Donald JM, Savva GM, Brayne C, Welzel AT, Forster G, Shankar GM, Selkoe DJ, Ince PG, Walsh DM: The presence of sodium dodecyl sulphate-stable Abeta dimers is strongly associated with Alzheimer-type dementia. Brain. 2010, 133: 1328-1341. 10.1093/brain/awq065.
Reed MN, Hofmeister JJ, Jungbauer L, Welzel AT, Yu C, Sherman MA, Lesne S, LaDu MJ, Walsh DM, Ashe KH, Cleary JP: Cognitive effects of cell-derived and synthetically derived Abeta oligomers. Neurobiol Aging. 2011, 32: 1784-1794. 10.1016/j.neurobiolaging.2009.11.007.
Freir DB, Fedriani R, Scully D, Smith IM, Selkoe DJ, Walsh DM, Regan CM: Abeta oligomers inhibit synapse remodelling necessary for memory consolidation. Neurobiol Aging. 2011, 32: 2211-2218. 10.1016/j.neurobiolaging.2010.01.001.
Kittelberger KA, Piazza F, Tesco G, Reijmers LG: Natural amyloid-beta oligomers acutely impair the formation of a contextual fear memory in mice. PLoS One. 2012, 7: e29940-10.1371/journal.pone.0029940.
Barry AE, Klyubin I, Mc Donald JM, Mably AJ, Farrell MA, Scott M, Walsh DM, Rowan MJ: Alzheimer’s disease brain-derived amyloid-ß-mediated inhibition of LTP in vivo is prevented by immunotargeting cellular prion protein. J Neurosci. 2011, 31: 7259-63. 10.1523/JNEUROSCI.6500-10.2011.
Li S, Hong S, Shepardson NE, Walsh DM, Shankar GM, Selkoe D: Soluble oligomers of amyloid beta protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron. 2009, 62: 788-801. 10.1016/j.neuron.2009.05.012.
Li S, Jin M, Koeglsperger T, Shepardson NE, Shankar GM, Selkoe DJ: Soluble Abeta oligomers inhibit long-term potentiation through a mechanism involving excessive activation of extrasynaptic NR2B-containing NMDA receptors. J Neurosci. 2011, 31: 6627-6638. 10.1523/JNEUROSCI.0203-11.2011.
Lesne S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH: A specific amyloid-beta protein assembly in the brain impairs memory. Nature. 2006, 440: 352-357. 10.1038/nature04533.
O'Nuallain B, Freir DB, Nicoll AJ, Risse E, Ferguson N, Herron CE, Collinge J, Walsh DM: Amyloid beta-protein dimers rapidly form stable synaptotoxic protofibrils. J Neurosci. 2010, 30: 14411-14419. 10.1523/JNEUROSCI.3537-10.2010.
Hartley DM, Zhao C, Speier AC, Woodard GA, Li S, Li Z, Walz T: Transglutaminase induces protofibril-like amyloid beta-protein assemblies that are protease-resistant and inhibit long-term potentiation. J Biol Chem. 2008, 283: 16790-16800. 10.1074/jbc.M802215200.
Harmeier A, Wozny C, Rost BR, Munter LM, Hua H, Georgiev O, Beyermann M, Hildebrand PW, Weise C, Schaffner W, et al: Role of amyloid-beta glycine 33 in oligomerization, toxicity, and neuronal plasticity. J Neurosci. 2009, 29: 7582-7590. 10.1523/JNEUROSCI.1336-09.2009.
Martins IC, Kuperstein I, Wilkinson H, Maes E, Vanbrabant M, Jonckheere W, Van Gelder P, Hartmann D, D'Hooge R, De Strooper B, et al: Lipids revert inert Abeta amyloid fibrils to neurotoxic protofibrils that affect learning in mice. EMBO J. 2008, 27: 224-233. 10.1038/sj.emboj.7601953.
Kuperstein I, Broersen K, Benilova I, Rozenski J, Jonckheere W, Debulpaep M, Vandersteen A, Segers-Nolten I, Van Der Werf K, Subramaniam V, et al: Neurotoxicity of Alzheimer's disease Abeta peptides is induced by small changes in the Abeta42 to Abeta40 ratio. EMBO J. 2010, 29: 3408-3420. 10.1038/emboj.2010.211.
O'Nuallain B, Klyubin I, Mc Donald JM, Foster JS, Welzel A, Barry A, Dykoski RK, Cleary JP, Gebbink MF, Rowan MJ, Walsh DM: A monoclonal antibody against synthetic Abeta dimer assemblies neutralizes brain-derived synaptic plasticity-disrupting Abeta. J Neurochem. 2011, 119: 189-201. 10.1111/j.1471-4159.2011.07389.x.
Parodi J, Sepulveda FJ, Roa J, Opazo C, Inestrosa NC, Aguayo LG: Beta-amyloid causes depletion of synaptic vesicles leading to neurotransmission failure. J Biol Chem. 2010, 285: 2506-2514. 10.1074/jbc.M109.030023.
Glabe CG: Structural classification of toxic amyloid oligomers. J Biol Chem. 2008, 283: 29639-29643. 10.1074/jbc.R800016200.
Haupt C, Leppert J, Ronicke R, Meinhardt J, Yadav JK, Ramachandran R, Ohlenschlager O, Reymann KG, Gorlach M, Fandrich M: Structural basis of beta-amyloid-dependent synaptic dysfunctions. Angew Chem Int Ed Engl. 2012, 51: 1576-1579. 10.1002/anie.201105638.
Caughey B, Lansbury PT: Protofibrils, pores, fibrils, and neurodegeneration: separating the responsible protein aggregates from the innocent bystanders. Annu Rev Neurosci. 2003, 26: 267-298. 10.1146/annurev.neuro.26.010302.081142.
Yamin G, Ono K, Inayathullah M, Teplow DB: Amyloid beta-protein assembly as a therapeutic target of Alzheimer's disease. Curr Pharm Des. 2008, 14: 3231-3246. 10.2174/138161208786404137.
Lambert MP, Velasco PT, Viola KL, Klein WL: Targeting generation of antibodies specific to conformational epitopes of amyloid beta-derived neurotoxins. CNS Neurol Disord Drug Targets. 2009, 8: 65-81. 10.2174/187152709787601876.
O'Nuallain B, Wetzel R: Conformational Abs recognizing a generic amyloid fibril epitope. Proc Natl Acad Sci U S A. 2002, 99: 1485-1490. 10.1073/pnas.022662599.
Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG: Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003, 300: 486-489. 10.1126/science.1079469.
Kayed R, Sokolov Y, Edmonds B, McIntire TM, Milton SC, Hall JE, Glabe CG: Permeabilization of lipid bilayers is a common conformation-dependent activity of soluble amyloid oligomers in protein misfolding diseases. J Biol Chem. 2004, 279: 46363-46366. 10.1074/jbc.C400260200.
Lasagna-Reeves CA, Castillo-Carranza DL, Sengupta U, Clos AL, Jackson GR, Kayed R: Tau oligomers impair memory and induce synaptic and mitochondrial dysfunction in wild-type mice. Mol Neurodegener. 2011, 6: 39-10.1186/1750-1326-6-39.
Emmanouilidou E, Melachroinou K, Roumeliotis T, Garbis SD, Ntzouni M, Margaritis LH, Stefanis L, Vekrellis K: Cell-produced alpha-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. J Neurosci. 2010, 30: 6838-6851. 10.1523/JNEUROSCI.5699-09.2010.
Huls S, Hogen T, Vassallo N, Danzer KM, Hengerer B, Giese A, Herms J: AMPA-receptor-mediated excitatory synaptic transmission is enhanced by iron-induced alpha-synuclein oligomers. J Neurochem. 2011, 117: 868-878. 10.1111/j.1471-4159.2011.07254.x.
Martin ZS, Neugebauer V, Dineley KT, Kayed R, Zhang W, Reese LC, Taglialatela G: alpha-Synuclein oligomers oppose long-term potentiation and impair memory through a calcineurin-dependent mechanism: relevance to human synucleopathic diseases. J Neurochem. 2012, 120: 440-452. 10.1111/j.1471-4159.2011.07576.x.
Forloni G, Angeretti N, Chiesa R, Monzani E, Salmona M, Bugiani O, Tagliavini F: Neurotoxicity of a prion protein fragment. Nature. 1993, 362: 543-546. 10.1038/362543a0.
Brown DR, Schmidt B, Kretzschmar HA: Role of microglia and host prion protein in neurotoxicity of a prion protein fragment. Nature. 1996, 380: 345-347. 10.1038/380345a0.
Crozet C, Beranger F, Lehmann S: Cellular pathogenesis in prion diseases. Vet Res. 2008, 39: 44-10.1051/vetres:2008021.
Vassallo N: Properties and pathogenicity of prion-derived peptides. Protein Pept Lett. 2009, 16: 230-238. 10.2174/092986609787601651.
Grabenauer M, Wu C, Soto P, Shea JE, Bowers MT: Oligomers of the prion protein fragment 106-126 are likely assembled from beta-hairpins in solution, and methionine oxidation inhibits assembly without altering the peptide's monomeric conformation. J Am Chem Soc. 2010, 132: 532-539. 10.1021/ja905595k.
Walsh P, Neudecker P, Sharpe S: Structural properties and dynamic behavior of nonfibrillar oligomers formed by PrP(106-126). J Am Chem Soc. 2010, 132: 7684-7695. 10.1021/ja100431q.
Mallucci GR: Prion neurodegeneration: starts and stops at the synapse. Prion. 2009, 3: 195-201. 10.4161/pri.3.4.9981.
Scott MR, Supattapone S, Nguyen HO, DeArmond SJ, Prusiner SB: Transgenic models of prion disease. Arch Virol Suppl. 2000, 113-124.
Han H, Weinreb PH, Lansbury PT: The core Alzheimer's peptide NAC forms amyloid fibrils which seed and are seeded by beta-amyloid: is NAC a common trigger or target in neurodegenerative disease?. Chem Biol. 1995, 2: 163-169. 10.1016/1074-5521(95)90071-3.
Kellett KA, Hooper NM: Prion protein and Alzheimer disease. Prion. 2009, 3: 190-194. 10.4161/pri.3.4.9980.
Eisele YS, Obermuller U, Heilbronner G, Baumann F, Kaeser SA, Wolburg H, Walker LC, Staufenbiel M, Heikenwalder M, Jucker M: Peripherally applied Abeta-containing inoculates induce cerebral beta-amyloidosis. Science. 2010, 330: 980-982. 10.1126/science.1194516.
Lee SJ, Desplats P, Sigurdson C, Tsigelny I, Masliah E: Cell-to-cell transmission of non-prion protein aggregates. Nat Rev Neurol. 2010, 6: 702-706. 10.1038/nrneurol.2010.145.
Bate C, Tayebi M, Williams A: Phospholipase A2 inhibitors protect against prion and Abeta mediated synapse degeneration. Mol Neurodegener. 2010, 5: 13-10.1186/1750-1326-5-13.
Frost B, Diamond MI: Prion-like mechanisms in neurodegenerative diseases. Nat Rev Neurosci. 2010, 11: 155-159.
Morales R, Estrada LD, Diaz-Espinoza R, Morales-Scheihing D, Jara MC, Castilla J, Soto C: Molecular cross talk between misfolded proteins in animal models of Alzheimer's and prion diseases. J Neurosci. 2010, 30: 4528-4535. 10.1523/JNEUROSCI.5924-09.2010.
Lauren J, Gimbel DA, Nygaard HB, Gilbert JW, Strittmatter SM: Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature. 2009, 457: 1128-1132. 10.1038/nature07761.
Chen S, Yadav SP, Surewicz WK: Interaction between human prion protein and amyloid-beta (Abeta) oligomers: role of N-terminal residues. J Biol Chem. 2010, 285: 26377-26383. 10.1074/jbc.M110.145516.
Freir DB, Nicoll AJ, Klyubin I, Panico S, Mc Donald JM, Risse E, Asante E, Farrow MA, Sessions RB, Saibil HR: Interaction between prion protein and toxic Aß assemblies can be therapeutically targeted at multiple sites. Nature Commun. 2011, 2: 336-10.1038/ncomms1341.
Solforosi L, Criado JR, McGavern DB, Wirz S, Sanchez-Alavez M, Sugama S, DeGiorgio LA, Volpe BT, Wiseman E, Abalos G, et al: Cross-linking cellular prion protein triggers neuronal apoptosis in vivo. Science. 2004, 303: 1514-1516. 10.1126/science.1094273.
Klohn PC, Farmer M, Linehan JM, O'Malley C, Fernandez de Marco M, Taylor W, Farrow M, Khalili-Shirazi A, Brandner S, Collinge J: PrP antibodies do not trigger mouse hippocampal neuron apoptosis. Science. 2012, 335: 52-10.1126/science.1215579.
Bate C, Williams A: Amyloid-beta-induced synapse damage is mediated via cross-linkage of cellular prion proteins. J Biol Chem. 2011, 286: 37955-37963. 10.1074/jbc.M111.248724.
You H, Tsutsui S, Hameed S, Kannanayakal TJ, Chen L, Xia P, Engbers JD, Lipton SA, Stys PK, Zamponi GW: Abeta neurotoxicity depends on interactions between copper ions, prion protein, and N-methyl-D-aspartate receptors. Proc Natl Acad Sci U S A. 2012, 109: 1737-1742. 10.1073/pnas.1110789109.
Renner M, Lacor PN, Velasco PT, Xu J, Contractor A, Klein WL, Triller A: Deleterious effects of amyloid beta oligomers acting as an extracellular scaffold for mGluR5. Neuron. 2010, 66: 739-754. 10.1016/j.neuron.2010.04.029.
Kessels HW, Nguyen LN, Nabavi S, Malinow R: The prion protein as a receptor for amyloid-beta. Nature. 2010, 466: E3-4. 10.1038/nature09217. discussion E4-5
Calella AM, Farinelli M, Nuvolone M, Mirante O, Moos R, Falsig J, Mansuy IM, Aguzzi A: Prion protein and Abeta-related synaptic toxicity impairment. EMBO Mol Med. 2010, 2: 306-314. 10.1002/emmm.201000082.
Chung E, Ji Y, Sun Y, Kascsak RJ, Kascsak RB, Mehta PD, Strittmatter SM, Wisniewski T: Anti-PrPC monoclonal antibody infusion as a novel treatment for cognitive deficits in an alzheimer's disease model mouse. BMC Neurosci. 2010, 11: 130-10.1186/1471-2202-11-130.
Gimbel DA, Nygaard HB, Coffey EE, Gunther EC, Lauren J, Gimbel ZA, Strittmatter SM: Memory impairment in transgenic Alzheimer mice requires cellular prion protein. J Neurosci. 2010, 30: 6367-6374. 10.1523/JNEUROSCI.0395-10.2010.
Balducci C, Beeg M, Stravalaci M, Bastone A, Sclip A, Biasini E, Tapella L, Colombo L, Manzoni C, Borsello T, et al: Synthetic amyloid-beta oligomers impair long-term memory independently of cellular prion protein. Proc Natl Acad Sci U S A. 2010, 107: 2295-2300. 10.1073/pnas.0911829107.
Cisse M, Sanchez PE, Kim DH, Ho K, Yu GQ, Mucke L: Ablation of cellular prion protein does not ameliorate abnormal neural network activity or cognitive dysfunction in the J20 line of human amyloid precursor protein transgenic mice. J Neurosci. 2011, 31: 10427-10431. 10.1523/JNEUROSCI.1459-11.2011.
Cisse M, Halabisky B, Harris J, Devidze N, Dubal DB, Sun B, Orr A, Lotz G, Kim DH, Hamto P, et al: Reversing EphB2 depletion rescues cognitive functions in Alzheimer model. Nature. 2011, 469: 47-52. 10.1038/nature09635.
Shankar GM, Leissring MA, Adame A, Sun X, Spooner E, Masliah E, Selkoe DJ, Lemere CA, Walsh DM: Biochemical and immunohistochemical analysis of an Alzheimer's disease mouse model reveals the presence of multiple cerebral Abeta assembly forms throughout life. Neurobiol Dis. 2009, 36: 293-302. 10.1016/j.nbd.2009.07.021.
Deshpande A, Kawai H, Metherate R, Glabe CG, Busciglio J: A role for synaptic zinc in activity-dependent Abeta oligomer formation and accumulation at excitatory synapses. J Neurosci. 2009, 29: 4004-4015. 10.1523/JNEUROSCI.5980-08.2009.
Kayed R, Canto I, Breydo L, Rasool S, Lukacsovich T, Wu J, Albay R, Pensalfini A, Yeung S, Head E, et al: Conformation dependent monoclonal antibodies distinguish different replicating strains or conformers of prefibrillar Abeta oligomers. Mol Neurodegener. 2010, 5: 57-10.1186/1750-1326-5-57.
Bieschke J, Herbst M, Wiglenda T, Friedrich RP, Boeddrich A, Schiele F, Kleckers D, Lopez del Amo JM, Gruning BA, Wang Q, et al: Small-molecule conversion of toxic oligomers to nontoxic beta-sheet-rich amyloid fibrils. Nat Chem Biol. 2012, 8: 93-101.
Acknowledgements
We thank Prof. Dominic Walsh for extensive collaboration. This research was supported by Science Foundation Ireland, the Health Research Board of Ireland and the European Commission Seventh Framework Programme (Grant Agreement MEMOLOAD 201159).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no conflict of interest to disclose.
Authors’ contributions
Both IK and MJR drafted and edited the manuscript. IK, NWH and WKC provided their experimental data. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Klyubin, I., Cullen, W.K., Hu, NW. et al. Alzheimer’s disease Aβ assemblies mediating rapid disruption of synaptic plasticity and memory. Mol Brain 5, 25 (2012). https://doi.org/10.1186/1756-6606-5-25
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1756-6606-5-25