
Abstract
Background
Although spargana, which are the plerocercoids of Spirometra erinacei, are of biological and clinical importance, expressed sequence tags (ESTs) from this parasite have not been explored. To understand molecular and biological features of this parasite, sparganum ESTs were examined by large-scale EST sequencing and multiple bioinformatics tools.
Methods
Total RNA was isolated from spargana and then ESTs were generated, assembled and sequenced. Many biological aspects of spargana were investigated using multi-step bioinformatics tools.
Results
A total of 5,634 ESTs were collected from spargana. After clustering and assembly, the functions of 1,794 Sparganum Assembled ESTs (SpAEs) including 934 contigs and 860 singletons were analyzed. A total of 1,351 (75%) SpAEs were annotated using a hybrid of BLASTX and InterProScan. Of these genes, 1,041 (58%) SpAEs had high similarity to tapeworms. In the context of the biology of sparganum, our analyses reveal: (i) a highly expressed fibronectin 1, a ubiquitous and abundant glycoprotein; (ii) up-regulation of enzymes related with glycolysis pathway; (iii) most frequent domains of protein kinase and RNA recognition motif domain; (iv) a set of helminth-parasitic and spargana-specific genes that may offer a number of antigen candidates.
Conclusions
Our transcriptomic analysis of S. erinacei spargana demonstrates biological aspects of a parasite that invades and travels through subcutaneous tissue in intermediate hosts. Future studies should include comparative analyses using combinations of transcriptome and proteome data collected from the entire life cycle of S. erinacei.
Similar content being viewed by others


Background
Spargana, the plerocercoid form of Spirometra erinacei, are the larvae of intestinal tapeworms of the order Diphyllobothriidea in the class Cestoda[1]. Sparganosis has been reported in many countries, including the United States and Europe[2]. Human sparganosis occasionally occurs by ingestion of water contaminated with Copepods that have been infected with procercoids or by invasion of plerocercoids from hosts such as frogs and snakes.
The ingested sparganum has the ability to invade various organs, which include eyes, subcutaneous tissues, abdominal walls, brains, spinal cords, lungs, and breasts, among others[3–5]. Human sparganosis can cause diverse symptoms, such as non-specific irritation, uncertain pain, apparent masses, and headaches. Although radiologic examinations have been introduced, using techniques such as ultrasonography, CT, and MR, it is difficult to confirm a correct diagnosis. Because expensive equipment and experts are necessary, this approach is not appropriate as a practical method for field diagnosis. Furthermore, sparganosis cannot even be deciphered by autopsy because of restrictions, which include many latent infections, unexpected locations of the worm in the body and a low predicted infection rate[6].
Serodiagnostic tests using sparganum antigen proteins could be good alternative techniques for diagnosing sparganosis. These tests include enzyme-linked immunosorbent assays (ELISA)[7] and immunoblotting[8]. Several antigenic proteases are reportedly present in spargana, including 31/36 kDa excretory-secretory (ES) proteins[9], a 27 kDa cathepsin S-like protease[10], and a 53 kDa neutral protease[11]. ES proteins in crude extracts have been shown to be highly specific and sensitive in sera from patients with sparganosis. However, preparation of sufficient amounts of ES proteins is labor-intensive and time-consuming[12]. Therefore, recombinant antigens were employed to overcome the disadvantages of ES protein preparation. Recently, multiple antigen mixtures using combinations of these antigenic proteins have been recommended because an absolute antigen with high sensitivity and specificity does not yet exist[13].
As mentioned above, the first definitive treatment is surgical resection of the worm from the infected tissues. The second choice for treating sparganosis is two drugs, praziquantel or mebendazole, which are also recommended for treatment of trematode or nematode infections, respectively[14, 15]. Although these drugs are currently orally administrated for treatment, low cure rates and high recurrence rates have already been observed[16, 17]. Because novel therapeutic targets against sparganosis are not studied, with the exception of these drugs, development of anti-helminthics should be actively encouraged.
Large-scale sequencing data can be applied to gene-based discovery of drug targets and diagnostic antigens[18]. Recently, genomes or transcriptomes from other cestode parasites have been sequenced and functionally analyzed, including data from Taenia solium[19–21], Echinococcus multilocularis[21], E. granulosus[21, 22] and Hymenolepis microstoma[21]. This genetic information has been applied to understanding a number of metabolic mechanisms used for parasite growth and during host-parasite interactions. Furthermore, monitoring fluctuations in gene expression is indispensable for finding drug targets, predicting secretory proteins, and elucidating evolutionary relationships[18, 21, 23]. Currently, however, knowledge regarding the genome or transcriptome of various developmental stages in S. erinacei is restricted to adult worms.
In this study, a major expressed sequence tags (ESTs) sequencing project on S. erinacei spargana was carried out to improve a basic genetic resource. This transcriptome profile is presented with the abundant transcripts, frequently occurring functional domains and antigen candidates.
Methods
Sample collection
Spargana of S. erinacei were collected from naturally infected Rhabdophis tigrinus snakes in Gyeong-sangnam-do province, South Korea. All worms were washed with physiological saline several times and either used directly for RNA preparation or stored at -70°C until use.
RNA isolation and cDNA library construction
After separating the mycelia from S. erinacei spargana, the worms were submerged in liquid nitrogen in pre-chilled grinding jars and a grinding ball on a bed of dry ice. Spargana in pre-chilled grinding jars were pulverized using a Mixer Mill MM301 (Retsch GmbH, Germany). Spargana were transferred to 15 ml polypropylene tubes filled with liquid nitrogen and stored at -80°C. Total RNA was extracted from the fragmented frozen tissues using TRI reagent (MRCgene, OH, USA). Total RNA was purified (100 μg) using the absolutely mRNA Purification Kit (Stratagene, CA, USA) according to the manufacturer’s instructions. To construct the cDNA library, a directional λ ZAP cDNA synthesis/Gigapack III gold cloning kit (Stratagene, CA, USA) was used. Reverse transcription of mRNA for first stand cDNA synthesis was primed from the poly-A tail using an oligo-dT linker primer containing an Xho I cloning site. Following second strand synthesis, EcoR I linkers were ligated to the 5′-termini. Xho I digestion released the Eco RI adapter and residual linker primer from the 3′ end of the cDNA. These two fragments were separated on a drip column containing Sepharose® CL-2B gel filtration medium. The fractionated cDNA (above 500 bp) was then precipitated and ligated into the ZAP Express vector (pBK-CMV). The primary library was produced by in vitro packaging of the ligation product with a ZAP Express cDNA Gigapack III Gold cloning Kit.
cDNA sequencing
cDNA clones were plated onto LB-kanamycin plates (Rectangle, 23.5 cm × 23.5 cm) with X-gal and IPTG for blue/white selection. White colonies were randomly and manually picked, inoculated into 15 384-well plates (Corning, NY, USA) containing 40 μl TB/kanamycin and incubated for 16 h at 37°C with fixation culture. Sequences of the cDNA inserts were determined from the 5′ end of clones using the BigDye Terminator Sequencing Kit ver. 3.1 (Applied Biosystems, Foster City, CA, USA) and a 3730XL DNA analyzer (Applied Biosystems).
EST cleaning and clustering
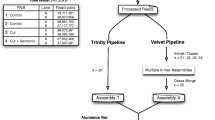
The ESTs were initially analyzed and annotated using PESTAS, an automated EST analysis platform[24]. In our study, the analysis pipeline consisted of three steps (Figure 1). In step I, EST trace data from S. erinacei sparganum were base-called from trace chromatogram data using Phred quality scores of 13[25, 26]. The sequences were then processed with Cross_Match (http://www.phrap.org), RepeatMasker (http://www.repeatmasker.org/) and SeqClean (http://seqclean.sourceforge.net/) to filter out sequences from vectors, E. coli, repetitive elements and mitochondrial DNA. Trimmed sequences over 100 bp in length were clustered and assembled into putative unique EST objects by TGICL[27] and CAP3[28], using the default options.

Main workflow for analysis. Outlay of analysis steps performed for Spirometra erinacei ESTs data. External programs used for analysis are shown where appropriate. ESTs were pre-processed and subjected to clustering and assembly (A). Singlets and contigs were examined for homology (B), screened for secretory antigen candidates (C) and compared with other species at the whole transcriptome scale (D).
Homology search and functional annotation
To assign putative functions to S. erinacei ESTs, we took into account the BLASTX best hit descriptions and subsequent alignments with E-value cutoffs below 1e-10 and compared them to the non-redundant (NR) protein database at NCBI. Because a large portion of these ESTs have not yet been annotated, we further characterized domains/families of the SpAEs using InterPro database version 27 (HMMPfam, HMMSmart, HMMTigr, HMMPanther and SuperFamily; flagged as true by InterProScan with E-value ≤ 1e-4)[29]. We also classified our SpAEs with Gene Ontology (GO) terms at the protein level using BLAST2GO (cut-off E-value ≤ 1e-10)[30]. These GO terms were further mapped and classified at the third level to two GO categories: ‘molecular function,’ and ‘biological process.’ Because some predicted proteins were assigned to more than one GO term, the percentages of each category add up to one hundred percent. SpAEs also were mapped to the Enzyme Commission (EC) database via BLAST2GO.
Comparative transcriptome analysis
Gene sequences of spargana were globally compared to those of other species using TBLASTX (E-value 1e-5) and the results were displayed using the SimiTri program (BLAST score cut-off score: 50)[31]. Sequences of the comparator species were downloaded from the GenBank databases.
Secretome analysis
From the ORFs inferred from SpAEs, secreted proteins were predicted using a combination of four programs (ORFpredictor[32], SignalP[33], TMHMM[33] and YLoc[34]) to minimize the number of false positive predictions. Firstly, we identified protein-coding regions of ORFs in SpAEs by starting exactly at the initiation codon encoding the amino acid methionine (Met) with ORFpredictor. Secondly, SignalP 3.0 was used to predict the presence of secretory signal peptides and signal anchors for each predicted SpAE protein, using both neural networks and Hidden Markov models with default option. To exclude erroneous predictions of putative transmembrane (TM) sequences as signal sequences, TMHMM, a membrane topology prediction program, was applied. We further validated the list of secreted proteins with extracellular localization using YLoc.
Results and discussion
Overview of sparganum EST analysis
Of the 5,760 clones sequenced, a total of 5,634 high-quality ESTs (an average read length of 687 bp) were obtained with a 97.8% sequencing success rate, after trimming vector contamination and low quality bases and eliminating trimmed sequences less than 100 bp in length. A total of 1,794 SpAEs (Sparganum Assembled ESTs, average read length of 715 bp) were obtained after clustering a set of 5,634 ESTs (Figure 1A). The set of SpAEs is comprised of 934 contigs and 860 singletons (Table 1). Average sequence lengths for the contigs and singletons were 764 bp and 661 bp, respectively. The contigs were mostly composed of two to six ESTs (Figure 2), with a maximum of 164 different ESTs in a single contig (Additional file1: Table S1). All trimmed ESTs were deposited into the NCBI GenBank with continuous accession numbers of HS514072-HS519705.

Distribution of ESTs within contigs after clustering the 5,634 sequences using CAP3.
Functional annotation of SpAEs
To identify likely S. erinacei sparganum genes through sequence similarity, BLASTX analyses and InterProScan domain searches were performed on all SpAEs against the NCBI NR protein databases and the InterProScan database (Figure 1B). The two alignment algorithms were used to annotate 1,351 SpAEs (75%), and most matches were to tapeworms, such as E. granulosus and H. microstoma (Additional file2: Figure S1). After removing redundant protein hits, 1,335 unique reference proteins were identified within public databases. Among them, 1,268 (95%) of the annotated SpAEs had E-values of ≤ 1e-10 (Additional file1: Table S1). In our study, 443 SpAEs (30%) did not share sequence similarity with any other predicted or known molecules in public databases. These SpAEs potentially represent novel genes with unknown functions in S. erinacei spargana.
Gene ontology
Annotation of EST-derived sparganum genes was implemented on the basis of existing annotation available in public databases. These annotations followed gene ontology (GO) vocabularies for organization into two categories representing biological processes and molecular functions[35]. In our study, 977 of the total 1,794 SpAEs could be assigned to biological process (BP) and molecular function (MF) GO classifications through BLAST2GO[30]. All of the SpAEs defined in the GO database could be assigned to more than one ontology. Of the 977 SpAEs mapped with GO terms below level 3, 669 SpAEs had BP annotation and 825 SpAEs had MF annotation. Among genes annotated with BPs, the most highly scored categories were Cellular macromolecule metabolic process (GO:0044260, 31.83%), Cellular protein metabolic process (GO:0044267, 24.51%), Gene expression (GO:0010467, 19.13%) and Translation (GO:0006412, 12.25%). The largest proportion of MFs for the SpAEs were involved in ATP binding (GO:0005524, 12.12%), Purine ribonucleoside binding (GO:0032550, 16.36%), Purine ribonucleoside triphosphate binding (GO:0035639, 16.36%) and Nucleoside phosphate binding (GO:1901265, 22.54%) (Table 2). Spargana grow into their adult stages in the final host. To achieve this developmental transition, various proteins, such as structural proteins or metabolic proteins, should be produced through translation. Both BP and MF exhibited high ranked GO categories that elucidated physiological features of spargana, including protein synthesis, protein transport, and protein regulation.
Highly abundant genes
We determined, as highly abundant genes, SpAEs with more than fourteen ESTs in one contig after exclusion of ribosomal RNA and mitochondrial genes (Table 3). In an attempt to characterize highly expressed genes, there were active components in the metabolism of the parasite, including fructose-bisphosphate aldolase (FBA) and glyceraldehyde-3-phosphate dehydrogenase. Their up-regulation may be required for high metabolic activity during development[18]. Plerocercoid growth factor/cysteine protease and signal peptidase complex subunit 3 also were found, of which cysteine proteinase has been previously investigated for their role in parasite-host relationship[36]. In our study, fibronectin 1 (FN1), which was represented by 164 ESTs, was the most frequently expressed gene. FN is a ubiquitous and abundant glycoprotein. FN consists of three discrete domains composed of FN1, FN2, and FN3. Interaction of FN with different receptors is important for mediating cellular adhesion and migration processes such as embryonic development and wound healing[37]. FN can also modulate host defenses by binding to immunoglobulin molecules like IgG and immobilizing them on a solid matrix[38]. Although FN functions are poorly studied in parasites, it is speculated that FN provides a structural basis for cell adhesion, transduces signals for cell proliferation and apoptosis, and serves for defenses against the host[38, 39].
A parasite should adapt to a variety of biological stresses in the host environment, including thermal shock, oxidative stress and other forms of stress[40]. Hence, proteins that allow spargana to survive stresses are important components for infection establishment. We found stress response-related proteins, such as HSP70, HSP40, HSP90, HSP71, HSP105, HSP60 and HSPA8. HSPs are highly conserved and abundant proteins in many parasitic organisms[21, 41, 42] and are essential for cellular viability and activity under both normal and stress conditions[43]. The top 3 most abundant genes are HSP70 (55 reads), HSP40 (47 reads) and HSP90 (24 reads). It has been previously observed that HSP70 and HSP80 in T. solium cysticerci were highly induced under temperature stress[44]. Recently, expansion of HSP70 was described in tapeworms and points out the importance of such proteins for the parasite life cycle. HSP40 gets involved in the prevention of protein aggregation and the regulation of protein refolding for parasitic development[45]. HSP90 functions downstream of the HSP70/HSP40-chaperone system and serves as an important determinant in regulating protein conformation and cell signal transduction[46].
Abundant domains
A comparison of SpAEs with the Pfam domain database[47] was performed to determine representation of protein families, domains, and functional sites in the sparganum. This analysis revealed matches to 614 unique protein domain families. The Pfam domain families with the most frequent representation in the SpAEs are presented in Table 4. These findings are similar with the result of Parkinson et al.[22], who showed that RNA recognition motif (PF00076), EF-hand domain pair (PF13499) and WD40 repeat (PF00400) were constantly abundant across the Lophotrochozoa. They also reported that dynein light chain (PF01221) and tetraspanin/peripherin (PF00335) appeared expanded in both cestode and trematode. In our study, the most abundant protein motifs were protein kinase domain (PF00069), followed by RNA recognition motif. Protein kinases mediate many other cellular processes including metabolism and transcription and protein kinase domains were consistently abundant in platyhelminthes except for Echinococcus species[22, 48]. Additionally, there were various functional domains that were involved in structural, regulatory and developmental activities.
Key enzymes
GO terms derived from the predicted proteins were mapped to Enzyme Commission (EC) numbers. In our study, a total of 162 SpAEs were assigned to 87 unique EC numbers. The top 10 highly represented EC numbers are shown in Table 5. The largest cluster corresponded to 36 ESTs for glyceraldehyde 3-phosphate dehydrogenase (GAPDH), which on the surface of Trichomonas vaginalis has been suggested may play a crucial role in providing the parasite with a survival advantage[49]. In addition, we found several enzymes related to glycolysis involving malate dehydrogenase, enolase and FBA. Most parasites utilize glucose and galactose as the main energy sources for a major anaerobic and a minor aerobic respiratory metabolism[50]. Glycolytic enzymes are crucial for the survival and pathogenicity of parasites and thereby have been considered as potential drug targets against protozoan parasites[51–54]. If the parasitic enzymes are highly conserved with human homologs, specificity between parasite and host can be solved using the ability of therapeutic chemistry, combined with new structural features that the enzyme catalytic domains show important parasite-specific structural differences[55, 56] The second-largest cluster was comprised of 35 ESTs for ATP dependent RNA helicase DDX 1 (DEAD box protein 1), which has been identified as essential for parasitic survival[57].
Diagnostic candidate genes based on secretome analysis
ES proteins or other proteins predicted to be expressed on the cell surface have been proposed as diagnostic candidates[58, 59]. Thus, proteins inferred from the sparganum transcriptome were screened for signal peptide and transmembrane domains to find potentially exported proteins. We conducted an analysis of open reading frames (ORFs) containing an N-terminal signal peptide by using multiple bioinformatic tools, such as ORFpredictor, SignalP, TMHMM, and YLoc. A total of 39 SpAEs contained ORFs with extracellular localization sequences (Table 6). The dataset was divided into sequences that were novel and sequences that were found across different phyla. Novel sequences constituted approximately 50% of the total. These genes with no previously identified homologs in other organisms could be particularly intriguing for the development of diagnostic candidates because the lack of host homologs improves the expectation of therapeutic safety and efficacy.
Transcriptome-wide comparison and parasitism
To investigate the relative similarity between spargana and four parasitic flatworms and a free-living one, TBLASTX was performed against other organisms with publicly available ESTs and the degree of similarity was figuratively displayed using SimiTri program[31]. These included Taenia solium (30,587 ESTs) and Echinococcus granulosus (10,091 ESTs), Clonorchis sinensis (13,305 ESTs)[60] and Schistosoma japonicum (24,796 ESTs) and Schmidtea mediterranea (78,720 ESTs). Spargana (1,794 SpAEs) was more close to T. solium than E. granulosus (Figure 3A). This result showed the phylogenetic closeness within Eucestoda of class Cestoda. Evolutionary relationships of tapeworms represent a monophyletic group based on small (SSU) and large (LSU) subunit ribosomal DNA sequences and morphological characteristics[61]. S. erinacei (Cestoda, Pseudophyllidea) is sister group to Taenia sp. (Cestoda, Cyclophyllidea) while E. granulosus (Cestoda, Cyclophyllidea) forms a group with Gyrocotyle rugosa (Gyrocotylidea)[62]. When compared to both C. sinensis and S. japonicum (Trematoda, Digenea), SpAEs were scattered across two flukes’ transcriptomes (Figure 3B). Comparison of Pseudophyllidea with Digenea encompasses diversity across the parasitic Neodermata including Cestoda and Trematoda[63].

Transcriptome-wide relative similarity between sparganum and other species. Spargana contigs and singlets were searched against the whole transcriptome using TBLASTX score (a cut-off of ≥50). The Venn diagrams show the number of spargana sequences associated with each dataset. Global similarity comparison of cestoda (A) and trematoda (B) with a free-living flatworm. Square tiles indicate genes, with the squares colored by their highest TBLASTX score to each of the databases: red ≥300; yellow ≥200; green ≥150, blue ≥100 and purple <100.
We identified 28 SpAEs, which were predicted to be helminth-parasitic genes in the intersection between cestode-parasitic genes (a) and trematode-parasitic genes (b) in the Figure 3 (Additional file3: Table S2). These proteins in parasitic helminth were absent from the corresponding molecules in the free-living S. mediterranea (Turbellaria, outside of Neodermata)[64]. Of these, 9 showed sequence similarity neither to a gene/protein of known function nor to an identifiable protein domain. Due to the presence of these gene products only within parasitic helminths, and although their full characterization is needed, they may be good candidates for the development of potentially novel parasitic helminth drug targets. From the BLAST analyses, 537 SpAEs did not have any homologs in the analyzed species (Additional file4: Table S3). These gene products can be explored as potential species-specific antigen candidates against sparganosis.
Conclusions
This study is the first to analyze and characterize the transcriptome of S. erinacei spargana. This project provides an all-inclusive overview and preliminary analyses for genomic research on S. erinacei spargana and is a useful starting point for gene discovery, new drug development, novel antigen identification, and comparative analyses of genomes. In addition, this study will help facilitate whole genome sequencing and annotation.
References
Kuchta R, Scholz T, Brabec J, Bray RA: Suppression of the tapeworm order pseudophyllidea (platyhelminthes: eucestoda) and the proposal of two new orders, bothriocephalidea and diphyllobothriidea. Int J Parasitol. 2008, 38: 49-55.
Cummings TJ, Madden JF, Gray L, Friedman AH, McLendon RE: Parasitic lesion of the insula suggesting cerebral sparganosis: case report. Neuroradiology. 2000, 42: 206-208.
Song T, Wang WS, Zhou BR, Mai WW, Li ZZ, Guo HC, Zhou F: CT and MR characteristics of cerebral sparganosis. AJNR Am J Neuroradiol. 2007, 28: 1700-1705.
Wiwanitkit V: A review of human sparganosis in Thailand. Int J Infect Dis. 2005, 9: 312-316.
Otranto D, Eberhard ML: Zoonotic helminths affecting the human eye. Parasit Vectors. 2011, 4: 41
Kong Y, Cho SY, Kang WS: Sparganum infections in normal adult population and epileptic patients in Korea: a seroepidemiologic observation. Korean J Parasitol. 1994, 32: 85-92.
Kim H, Kim SI, Cho SY: Serological diagnosis of human sparganosis by means of micro-ELISA. Kisaengchunghak Chapchi. 1984, 22: 222-228.
Choi SH, Kang SY, Kong Y, Cho SY: Antigenic protein fractions reacting with sera of sparganosis patients. Kisaengchunghak Chapchi. 1988, 26: 163-167.
Cho SY, Chung YB, Kong Y: Component proteins and protease activities in excretory-secretory product of sparganum. Kisaengchunghak Chapchi. 1992, 30: 227-230.
Kong Y, Chung YB, Cho SY, Kang SY: Cleavage of immunoglobulin G by excretory-secretory cathepsin S-like protease of spirometra mansoni plerocercoid. Parasitology. 1994, 109 (Pt 5): 611-621.
Kong Y, Kang SY, Kim SH, Chung YB, Cho SY: A neutral cysteine protease of Spirometra mansoni plerocercoid invoking an IgE response. Parasitology. 1997, 114 (Pt 3): 263-271.
Choi MH, Park IC, Li S, Hong ST: Excretory-secretory antigen is better than crude antigen for the serodiagnosis of clonorchiasis by ELISA. Korean J Parasitol. 2003, 41: 35-39.
Lee JY, Kim TY, Gan XX, Kang SY, Hong SJ: Use of a recombinant clonorchis sinensis pore-forming peptide, clonorin, for serological diagnosis of clonorchiasis. Parasitol Int. 2003, 52: 175-178.
Kim TI, Yoo WG, Li S, Hong ST, Keiser J, Hong SJ: Efficacy of artesunate and artemether against clonorchis sinensis in rabbits. Parasitol Res. 2009, 106: 153-156.
Seah SK: Mebendazole in the treatment of helminthiasis. Can Med Assoc J. 1976, 115: 777-779.
Chai J, Yu J, Lee S, SI K, Cho S: Ineffectiveness of praziquantel treatment for human sparganosis (a case report). Seoul J Med. 1988, 29: 397-399.
Lee S, Chai J, Sohn W, Hong S, Lee K: In vitro and in vivo effects of praziquantel on sparganum. Seoul J Med. 1986, 27: 135-142.
Lizotte-Waniewski M, Tawe W, Guiliano DB, Lu W, Liu J, Williams SA, Lustigman S: Identification of potential vaccine and drug target candidates by expressed sequence tag analysis and immunoscreening of onchocerca volvulus larval cDNA libraries. Infect Immun. 2000, 68: 3491-3501.
Aguilar-Diaz H, Bobes RJ, Carrero JC, Camacho-Carranza R, Cervantes C, Cevallos MA, Davila G, Rodriguez-Dorantes M, Escobedo G, Fernandez JL, Fragoso G, Gaytan P, Garciarubio A, Gonzalez VM, Gonzalez L, Jose MV, Jimenez L, Laclette JP, Landa A, Larralde C, Morales-Montor J, Morett E, Ostoa-Saloma P, Sciutto E, Santamaria RI, Soberon X, de la Torre P, Valdes V, Yanez J: The genome project of Taenia solium. Parasitol Int. 2006, 55 (Suppl): S127-S130.
Almeida CR, Stoco PH, Wagner G, Sincero TC, Rotava G, Bayer-Santos E, Rodrigues JB, Sperandio MM, Maia AA, Ojopi EP, Zaha A, Ferreira HB, Tyler KM, Davila AM, Grisard EC, Dias-Neto E: Transcriptome analysis of taenia solium cysticerci using open reading frame ESTs (ORESTES). Parasit Vectors. 2009, 2: 35-
Tsai IJ, Zarowiecki M, Holroyd N, Garciarrubio A, Sanchez-Flores A, Brooks KL, Tracey A, Bobes RJ, Fragoso G, Sciutto E, Aslett M, Beasley H, Bennett HM, Cai J, Camicia F, Clark R, Cucher M, De Silva N, Day TA, Deplazes P, Estrada K, Fernandez C, Holland PW, Hou J, Hu S, Huckvale T, Hung SS, Kamenetzky L, Keane JA, Kiss F: The genomes of four tapeworm species reveal adaptations to parasitism. Nature. 2013, 496: 57-63.
Parkinson J, Wasmuth JD, Salinas G, Bizarro CV, Sanford C, Berriman M, Ferreira HB, Zaha A, Blaxter ML, Maizels RM, Fernandez C: A transcriptomic analysis of echinococcus granulosus larval stages: implications for parasite biology and host adaptation. PLoS Negl Trop Dis. 2012, 6: e1897-
Rosenzvit MC, Zhang W, Motazedian H, Smyth D, Pearson M, Loukas A, Jones MK, McManus DP: Identification of membrane-bound and secreted proteins from echinococcus granulosus by signal sequence trap. Int J Parasitol. 2006, 36: 123-130.
Nam SH, Kim DW, Jung TS, Choi YS, Choi HS, Choi SH, Park HS: PESTAS: a web server for EST analysis and sequence mining. Bioinformatics. 2009, 25: 1846-1848.
Ewing B, Green P: Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res. 1998, 8: 186-194.
Ewing B, Hillier L, Wendl MC, Green P: Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res. 1998, 8: 175-185.
Pertea G, Huang X, Liang F, Antonescu V, Sultana R, Karamycheva S, Lee Y, White J, Cheung F, Parvizi B, Tsai J, Quackenbush J: TIGR gene indices clustering tools (TGICL): a software system for fast clustering of large EST datasets. Bioinformatics. 2003, 19: 651-652.
Huang X, Madan A: CAP3: A DNA sequence assembly program. Genome Res. 1999, 9: 868-877.
Hunter S, Apweiler R, Attwood TK, Bairoch A, Bateman A, Binns D, Bork P, Das U, Daugherty L, Duquenne L, Finn RD, Gough J, Haft D, Hulo N, Kahn D, Kelly E, Laugraud A, Letunic I, Lonsdale D, Lopez R, Madera M, Maslen J, McAnulla C, McDowall J, Mistry J, Mitchell A, Mulder N, Natale D, Orengo C, Quinn AF: InterPro: the integrative protein signature database. Nucleic Acids Res. 2009, 37: D211-D215.
Conesa A, Gotz S, Garcia-Gomez JM, Terol J, Talon M, Robles M: Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005, 21: 3674-3676.
Parkinson J, Blaxter M: SimiTri–visualizing similarity relationships for groups of sequences. Bioinformatics. 2003, 19: 390-395.
Min XJ, Butler G, Storms R, Tsang A: OrfPredictor: predicting protein-coding regions in EST-derived sequences. Nucleic Acids Res. 2005, 33: W677-W680.
Chen Y, Yu P, Luo J, Jiang Y: Secreted protein prediction system combining CJ-SPHMM, TMHMM, and PSORT. Mamm Genome. 2003, 14: 859-865.
Briesemeister S, Rahnenfuhrer J, Kohlbacher O: YLoc--an interpretable web server for predicting subcellular localization. Nucleic Acids Res. 2010, 38 (Suppl): W497-W502.
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G: Gene ontology: tool for the unification of biology. The gene ontology consortium. Nat Genet. 2000, 25: 25-29.
Kang JM, Lee KH, Sohn WM, Na BK: Identification and functional characterization of CsStefin-1, a cysteine protease inhibitor of clonorchis sinensis. Mol Biochem Parasitol. 2011, 177: 126-134.
Pankov R, Yamada KM: Fibronectin at a glance. J Cell Sci. 2002, 115: 3861-3863.
Rostagno AA, Frangione B, Gold LI: Biochemical characterization of the fibronectin binding sites for IgG. J Immunol. 1989, 143: 3277-3282.
Leiss M, Beckmann K, Giros A, Costell M, Fassler R: The role of integrin binding sites in fibronectin matrix assembly in vivo. Curr Opin Cell Biol. 2008, 20: 502-507.
Young RA, Elliott TJ: Stress proteins, infection, and immune surveillance. Cell. 1989, 59: 5-8.
Newport G, Culpepper J, Agabian N: Parasite heat-shock proteins. Parasitol Today. 1988, 4: 306-312.
Yoo WG, Kim DW, Ju JW, Cho PY, Kim TI, Cho SH, Choi SH, Park HS, Kim TS, Hong SJ: Developmental transcriptomic features of the carcinogenic liver fluke, clonorchis sinensis. PLoS Negl Trop Dis. 2011, 5: e1208-
Hedstrom R, Culpepper J, Harrison RA, Agabian N, Newport G: A major immunogen in schistosoma mansoni infections is homologous to the heat-shock protein Hsp70. J Exp Med. 1987, 165: 1430-1435.
Vargas-Parada L, Solis CF, Laclette JP: Heat shock and stress response of taenia solium and T. Crassiceps (cestoda). Parasitology. 2001, 122: 583-588.
Coskun KA, Ozgur A, Otag B, Mungan M, Tutar Y: Heat shock protein 40-Gok1 isolation from toxoplasma gondii RH strain. Protein Peptide Letters. 2013, 20: 1294-1301.
Fabbri E, Valbonesi P, Franzellitti S: HSP expression in bivalves. Invertebr Surviv J. 2008, 5: 135-161.
Sonnhammer EL, Eddy SR, Durbin R: Pfam: a comprehensive database of protein domain families based on seed alignments. Proteins. 1997, 28: 405-420.
Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S: The protein kinase complement of the human genome. Science. 2002, 298: 1912-1934.
Lama A, Kucknoor A, Mundodi V, Alderete JF: Glyceraldehyde-3-phosphate dehydrogenase is a surface-associated, fibronectin-binding protein of trichomonas vaginalis. Infect Immun. 2009, 77: 2703-2711.
Bryant C: The regulation of respiratory metabolism in parasitic helminths. Adv Parasitol. 1978, 16: 311-331.
Wanidworanun C, Nagel RL, Shear HL: Antisense oligonucleotides targeting malarial aldolase inhibit the asexual erythrocytic stages of plasmodium falciparum. Mol Biochem Parasitol. 1999, 102: 91-101.
Chan M, Sim TS: Functional characterization of an alternative [lactate dehydrogenase-like] malate dehydrogenase in plasmodium falciparum. Parasitol Res. 2004, 92: 43-47.
Galkin A, Kulakova L, Melamud E, Li L, Wu C, Mariano P, Dunaway-Mariano D, Nash TE, Herzberg O: Characterization, kinetics, and crystal structures of fructose-1,6-bisphosphate aldolase from the human parasite, Giardia lamblia. J Biol Chem. 2007, 282: 4859-4867.
Caceres AJ, Michels PA, Hannaert V: Genetic validation of aldolase and glyceraldehyde-3-phosphate dehydrogenase as drug targets in trypanosoma brucei. Mol Biochem Parasitol. 2010, 169: 50-54.
de Koning HP, Gould MK, Sterk GJ, Tenor H, Kunz S, Luginbuehl E, Seebeck T: Pharmacological validation of trypanosoma brucei phosphodiesterases as novel drug targets. J Infect Dis. 2012, 206: 229-237.
Wang H, Yan Z, Geng J, Kunz S, Seebeck T, Ke H: Crystal structure of the leishmania major phosphodiesterase LmjPDEB1 and insight into the design of the parasite-selective inhibitors. Mol Microbiol. 2007, 66: 1029-1038.
Young ND, Jex AR, Li B, Liu S, Yang L, Xiong Z, Li Y, Cantacessi C, Hall RS, Xu X, Zerlotini A, Oliveira G, Hofmann A, Zhang G, Fang X, Kang Y, Campbell BE, Loukas A, Ranganathan S, Rollinson D, Rinaldi G, Brindley PJ, Yang H, Wang J, Wang J, Gasser RB: Whole-genome sequence of schistosoma haematobium. Nat Genet. 2012, 44: 221-225.
Dalton JP, Brindley PJ, Knox DP, Brady CP, Hotez PJ, Donnelly S, O’Neill SM, Mulcahy G, Loukas A: Helminth vaccines: from mining genomic information for vaccine targets to systems used for protein expression. Int J Parasitol. 2003, 33: 621-640.
Geary JF, Satti MZ, Moreno Y, Madrill N, Whitten D, Headley SA, Agnew D, Geary TG, Mackenzie CD: First analysis of the secretome of the canine heartworm, dirofilaria immitis. Parasit Vectors. 2012, 5: 140-
Venn diagrams.http://pathod.cdc.go.kr/index.jsp.
Olson PD, Littlewood DT, Bray RA, Mariaux J: Interrelationships and evolution of the tapeworms (platyhelminthes: cestoda). Mol Phylogenet Evol. 2001, 19: 443-467.
Campos A, Cummings MP, Reyes JL, Laclette JP: Phylogenetic relationships of platyhelminthes based on 18S ribosomal gene sequences. Mol Phylogenet Evol. 1998, 10: 1-10.
Littlewood DTJ: Parasitic flatworms: molecular biology, biochemistry, immunology and physiology. 2006, Wallingford (UK): CABI Publishing, 1-36.
Carranza S, Baguna J, Riutort M: Are the platyhelminthes a monophyletic primitive group? An assessment using 18S rDNA sequences. Mol Biol Evol. 1997, 14: 485-497.
Acknowledgements
This work was supported by grant 2006-N54002-00 and the Pathogenic Proteome Management Program (4800-4847-300) from the Korea National Institute of Health, Korea Centers for Disease Control and Prevention.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The author declare that he have no competing interests.
Authors’ contributions
DWK and JWJ contributed to the conception and design of the project, DWK, MRL and WGY conducted laboratory experiments (EST libraries), designed bioinformatics scripts, performed analyses and interpretations of the data and drafted the manuscript. SHC and WJL helped conceive the project and participated in its coordination. YJK and HWY conducted laboratory experiments (EST libraries) and helped design bioinformatics scripts. All authors read, helped to edit, and approved the final manuscript.
Dae-Won Kim, Won Gi Yoo, Myoung-Ro Lee contributed equally to this work.
Electronic supplementary material
13071_2014_1634_MOESM1_ESM.xlsx
Additional file 1: Table S1.: Functional annotation of 1,794 SpAEs. This file contains BLASTX hits for each SpAE through BLAST2GO. (XLSX 255 KB)
13071_2014_1634_MOESM3_ESM.xls
Additional file 3: Table S2.: SpAEs of S. erinacei spargana of potential genes associated with helminth parasitism. (XLS 105 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Kim, DW., Yoo, W.G., Lee, MR. et al. Transcriptome sequencing and analysis of the zoonotic parasite Spirometra erinacei spargana (plerocercoids). Parasites Vectors 7, 368 (2014). https://doi.org/10.1186/1756-3305-7-368
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1756-3305-7-368