
Abstract
Background
The apicomplexan parasite Toxoplasma gondii can infect and replicate in virtually any nucleated cell in many species of warm-blooded animals; T. gondii has elaborate mechanisms to counteract host-cell apoptosis in order to maintain survival and breed in the host cells.
Methods
Using microarray profiling and a combination of conventional molecular approaches, we investigated the levels of microRNAs (miRNAs ) in human macrophage during T. gondii infection. We used molecular tools to examine Toxoplasma-upregualted miRNAs to revealed potential signal transducers and activators of transcription 3(STAT3) binding sites in the promoter elements of a subset of miRNA genes. We analysed the apoptosis of human macrophage with the functional inhibition of the STAT3-binding miRNAs by flow cytometry.
Results
Our results demonstrated differential alterations in the mature miRNA expression profile in human macrophage following T. gondii infection. Database analysis of Toxoplasma-upregulated miRNAs revealed potential STAT3 binding sites in the promoter elements of a subset of miRNA genes. We demonstrated that miR-30c-1, miR-125b-2, miR-23b-27b-24-1 and miR-17 ~ 92 cluster genes were transactivated through promoter binding of the STAT3 following T. gondii infection. Importantly, functional inhibition of selected STAT3-binding miRNAs in human macropahges increased apoptosis of host cells.
Conclusions
A panel of miRNAs is regulated through promoter binding of the STAT3 in human macrophage and these miRNAs are involved in anti-apoptosis in response to T. gondii infection.
Similar content being viewed by others


Background
As a typical intracellular protozoan parasite of warm-blooded vertebrates, Toxoplasma gondii has elaborate mechanisms to counteract host-cell apoptosis in order to maintain survival and breed in the host cells [1–4]. Upon invasion of T. gondii, the parasite uses a specialized set of secretory organelles to inject parasite-derived effector molecules into its host cell [5]. These effector molecules can specifically modulate host cell gene expression to improve the ability of the parasite to infect and proliferate, via inhibition of host immune responses, and change their gene expression to escape immunologic surveillance [6]. Quantitative analysis of the host mRNAs and proteome during T. gondii infection showed that upwards of 15% of mRNAs and upregulation of 213 protein spots display altered abundance relative to uninfected cells, including crucial mRNAs and proteins that were associated with the apoptotic pathway [7, 8].
MicroRNAs (miRNAs) are endogenous small (19 ~ 24 nt long) noncoding RNAs that regulate gene expression in a sequence specific manner. This is primarily accomplished through binding to 3′UTR of target mRNAs, either targeting the transcripts for degradation or blocking their translation [9]. However, molecular mechanisms underlying miRNA gene transcriptional regulation are largely unclear. Recent studies on expression of miRNA genes have revealed potential transcriptional regulation by transcription factors, such as p53, NF-κB and MAPK [6, 10, 11]. Recently, the signal transducer and activator of the transcription (STAT) signaling pathway has emerged as a major target of exploitation by Toxoplasma. Infection of mouse bone marrow-derived macrophage induces rapid and sustained activation of signal transducers and activators of transcription 3(STAT3) [12, 13]. Although a number of miRNAs have been shown to be important components of the STAT3 in various cell types [14–16], the potential interaction between miRNAs and STAT3 in human macrophage, in which the activation of STAT3 by infection with Toxoplasma may result in distinct biological consequences, has not yet been established.
In this study, we provide evidence that Toxoplasma-host cell interactions counteract the death of parasite-infected cells through upregulation of STAT3-binding miRNAs in human macrophage. The array analysis of miRNA exparession revealed significant alterations in miRNA expression in human macrophage following Toxoplasma infection. We report here that STAT3 mediated a prosurvival pathway by upregulation the miRNAs, leading to inhibition of host cells with Toxoplasma infection. Thus, the role of STAT3-binding miRNAs is postulated to be an important apparatus in Toxoplasma biology.
Methods
Parasites
The T. gondii strain, named TgCtwh3 with the atypical genotype China 1 (ToxoDB#9) and high virulence to mice as previously identified [17, 18], was kept in the laboratory by mouse passage. Tachyzoites were maintained by twice weekly passage on HF (human fibroblast cells) in culture medium (DMEM with 10% FCS, 100U/ml penicillin and 100 μg/ml streptomycin, GIBCO).
Human macrophage separation, culture and determination
Human PBMCs for in vitro parasite infection assays were isolated from the whole blood of 6 healthy individuals with written informed consent (2 female and 4 males, mean age 22+/-3 yrs). Ethical permission was obtained from the Institutional Review Board (IRB) of the Institute of Biomedicine at Anhui Medical University (Permit Number: AMU26-08610), which records and regulates all research activities in the school. Heparinized fresh whole blood (10 IU heparin/ml) was centrifuged against a Ficoll-Paque density gradient (density:1.077 g/ml; Solarbio) for 30 min at 2500 rpm. Peripheral blood mononuclear cells (PBMCs) were aspirated and washed in PBS before culture. The PBMCs were cultured at 37°C in 5% CO2 atmosphere at a density of 1 × 106 cells/well in DMEM medium supplemented with 10% FCS. PBMCs were used as host macrophage after induction to differentiate with 1000U/ml recombinant human GM-CSF (Preprotech) for 48 h. Macrophages were stained with FITC-conjugated anti-CD14 to determine the purity of CD14+ cells. Control and infected cells were stained with Wright-Giemsa for 8 min, rinsed with distilled water and air dried. Cell morphology was observed by light microscopy. THP-1 cells (Cell bank, CAS) were cultured in DMEM medium supplemented with 10% FCS, and treated with 20 nmol of phorbol 12-myristate 13-acetate (PMA, Sigma) to induce THP-1 cells differentiation into macrophage-like THP-1 cells [19].
MiRCURYTM LNA array analysis of miRNAs
The Exiqon (Vedbaek, Denmark) mercury LNA microRNA arrays service were used to process the samples were used. Briefly, human macrophage were exposed to TgCtwh3 for 24 h. Total RNAs from macrophage were harvested using TRIzol (Invitrogen) and miRNeasy min kit (QIAGEN) according to manual instructions. After having passed RNA quantity measurement using the NanoDrop 1000, the samples were labeled using the miRCURYTM Hy3TM/Hy5TM Power labeling kit and hybridized on the miRCURYTM LNA Array (v.16.0). Following the washing steps the slides were scanned using the Axon GenePix 4000B microarray scanner (Axon Instruments, Foster City, CA). Scanned images were then imported into GenePix Pro 6.0 software (Axon) for grid alignment and data extraction. Replicated miRNAs were averaged and miRNAs that intensities are ≥50 in all samples were chosen for calculating a normalization factor. Expressed data were normalized using the Median normalization. After normalization, differentially expressed miRNAs were identified through Volcano Plot filtering. Hierarchical clustering was performed using MEV software (v4.6, TIGR).
qRT-PCR analysis
Macrophage were exposed to TgCtwh3 or LPS (1 μg/ml) as a control for different duration (6,12 and 24 h). For qPCR analysis of mature miRNAs, total RNA was reverse-transcribed from 0.05 μg total RNAs and determined with All-in-oneTM miRNA qRT-PCR detection kit (GeneCopoeia) by Applied Biosystems 7500 real-time PCR System (Applied Biosystems). The specific primers used in this reaction were as follows: miR-19b; miR-19a; miR-20a; miR-27b; miR-30c; miR-125 (all from GeneCopoeia).
For analysis of pri-miRNAs, total RNA was isolated from cells with Trizol reagent (Ambion). RNAs were treated with DNA-free TM Kit (Ambion) to remove any remaining DNA. Comparative real-time PCR was performed by using the SYBR Green PCR Master Mix (Applied Biosystems). Specific primers for pri-miRNAs were listed in Additional file 1: Table S1.
All reactions were run in triplicate. The Ct values were analyzed using the comparative Ct (ΔΔCt) method. Normalization was performed by using the small housekeeping U6 relative to the control (non-treated cells).
Protein extraction and western blotting
To obtain total cellular lysates, transfected cells and controls were lysed in ice-cold cell lysis buffer [20 nM tris, 250 mM NaCl, 2 Mm EDTA, 0.1% Triton X-100, 0.01 mg/ml aprotinin, 0.003 mg/ml leupetin, 0.4 mM phenylmethylsulfonyl fluoride, and 4 mM NaVO4] (Sigma). The protein concentration was determined with the Bradford protein assay kit (BESTBIO) using a c-globulin standard curve. Proteins were separated by standard SDS-PAGE and transferred onto nitrocellulose membranes (Beyotime). Nonspecific binding sites were blocked using 5% dry skimmed milk, 0.2% Tween-20 in PBS (pH 7.4) for overnight at 4°C and then incubated with primary antibodies to STAT3 and GAPDH (Proteintech).
Plasmid construction and transcriptional reporter assays
For construction of a constitutively active form of human STAT3, named STAT3-C, the amino acid residues at A661 and N663 were mutated to cysteine [20], the sequences were chemically synthesized and cloned into pEZ-M02 (GeneCopeia). STAT3-WT:3′-GATGCT ACCA AT ATCCTGGT-5′; STAT3-C: 3′-GATTGC ACCTGC ATCCTGGT-5′pEZ-M02-STAT3-WT (200 ng), pEZ-M02-STAT3-C (300 ng) or empty vector was individually cotransfected into THP-1 cells, together with appropriate miR-17 ~ 92 gene cluster promoter reporter plasmids (200 ng) by using Effectene Transfection Reagent (Qiagen). In each transfection, cells were also cotransfected with pRL-CMV (Renilla luciferase reporter plasmid, Promega). Firefly and Renilla luciferases activity was assayed using a Dual-Luciferase Reporter Assay System (Promega). The magnitude of activation values was measured relatively to the levels of Firefly luciferase activity in THP-1 cells transfected with empty vectors and normalized by Renilla luciferase activity. A mutation was introduced into the STAT3 binding site (STAT3-BR-1) using the following primers: sense 5′ - TAT GTC CT T GAG AAT TCC GGA ATT TCC TG - 3′; anti-sense 5′ - TCT CAA GG A CAT AAT TGT TAA AAG TGA GG - 3′ (alterations underlined) [20]. The correct sequence of each insert was confirmed by sequencing. SiRNA. THP-1 cells were transiently transfected with STAT3 siRNA or siRNA-control (40 nM, Santa Cruz) for 24 h using SuperFectin II in vitro siRNA Transfection Reagent (PuFei Biotech) according to the manufacturer’s protocols.
Flow cytometry
Cell death following induction of apoptosis and infection with Toxoplasma was quantified by flow cytometry. Annexin-V/propidium iodide (PI) double assay was performed using the Annexin V-EGFP Apoptosis Detection Kit (BESTBIO). Infected or non-infected macrophages were washed twice with PBS. 1 × 106 cells were resuspended in 400 μl binding buffer and stained with 5 μl Annexin V-EGFP according to the manual’s recommendations. 10 μl PI was added and allowed to incubate with cells for 5 min at 4°C in the dark. Then cells were analyzed using a FACSCalibur flow cytometer (BD Biosciences).
Results
Toxoplasma infection induces alterations in miRNA expression in macrophage in vitro
Human macrophage were separated from whole blood and cultured after 48 h (Additional file 2: Figure S1A). Cells were stained with FITC-conjugated CD14 antibody and subjected to flow cytometry to determine the purity of macrophage. The percentages of CD14+ cells were 94.7 ± 0.60% after induction with GM-CSF in PBMCs (Additional file 2: Figure S1B). To determine if the parasites could infect macrophage, cells of control and infected cells were examined using Wright-Giemsa (Additional file 2: Figure S1C). After 24 h of infection, the tachyzoites can be seen in the cytoplasm of macrophage (Additional file 2: Figure S1D).
To globally assess miRNA expression in human macrophage following Toxoplasma infection, we performed a microarray analysis of mature miRNA expression in macrophage. We profiled the levels of miRNAs extracted at 24 h from uninfected and tachyzoites-infected human macrophage using miRCURYTM LNA Array (v.16.0). A total of 17 miRNAs were upregulated (p< = 0.20) following Toxoplasma infection (Figure 1A). Of the miRNAs expressed, miR-20a, miR-125, miR-19a, miR-19b, miR-27b and miR-30c expression were significantly increased (p< = 0.05) in human macropahge after exposure to Toxoplasma infection for 24 h (Figure 1A).

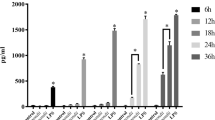
Expression profiling of mature miRNAs in macrophage following Toxoplasma infection and LPS stimulation. (A) miRNA expression profile in human macrophage following Toxoplasma infection. The left panel shows a heat-map of selected miRNAs indicating alteration in expression in macrophages following Toxoplasma infeciton.The filtered miRNA array data were subjected to unsupervised hierarchical clustering analysis. The metric was set as the Euclidean distance. The right panel shows expression miRNAs in macrophage following Toxoplasma infection. The fold change contains the ratio of mormalized intensities between Toxoplasma-infected macrophage (Group T) and non-infected macrophage (Group C). The p values are from the T test. (B) Altered expression of selected miRNAs confirmed by qRT-PCR. The amount of mature miRNAs was obtained by normalizing to the level of snRNA RNU6B in the samples. Graphics indicate those miRNAs showing an increased expression (including those significant change when p< = 0.05) in cells after treatment with LPS (n = 3) or exposure to Toxoplasma (n = 3). *, p,0.05 vs. the non-treated control.
To validate the microarray data and to specifically measure the effects of Toxoplasm a infection on miRNAs, qRT-PCR analysis using primers for mature miRNAs was performed to assess the kinetics of miRNAs in human macrophage following Toxoplasma infection. Increased expression of miR-20a, miR-125, miR-19a, miR-19b, miR-27b and miR-30c were noted in human macrophage at 6 h and 12 h postinfection, the abundance of these miRNAs significantly increased by ~23.5-fold at 24 h postinfection. The qRT-PCR analysis of miRNAs was also performed on human macrophage treated with LPS (1 mg/ml for 6 h, 12 h and 24 h) in order to determine the specificity of upregulation and expression of these miRNAs in Toxoplasma-infected cells. The results showed that increased expression of miRNAs was identified in Toxoplasma-infected cells but not in cells exposed to LPS at 24 h (p < =0.05; Figure 1B). No LPS contamination in the Toxoplasma preparation was detected using the Limulus Amebocyte Lysate (LAL) test kit (Bio-Whittaker) (data not shown).
Database analysis of upregulated miRNAs in human macropahge following Toxoplasma infection reveals potential STAT3 binding sites In their promoter elements
Differential alterations in the mature miRNA expression profile of Toxoplsma-infected human macropahge suggest that miRNA gene expression is finely controlled in macropahge in response to Toxoplasma infection. One potential mechanism for selectively altering miRNA levels is through activation of distinct intracellular signaling pathways and nuclear transcription factors. Based on TFSEARCH (http://www.cbrc.jp/research/db/TFSEARCH.html) and MOTIF (http://motif.genome.jp/) database searches, many of these miRNA genes have putative STAT3 binding sites in their potential promoter elements (Table 1). We hypothesized that activation of the STAT3 pathway is involved in the transcription of selected miRNAs upregulated by Toxoplasma. Several miRNAs upregulated in human macropahge following Toxoplasma infection are cluster miRNAs; e.g., miR-19a, miR-19b and miR-20a are from the miR-17 ~ 92 gene cluster. So we focused on determining whether STAT3 binds to the promoter and transactivates the miRNA genes upregulated by Toxoplasma infection in human macropahge.
Differential expression of primary transcripts of Toxoplasma-upregulated mature miRNAs in human macrophage
We then analyzed the kinetics of alterations of the primary transcripts (pri-miRNAs) for select mature miRNAs upregulated by Toxoplasma as listed in Table 1. Human macropahge were exposed to Toxoplasma for various periods of time and pri-miRNAs of interests were quantified by qRT-PCR (primers listed in Additional file 1: Table S1). Expression of pri-miR-30c-1, pri-miR-125b-2, pri-miR-23b-27b-24-1 and pri-miR-17 ~ 92 showed a time-dependent increase in cells following Toxoplasma infection (p< = 0.05, Figure 2). In contrast, for pri-miR-30c-2, pri-miR-125b-1, no significant increase was detected in cells after exposure to Toxoplasma infection (p > 0.05, Figure 2), suggesting a differential expression of the primary transcripts of Toxoplasma-upregulated miRNAs.

Differential expression of primary transcripts of Toxoplasma -upregulated mature miRNAs in human macrophage. Human macrophage were exposed to Toxoplasma for 6 h, 12 h and 24 h and primary transcripts (pri-miRNAs) of select miRNAs were quantified by qRT-PCR. The amount of pri-miRNAs was obtained by normalizing to the level of GAPDH in the samples. Data are expressed as the amount of pri-miRNAs in the infected samples relative to the control uninfected samples and representative of three independent experiments. *, p,0.05 vs. the non-infected control.
Promoter binding of STAT3 is required for the transcription of select miRNA genes induced by Toxoplasma in human macrophage
To test whether STAT3 is involved in Toxoplasma-induced transactivation of pri-miR-17 ~ 92 we exposed human macrophage to Toxoplasma infection in the presence of siRNA-STAT3, that prevents expression of the STAT3 (Figure 3A). siRNA-STAT3 blocked the Toxoplasma-induced increase of pri-miR-17 ~ 92 (Figure 3B). To further test the potential transactivation of miR-17 ~ 92 gene by STAT3, luciferase reporter gene constructs was used. Toxoplasma infection increased luciferase activity in cells transfected with the luciferase constructs that encompassed the binding site for STAT3 in the promoter region of miR-17 ~ 92 gene. A mutant of the STAT3 binding site blocked Toxoplasma-induced luciferase activity (Figure 3C). In addition, siRNA-STAT3 significantly inhibited the increase of luciferase activity induced by Toxoplasma infection (Figure 3D). Moreover, STAT3-associated transactivation of the miR-17 ~ 92 promoter was also confirmed by the upregulation of luciferase activity after STAT3 overexpression in the cells (Figure 3D). Together, these data demonstrate that STAT3 binding to the promoter element of the miR-17 ~ 92 gene mediates miRNAs (miR-17-5p, miR-18a, miR-19a, miR-20a, miR-19b and miR-92a) upregulation in human macrophage in response to Toxoplasma infection.

Promoter binding of STAT3 transactivates miR-17 ~ 92 gene to increase mature miR-17 ~ 92 miRNAs expression following Toxoplasma infection. (A) Western blot analysis showing stable knock-down of STAT3 by siRNA. (B) STAT3-dependent upregulation of pri-miR-17 ~ 92 in human macrophage following Toxoplasma infection. Data are presented as the relative expression level of pri-miR-17 ~ 92 in THP-1 cells following Toxoplasma infection in the presence or absence of siRNA as assessed by qRT-PCR. (C) A schematic illustration of pGL-based reporter constructs was used in dual luciferase assays to examine the transcriptional activity of the STAT3-BR in response to TgCtwh3 infection. Dotted lines indicated deleted region. (D) THP-1 cells were cotransfected with the indicated reporter constructs and Renilla luciferase plasmids. Twenty-four hours later, transcriptional activity was determined by luciferase assays. THP-1 cells with or without STAT3 knock-down by siRNA were cotransfected with the indicated reporter constructs and Renilla luciferase plasmids. Twenty-four hours later, cells were exposed to TgCtwh3 for another 24 h, followed by measurement of reporter activity by using luciferase assays. *, p,0.05 vs. non-treated cells (in B) or cells transfected with a control vector (in D) .Values are presented as mean ± SD (n =3 in A, B and D)
Functional inhibition of selected STAT3-dependent miRNAs increase apoptosis of human macrophage with Toxoplasma infection
To test whether miRNAs are involved in human macrophage inhibition of apoptosis with Toxoplasma infection, we assessed apoptosis over time in cultured macrophage transfected with various anti-miRs thereby inhibiting function of specific Toxoplasma-upregulated miRNAs. MiRNA inhibitors are commercially available, chemically modified single stranded nucleic acids designed to specifically bind to and inhibit endogenous miRNAs. Cells were transfected with specific anti-miRs (30 nM, Ambion) or a mixture of anti-miRs to miR-17-5b and miR-20a (a total of 30 nM with 15 nM for each), and then exposed to Toxoplasma for 24 h. The percentages of parasite-positive macrophage following exposure to Toxoplasma for 24 h was similar in all cultures, to those transfected with the specific anti-miRs (p > 0.05, Figure 4A), suggesting that those transfected with anti-miRs did not affect the number of parasite-infected macrophage. Cells transfected with anti-miRs to miR-17-5b and miR-20a, as well as a mixture of two anti-miRs, displayed a decreased the number of Annexin V-/PI- macrophage (viable cells) as compared to control cells. In addition, these anti-miRs increased the number of Annexin V+/PI- macrophage (early apoptosis) (p < = 0.05, Figure 4B).

Functional inhibition of selected STAT3-dependent miRNAs in human macrophage increases apoptosis of cells in vitro. (A) Percentages of parasite-positive macrophage (Wright-Giemsa stain as shown in Additional file 2: Figure S1) after transfection with specific anti-miRs (black bars) or untreated (white bars). (B) Transfection of cells with anti-miRs on apoptosis of human macrophage for 24 h after initial exposure to the parasite. Dot plots depict the amounts of macrophage stained with Annexin V and/or PI; numbers indicate percentages of cells in each quadrant from a representative experiment. (C) Percentage of cells with Toxoplasma after transfected with anti-miR-17-5p, anti-miR-20a, or ant-miR-17-5p and anti-miR-20a. *, p,0.05 vs. the non-transfected with Toxoplasma infection control.Values are presented as mean ± SD (n = 3 in A-B).
Discussion
There is evidence that miRNAs play a critical role in the regulation of apoptosis [21, 22] A better understanding miRNA expression changes in human macrophage following Toxoplasma infection will provide new insights in miRNA-associated apoptosis. we report significant alterations in miRNA expression profiles in human macrophage following Toxoplasma infection. Our analysis of miRNAs upregulated by Toxoplasma in human macrophage revealed that miR-30c-1, miR-125b-2, miR-23b-27b-24-1 and miR-17 ~ 92 genes are transactivated via potential promoter binding of the STAT3.
These data provide several insights relevant to miRNA expression regulation in human macrophage following Toxoplasma infection. First, transactivation of miRNA genes that produce the same mature miRNA can be differentially controlled. Specifically, both mir-125b-1 and mir-125b-2 genes can produce mature miR-125b, but only transactivation of mir-125b-2 gene was detected in cells following Toxoplasma infection. In this study, we demonstrated that promoter binding of the STAT3 is required for transactivation of the miR-30c-1, miR-125b-2, miR-23b-27b-24-1 and miR-17 ~ 92 genes in cells following Toxoplasma infection. Given the complexity and variability in the gene structure for each miRNA, it is obvious that multiple mechanisms are involved in the transcriptional regulation of human miRNA genes [23–25]. Therefore, transcription of miRNA genes is expected to be a dynamic process in response to the constant alterations in intracellular signals. miRNA expression thus reflects the final integrated result of multiple interrelated signals on miRNA transcription. In this regard, other transcription factors, such as NF-kB, AP-1, c-myc, C/EBPa, may also be involved in the transcriptional regulation of miRNA genes in human macrophage in response to Toxoplasma infection. Future studies will focus on whether the other transcription factors are involved in the Toxoplasma-induced upregulation or downregulation of miRNA expression. Secondly, transactivation of genes of cluster miRNAs or as introns in other gene alleles may be controlled by the same promoter element. Of note, miR-19b, miR-19a and miR-20a are cluster gene miRNAs and co-transcribed with a host gene, C13orf25 [26]. Toxoplamsa infection upregulates expression of the mature forms of these three miRNAs, as well as pri-miR-17 ~ 92 and the host gene transcript, C13orf25. Our data are consistent with recent studies demonstrating transcriptional control of genes that code cluster miRNAs or that encode both miRNAs and other host transcripts [25]. Finally, the miR-17 ~ 92 gene appear to attenuate apoptotic responsiveness by targeting several mRNAs encoding pro-apoptotic effectors and favor progression from G1 to S-phase by targeting mRNAs that encode negative regulators of the cell cycle [27, 28]. In our study, we found promotion of apoptosis of human macrophage with Toxoplasma infection via inhibition of miR-17 ~ 92 gene.
Previous investigation showed that a single amino acid substitution in the kinase domain of Toxoplasma ROP16 in difference strains determined STAT3 activation between type I and type II. Significantly, we found that TgCtwh3 strain with atypical genotype China 1 (ToxoDB#9) has the same amino acid substitution of the kinase domain of ROP16 at 503 bp (503 L, data not shown) as that of TGGT1 strain (503 L) with archetypical type I, which might account for the identical way of activation to STAT3 as type I [12]. Although Toxoplasma can infect any kind of nucleated cells, macrophage and related mononuclear phagocytes are its preferred target in vivo, and the parasite seems to have multiple ways of avoiding being killed [29, 30]. Therefore, macrophage-centric research is essential to understand the basic strategies of the parasite-host interaction [31]. miRNAs have been identified in both mammalian and nonmammalian cells including virus and parasites [32, 33]. Toxoplasma could increased the levels of miRNA in HFF cells [34], however, expression of miRNAs in Toxoplasma has not yet been examined and whether Toxoplsma-derived miRNAs can be localized in human macrophage is unknown. Nevertheless, the probes used in the microarray analysis in this study are human-miRNA specific with minimal cross-interaction with known miRNAs from other species.
Conclusions
In summary, this first miRNA profiling in human macrophage in response to Toxoplasma infection in vitro revealed significant alterations in cellular miRNA expression. The mechanism by which Toxoplasma induces upregulation of a panel of miRNAs in human macrophage involves transactivation of miRNA genes through promoter binding of the STAT3. In addition, functional inhibition of the upregulated miRNAs increases apopotosis of macrophage with Toxoplasma infection thereby implicating these miRNAs in host cell help parasite survival. These data demonstrate a better understanding of miRNA expression in human macrophage following Toxoplasma infection will play a critical role in inhibition of apoptosis of host cells and it may provide new insights into relationship between the intracellular parasite and its hose cells.
References
Song KJ, Ahn HJ, Nam HW: Anti-apoptotic effects of SERPIN B3 and B4 via STAT6 activation in macrophages after infection with Toxoplasma gondii. Korean J Parasitol. 2012, 50: 1-6. 10.3347/kjp.2012.50.1.1.
Persson EK, Agnarson AM, Lambert H, Hitziger N, Yagita H, Chambers BJ, Barragan A, Grandien A: Death receptor ligation or exposure to perforin trigger rapid egress of the intracellular parasite Toxoplasma gondii. J Immunol. 2007, 179: 8357-8365.
Luder CG, Gross U: Apoptosis and its modulation during infection with Toxoplasma gondii: molecular mechanisms and role in pathogenesis. Curr Top Microbiol Immunol. 2005, 289: 219-237.
Hippe D, Weber A, Zhou L, Chang DC, Hacker G, Luder CG: Toxoplasma gondii infection confers resistance against BimS-induced apoptosis by preventing the activation and mitochondrial targeting of pro-apoptotic Bax. J Cell Sci. 2009, 122: 3511-3521. 10.1242/jcs.050963.
Saeij JP, Coller S, Boyle JP, Jerome ME, White MW, Boothroyd JC: Toxoplasma co-opts host gene expression by injection of a polymorphic kinase homologue. Nature. 2007, 445: 324-327. 10.1038/nature05395.
Zhou R, Hu G, Liu J, Gong AY, Drescher KM, Chen XM: NF-kappaB p65-dependent transactivation of miRNA genes following Cryptosporidium parvum infection stimulates epithelial cell immune responses. PLoS Pathog. 2009, 5: e1000681-10.1371/journal.ppat.1000681.
Newmeyer DD, Ferguson-Miller S: Mitochondria: releasing power for life and unleashing the machineries of death. Cell. 2003, 112: 481-490. 10.1016/S0092-8674(03)00116-8.
Blader IJ, Manger ID, Boothroyd JC: Microarray analysis reveals previously unknown changes in Toxoplasma gondii-infected human cells. J Biol Chem. 2001, 276: 24223-24231. 10.1074/jbc.M100951200.
He L, Hannon GJ: MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet. 2004, 5: 522-531. 10.1038/nrg1379.
Liu X, Fang H, Chen H, Jiang X, Fang D, Wang Y, Zhu D: An artificial miRNA against HPSE suppresses melanoma invasion properties, correlating with a down-regulation of chemokines and MAPK phosphorylation. Plos one. 2012, 7: e38659-10.1371/journal.pone.0038659.
Jin L, Hu WL, Jiang CC, Wang JX, Han CC, Chu P, Zhang LJ, Thorne RF, Wilmott J, Scolyer RA: MicroRNA-149*, a p53-responsive microRNA, functions as an oncogenic regulator in human melanoma. Proc Natl Acad Sci USA. 2011, 108: 15840-15845. 10.1073/pnas.1019312108.
Yamamoto M, Standley DM, Takashima S, Saiga H, Okuyama M, Kayama H, Kubo E, Ito H, Takaura M, Matsuda T: A single polymorphic amino acid on Toxoplasma gondii kinase ROP16 determines the direct and strain-specific activation of Stat3. J Exp Med. 2009, 206: 2747-2760. 10.1084/jem.20091703.
Butcher BA, Fox BA, Rommereim LM, Kim SG, Maurer KJ, Yarovinsky F, Herbert DR, Bzik DJ, Denkers EY: Toxoplasma gondii rhoptry kinase ROP16 activates STAT3 and STAT6 resulting in cytokine inhibition and arginase-1-dependent growth control. PLoS Pathog. 2011, 7: e1002236-10.1371/journal.ppat.1002236.
Sugimura K, Miyata H, Tanaka K, Hamano R, Takahashi T, Kurokawa Y, Yamasaki M, Nakajima K, Takiguchi S, Mori M, Doki Y: Let-7 expression is a significant determinant of response to chemotherapy through the regulation of IL-6/STAT3 pathway in esophageal squamous cell carcinoma. Clin Cancer Res. 2012, 18: 5144-5153. 10.1158/1078-0432.CCR-12-0701.
Chen Q, Wang H, Liu Y, Song Y, Lai L, Han Q, Cao X, Wang Q: Inducible microRNA-223 down-regulation promotes TLR-triggered IL-6 and IL-1beta production in macrophages by targeting STAT3. Plos one. 2012, 7: e42971-10.1371/journal.pone.0042971.
Cai B, Li J, Wang J, Luo X, Ai J, Liu Y, Wang N, Liang H, Zhang M, Chen N: microRNA-124 regulates cardiomyocyte differentiation of bone marrow-derived mesenchymal stem cells via targeting STAT3 signaling. Stem Cells. 2012, 30: 1746-1755. 10.1002/stem.1154.
Wang L, Chen H, Liu D, Huo X, Gao J, Song X, Xu X, Huang K, Liu W, Wang Y: Genotypes and mouse virulence of Toxoplasma gondii isolates from animals and humans in China. Plos one. 2013, 8: e53483-10.1371/journal.pone.0053483.
Chen ZW, Gao JM, Huo XX, Wang L, Yu L, Halm-Lai F, Xu YH, Song WJ, Hide G, Shen JL, Lun ZR: Genotyping of Toxoplasma gondii isolates from cats in different geographic regions of China. Vet Parasitol. 2011, 183: 166-170. 10.1016/j.vetpar.2011.06.013.
Kramer PR, Wray S: 17-Beta-estradiol regulates expression of genes that function in macrophage activation and cholesterol homeostasis. J Steroid Biochem Mol Biol. 2002, 81: 203-216. 10.1016/S0960-0760(02)00065-1.
Brock M, Trenkmann M, Gay RE, Michel BA, Gay S, Fischler M, Ulrich S, Speich R, Huber LC: Interleukin-6 modulates the expression of the bone morphogenic protein receptor type II through a novel STAT3-microRNA cluster 17/92 pathway. Circ Res. 2009, 104: 1184-1191. 10.1161/CIRCRESAHA.109.197491.
Liu Y, Han Y, Zhang H, Nie L, Jiang Z, Fa P, Gui Y, Cai Z: Synthetic miRNA-mowers targeting miR-183-96-182 cluster or miR-210 inhibit growth and migration and induce apoptosis in bladder cancer cells. Plos one. 2012, 7: e52280-10.1371/journal.pone.0052280.
Salim H, Akbar NS, Zong D, Vaculova AH, Lewensohn R, Moshfegh A, Viktorsson K: Zhivotovsky B: miRNA-214 modulates radiotherapy response of non-small cell lung cancer cells through regulation of p38MAPK, apoptosis and senescence. Br J Cancer. 2012, 107: 1361-1373. 10.1038/bjc.2012.382.
Saini HK, Griffiths-Jones S, Enright AJ: Genomic analysis of human microRNA transcripts. Proc Natl Acad Sci U S A. 2007, 104: 17719-17724. 10.1073/pnas.0703890104.
Loffler D, Brocke-Heidrich K, Pfeifer G, Stocsits C, Hackermuller J, Kretzschmar AK, Burger R, Gramatzki M, Blumert C, Bauer K: Interleukin-6 dependent survival of multiple myeloma cells involves the Stat3-mediated induction of microRNA-21 through a highly conserved enhancer. Blood. 2007, 110: 1330-1333. 10.1182/blood-2007-03-081133.
Chang TC, Yu D, Lee YS, Wentzel EA, Arking DE, West KM, Dang CV, Thomas-Tikhonenko A, Mendell JT: Widespread microRNA repression by Myc contributes to tumorigenesis. Nat Genet. 2008, 40: 43-50. 10.1038/ng.2007.30.
Rinaldi A, Poretti G, Kwee I, Zucca E, Catapano CV, Tibiletti MG, Bertoni F: Concomitant MYC and microRNA cluster miR-17-92 (C13orf25) amplification in human mantle cell lymphoma. Leuk Lymphoma. 2007, 48: 410-412. 10.1080/10428190601059738.
Xiao C, Rajewsky K: MicroRNA control in the immune system: basic principles. Cell. 2009, 136: 26-36. 10.1016/j.cell.2008.12.027.
Mendell JT: miRiad roles for the miR-17-92 cluster in development and disease. Cell. 2008, 133: 217-222. 10.1016/j.cell.2008.04.001.
Zhao ZJ, Zhang J, Wei J, Li Z, Wang T, Yi SQ, Shen JL, Yang TB, Hide G, Lun ZR: Lower expression of inducible nitric oxide synthase and higher expression of arginase in rat alveolar macrophages are linked to their susceptibility to Toxoplasma gondii infection. Plos one. 2013, 8: e63650-10.1371/journal.pone.0063650.
Li Z, Zhao ZJ, Zhu XQ, Ren QS, Nie FF, Gao JM, Gao XJ, Yang TB, Zhou WL, Shen JL: Differences in iNOS and arginase expression and activity in the macrophages of rats are responsible for the resistance against T. gondii infection. Plos one. 2012, 7: e35834-10.1371/journal.pone.0035834.
Murray PJ: Macrophages as a battleground for toxoplasma pathogenesis. Cell Host Microbe. 2011, 9: 445-447. 10.1016/j.chom.2011.05.010.
Xue X, Sun J, Zhang Q, Wang Z, Huang Y, Pan W: Identification and characterization of novel microRNAs from Schistosoma japonicum. Plos one. 2008, 3: e4034-10.1371/journal.pone.0004034.
Winter J, Jung S, Keller S, Gregory RI, Diederichs S: Many roads to maturity: microRNA biogenesis pathways and their regulation. Nat Cell Biol. 2009, 11: 228-234. 10.1038/ncb0309-228.
Michael T, Zeiner KLN J, Gusti M: Toxoplasma gondii infection specifically increases the levels of key host microRNAs. Plos one. 2010, 5: e8742-e8750. 10.1371/journal.pone.0008742.
Acknowledgements
This study was founded by the National Natural Science Foundation of China (Grants No. 81101272) and Natural Basic Research Program of China (Grant No. 2010CB530001).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
JLS, YHC and LJ conceived and designed the study. YHC, HC and YBY performed the experiments. YHC and HC analyzed the data. YHC and JLS drafted the manuscript. All authors read and approved the final manuscript.
Electronic supplementary material
13071_2013_1137_MOESM2_ESM.bmp
{kind=link}
Additional file 2: Figure S1: Determination of macrophage from PBMCs. PBMCs-derived macrophage were separated from whole blood and stained with FITC-conjugated CD14 antibody to determine the purity of macrophage. (A) PBMCs-derived macrophage under microscope (×200). (B) CD14 expression of PBMCs-derived macrophage. (C) PBMCs-derived macrophage seen by microscopy (Wright-Giemsa, ×1000). (D) PBMCs-derived macrophage with TgCtwh3 for 24 h seen by microscopy (Wright-Giemsa, ×1000). (BMP 4 MB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Cai, Y., Chen, H., Jin, L. et al. STAT3-dependent transactivation of miRNA genes following Toxoplasma gondii infection in macrophage. Parasites Vectors 6, 356 (2013). https://doi.org/10.1186/1756-3305-6-356
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1756-3305-6-356