
Abstract
Background
Phlebotomus chinensis is a primary vector of visceral leishmaniasis; it occurs in various biotopes with a large geographical distribution, ranging from Yangtze River to northeast China. Phlebotomus sichuanensis, a species closely related to P. chinensis in high altitude regions, has a long term disputation on its taxonomic status. Both species occur in the current epidemic regions and are responsible for the transmission of leishmaniasis. Population genetic analysis will help to understand the population structure and infer the relationship for morphologically indistinguishable cryptic species. In this study, microsatellite markers were used for studying the genetic differentiation between P. chinensis and P. sichuanensis.
Methods
Sandflies were collected in 6 representative localities in China in 2005-2009. Ten microsatellite loci were used to estimate population genetic diversity. The intra-population genetic diversity, genetic differentiation and effective population size were estimated.
Results
All 10 microsatellite loci were highly polymorphic across populations, with high allelic richness and heterozygosity. Hardy-Weinberg disequilibrium was found in 23 out of 60 (38.33%) comparisons associated with heterozygote deficits, which was likely caused by the presence of null allele and the Wahlund effect. Bayesian clustering analysis revealed three clusters. The cluster I included almost all specimens in the sample SCD collected at high altitude habitats in Sichuan. The other two clusters were shared by the remaining 5 populations, SCJ in Sichuan, GSZ in Gansu, SXL and SXX in Shaanxi and HNS in Henan. The diversity among these 5 populations was low (FST = -0.003-0.090) and no isolation by distance was detected. AMOVA analysis suggested that the variations were largely derived from individuals within populations and among individuals. Consistently, the analysis of ribosomal DNA second internal transcribed spacer (ITS2) sequence uncovered three types of variants, which corresponded with the three gene pools revealed by microsatellites.
Conclusions
The data suggested that the SCD population carried a distinct gene pool, which was differentiated from the other populations. The high altitude ecological habitats, distinctive ITS2 and herein divergence inferred by microsatellite loci support the species status of P. sichuanensis. The P. chinensis populations did not have a significant divergence from each another.
Similar content being viewed by others

Background
Phlebotomine sandflies are small insects of great medical and veterinary relevance. In China, phlebotomine sandfly fauna includes over 40 species in five genera, Phlebotomus Rondani & Berte, Sergentomyia France & Farrot, Idiophlebotomus Quate & Fairchild, Grassomyia Theodor and Chinius Leng [1, 2]. In the genus Phlebotomus, P. chinensis Newstead, P. sichuanensis Leng & Yin, P. longiductus Nitzulesc, P. wuii Yang & Xiong and P. alexandri Sinton are vectors of leishmaniasis [2]. Visceral leishmaniasis (VL) is caused by Leishimania donovani, which has been and still is a serious threat to public health in the endemic areas in China [3, 4]. According to a report of VL surveillance in the period 2005 to 2010, most cases (97.7%) occurred in Xinjiang, Gansu and Sichuan [3].
Phlebotomus chinensis is a prevalent species with wide geographical distribution, having records in 20 provinces ranging from Yangtze River to the northeast China [1]. In 1983, Leng and Yin described a new species, P. sichuanensis, based on the study of a large number of sandfly specimens that were collected in the area of 29-33°N, 102-106°E in Sichuan province in 1976-1980 [5]. The species was described based upon the morphological comparison with the specimens of P. chinensis collected in the type locality. Various morphometric and morphostructural characters were used for distinguishing P. sichuanensis from P. chinensis[5, 6]. Male ascoid formula in P. sichuanensis is 2/3-8, 1/9-15, while it is 2/3-15 in P. chinensis. Phlebotomus sichuanensis is mainly distributed in the regions from 900 m to 2800 m in Gansu, Sichuan, Yunnan and Tibet [2, 6, 7] (see the map in Figure 1 in the ref. [6]). However, Xiong et al. (1990) argued that the morphological difference did not suffice to support P. sichuanensis as an independent species [8]. The body size of sandflies at high altitudes (above 2000 m) was larger than that of the sandflies at low altitudes (below 1600 m). The male ascoid formula 2/3-8, 1/9-15 was found in a high percentage (96.6%) in large size sandflies collected above 2000 m, and the formula 2/3-15 was found in small size sandflies collected at 1600 m. They considered the differences in body size and male ascoid formula as signs of different ecological types other than characters for taxonomic identification. Therefore, Xiong et al. (1990) referred to P. sichuanensis as a large type of P. chinensis, and synonymized P. sichuanensis to P. chinensis[8]. Epidemiologically, both sandflies are competent vectors of VL. Natural infection of L. donovani was detected in sandflies collected in high and low altitude regions [9]. In addition, sandflies from both collections were equally susceptible to Leishmania in experimental feeding on Chinese hamsters infected with L. donovani[8, 9]. The vector importance of sandflies in leishmaniasis endemic areas urges the necessity to resolve the taxonomic dispute of P. sichuanensis. Apparently, non-morphological, convincing and distinctive taxonomic markers are needed for solving the identity issue.

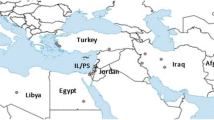
Collection localities of Phlebotomus sandflies in China. The diamond code indicates the field population. The number in the bracket is the altitude.
Molecular markers, such as mitochondrial cytochrome b (Cyt b), ND1, ND4, the second internal transcribed spacer (ITS2) of ribosomal DNA and microsatellite markers, have been widely used for sandfly studies in taxonomic discrimination and inference of phylogenetic relationship [10–16], as well as population genetics studies [17–24]. In an effort to apply molecular data to address the taxonomic conflict of P. sichuanensis, we conducted six sandfly collections in 2005-2009, where P. chinensis and P. sichuanensis occur. Among them, sample SCD was collected in habitats at high altitudes (above 2000 m), a typical locality of P. sichuanensis. Previously we have reported the sequence variation of the ITS2 and Cyt b generated from these samples [25]. Based on the ITS2 and Cyt b variations, P. chinensis was clearly separated from P. longiductus, P. perfiliewi, P. pernicious, P. ariasis, P. duboscqi and P. alexandri. The partial ITS2 sequences of SCD specimens showed distinctive variation from that of P. chinensis[25].
Microsatellites are highly polymorphic genetic markers that evolve much faster than mitochondrial or nuclear genes, which are particularly useful for resolving the structure of populations at a finer geographical and evolutionary scale. In this study, we conducted population genetic analysis of the same six sandfly collections reported in the paper above [25], based on 10 microsatellite loci. The population genetic analysis would help to understand the population structure and infer the relationship for morphologically indistinguishable cryptic species.
Methods
Sandfly collections and species identification
There are four species in the subgenus Adlerius in China, P. chinensis, P. sichuanensis, P. longiductus and P. fengi Leng & Zhang. The distribution region of P. longiductus and P. fengi is limited to Xingjiang and Yunnan. In this study, wild adult sandflies were collected in 2005 and 2009 from six locations in the range from 104°15′N to 110°60′ N, and 33°18′E to 36°18′ E in Sichuan, Gansu, Shaanxi, Henan provinces (Table 1, Figure 1). These collection sites corresponded to the current epidemic regions of leishmaniasis (see the VL distribution map in Figure 1 in ref. [3]). The sandfly specimens were collected by using human landing catches at livestock corrals or mountain caves, under the NSFC ethical guidelines for biomedical research involving living animals and human subjects. The collections in livestock corrals were conducted with the consent of the owners. The specimens were kept individually in tubes filled with silica gel at 4°C until dissection was performed. The specimens from the six populations were identified as P. chinensis sensu lato based on the morphological characters of the female or male external genitalia, the structure of the pharyngeal armature and the spermatheca [26] (see Results).
Microsatellite genotyping
Following morphological identification, the genomic DNA was extracted from individual adults from dissected tissue using the Insect Tissue DNA Extraction Kit (Biyuntian, Jiangsu China), using the manufacturer’s protocol. Ten microsatellite loci [27], GA1 (GenBank accession number FJ919882), GA5 (FJ919907), GA13 (FJ919884), GA24 (FJ919885), GA63 (FJ919932), GA76 (FJ919890), GA109 (FJ919900), AAT3 (FJ919917), HN54 (FJ919923) and TG1 (FJ919894), were used for genotyping. Microsatellite loci were amplified according to the detailed protocols described by Schuelke [28]. Each PCR reaction contained three primers: a sequence-specific forward primer with a M13(-21) adapter (5′- TGT AAA ACG ACG GCC AGT -3′) conjugated at its 5′ end, a sequence-specific reverse primer, and a universal FAM-labeled M13(-21) primer. Amplified fragments were separated by capillary electrophoresis in an automatic sequencer (ABI 3770, Applied Biosystems, Foster City, CA) and size was scored using GENOTYPER 3.7 software (Applied Biosystems, Foster City, CA).
Data analysis
Genetic diversity within samples and overall was measured at each locus by estimates of number of alleles A, allele richness Rs, inbreeding coefficient FIS, and observed heterozygosity H o [29], using the software FSTAT 2.9.3.2 [30]. Within each locality the frequency of null alleles was determined using the Brookfield 2 estimate [31], and the allele and genotype frequencies were then adjusted accordingly by using MICRO-CHECKER 2.2.3 [32]. To determine if the null alleles impacted the population genetic analyses, we performed these analyses both before and after the dataset were adjusted for estimated null allele frequencies. This treatment did not significantly change the degree or statistical significance of the estimated parameters. Genotypic frequencies were tested against Hardy-Weinberg equilibrium (HWE) for each locus in pooled populations and in each sample. Statistical significance was assessed by the exact probability test available in GENEPOP 3.2 [33]. Linkage disequilibrium (LD) between loci was tested by exact tests on contingency tables, also available in GENEPOP.
A Bayesian approach was used to infer the number of clusters (K) in the data set without prior information of the sampling locations, implemented with STRUCTURE 2.2 [34]. A model where the allele frequencies were correlated within populations was assumed (λ was set at 1, the default value). The software was run with the option of admixture, allowing for some mixed ancestry within individuals, and α was allowed to vary. Twenty independent runs were carried out for each value of K (K = 1 to 6), with a burn-in period of 1,000,000 chains and 1,000,000 Markov chains Monte Carlo replications. The method of Evanno was used to determine the most likely number of clusters [35]. This approach uses an ad hoc quantity, ΔK, based on the second order rate of change of the likelihood function between successive values of K.
Genetic differentiation was estimated by calculating FST between pairs of populations using ARLEQUIN 3.0 [36] and GENEPOP [33]. The number of migrants per population per generation (N m) between localities was estimated from pairwise FST[37]. An analysis of molecular variance (AMOVA) was used to examine the distribution of genetic variation in ARLEQUIN using FST. We focused on estimates of FST performed under the infinite alleles model (IAM) because this model is considered more reliable when fewer than 20 microsatellites are used [38]. The significance for all calculations was assessed by 10,000 permutations and the P-values. The isolation by distance model was investigated as a potential explanation for the observed population differentiation. The significance of the regression of genetic differences on geographic distance between sample pairs was tested using a Mantel test [39] with 100,000 permutations in GENEPOP.
The long-term effective population size (Ne) was estimated using NEESTIMATOR 1.3 [40] based on the heterozygote excess and linkage disequilibrium models.
Results
Species identification
Wild sandflies were collected at 6 locations in China (Figure 1), a total of 158 specimens including 93 females and 65 males were used in the study (Table 1). In these locations, the only documented species in the subgenus Adlerius were P. chinensis and P. sichuanensis[5, 6, 26]. The morphologic characters of P. chinensis were as follows: transverse ridges on the pharyngeal armature, ellipsoid shape to spermatheca, number of spermatheca segments 13 or 14, length of spermatheca duct longer than 2.5 times spermatheca, and the length ratio of genital filament to pompetta 1 : 5.6. Based on the above key criteria, all 158 specimens were identified as P. chinensis s. l. There are no absolute morphological characters to distinguish P. chinensis with P. sichuanensis, however, it was noted that the morphology of 26 specimens in the SCD population conformed to that of P. sichuanensis as described in [5, 6].
Genetic variability within populations
Ten microsatellite loci were genotyped for these specimens. The number of alleles (A) per locus ranged from 2.3 at locus HN54 to 9.83 at locus GA76. As an exception, only one allele was detected at locus GA1 in sample GSZ (Table 2). The minimum mean number of alleles of all loci was 5.5 in sample SXL, and the maximum was 7.1 in sample HNS. The average allelic richness per locus (Rs) across 6 samples was 3.72, ranging from 3.145 in SCD to 3.849 in HNS. The average observed heterozygosity (HO) across all samples ranged from 0.236 (TG1) to 0.77 (GA109), the minimum was in SXL (0.375), the maximum in HNS (0.503). The mean expected heterozygosity (HE) was 0.619, with a range of from 0.49 (SCD) to 0.693 (HNS). Putting these statistics together, the HNS population showed the highest diversity, whereas the SCD population showed the lowest diversity.
The Hardy-Weinberg exact tests were performed for the 10 loci. No locus was in HWE for all the samples assayed, except HN54. At the population level, 23 out of 60 (38.33%) comparisons did not conform to Hardy-Weinberg expectations, and the deviations were associated with positive inbreeding coefficient (FIS), reflecting heterozygosity deficits (Table 2). Significant deviation from HWE varied across loci in a population-dependent manner. The GSZ population had the highest number of loci in departure from HWE (6 of 10), while the SXX population had the fewest (1 of 10) (Table 2). In all samples, some specimens failed to amplify at one locus while they succeeded to amplify at the remaining loci, suggesting the presence of null alleles. Estimates of the frequency of null alleles are given in Table 2. The highest r value was at the locus HN54.
Fisher’s exact tests were conducted for LD within each of the six populations. Out of 270 comparisons only 17 pairs (6.30%) were at LD (P < 0.01). The pair of GA10 and HN54 appeared at LD in all populations, except in the SCD population.
Genetic differentiation among populations
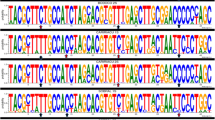
The significant deviations from HWE with heterozygote deficiency and the presence of LD suggest the presence of population subdivision within samples (Wahlund effect). We therefore examined if there were different gene pools in these samples. The Bayesian cluster analysis divided specimens into three gene clusters (posterior probability of Bayesian clustering Ln (D) likelihood score optimal for K = 3 clusters) (Figure 2). Sample SCD was clearly distinct from the others; almost all individuals belonged to cluster I (red). The rest of the five samples were mixed with the individuals from the other two gene pools. This pattern was consistent with the pattern inferred from ITS2 sequence comparison [25]. ITS2 sequences of those individuals showed three major types of variants. Based on the alignment of ITS2 sequences (Figure 3), phylogenetic analysis separated those variants into three clades (Figure 4). The SCD collection formed a single clade, the other five collections were clustered into one clade with two sister branches. Interestingly, each of these five collections was mixed with the two major types of ITS variants. In the analysis we also included the ITS2 sequences (PopSet 290794958) available in the NCBI deposited by Gu and Zhang. These specimens were collected in Dongshan, Sichuan; Wen County, Gansu; Sanmenxia, Henan; Yichuan, Shaanxi. Intriguingly, the three major types of ITS2 sequences were found in the collection (Figure 3). The sequence GU385746 was from a specimen collected in Dongshan, Sichuan, where the SCD was sampled in this study. The independent data added a strong support for the presence of distinct genetic clusters in the sandflies.

Bayesian cluster analysis using STUCTURE. Graphical representation of the data set for the most likely K (K = 3), where each color corresponds to a suggested cluster and each individual is represented by a vertical bar. The X-axis corresponds to population codes. The Y-axis presents the probability of assignment of an individual to each cluster.

The ITS2 sequence alignment of P. chinensis and P. sichuanensis . Presented partial sequences show the variations. There are three major types of variants.

The UPGMA tree of Phlebotomus sandflies inferred by rDNA-ITS2 sequences. The phylogram was generated using MEGA 5.10. The bootstrap values (1000 replications) are shown on the branch. The sequence id is presented by the Genbank accession numbers followed by the species identity. Population id is indicated next to the clades.
The gene flow and FST were estimated in a location based manner for the six samples, which is presented in Table 3. The SCD population diverged from the other five populations. The pairwise FST value was greater and the N m value was lower between SCD and the others than the pairwise comparisons among the other five populations. Therefore, the SCD represented a population diverged from the other five P. chinensis populations in the study. Tests of isolation by distance were performed for five populations except SCD. No statistically significant correlations were detected between genetic differentiation and geographic distances based on the Mantel test (R2=0.2846, P=0.10). The results suggested that geographic distance did not significantly contribute to the genetic differentiation observed in the P. chinensis populations.
In the hierarchical AMOVA, both the “among individuals within populations” and “among individuals” variance components were considerably high (83.35%), suggesting that the total variances largely came from individual variations (Table 4).
Effective population size
Estimates of long-term Ne varied considerably depending on the model used. Under the heterozygote excess model, all of the Ne estimates were infinity. Under the linkage disequilibrium model, diverse Ne values were detected across populations (Table 5). The six populations showed variability of Ne, from 5.7 (SXL) to 264.4 (SCJ).
Discussion
Phlebotomus chinensis s. l. is the major vector for VL transmission in the endemic regions except Xinjiang in China. The sandflies in these regions have two major types of ecological habitats. In the open flatland regions with an altitude <900 m, such as the vast plain area north of the Yangtze River, the sandflies are found in the indoor shelters such as human dwellings and domestic animal sheds. The transmission of VL is largely from human to human. In the mountain areas, the sandflies are largely exophilic, breeding in a variety of wild habitats, such as various caves and rodent burrows. If human dwellings are available, the sandflies feed on humans or domestic animals. The dogs are the main animal hosts of L. donovani, and VL is largely transmitted between dogs and humans. In the areas with an altitude >2000 m, usually there are no human dwellings. The sandflies feed on wild animals for breeding, and transmit Leishmania among wild animals [3, 41]. Phlebotomus sichuanensis[5, 6], or a large type of P. chinensis[8, 42] were mostly found in the high altitude areas in Sichuan, Yunnan, Shaanxi, Gansu and Tibet [6]. The current epidemiological status of leishmaniasis in China was reviewed recently. In brief, most cases of leishmaniasis occurred in Xingjiang, Gansu and Sichuan [3]. In past decades, the density of sandflies dropped drastically, most likely due to the wide applications of insecticides and environmental changes. In this study, 6 collection sites were chosen in the Loess Plateau, hills and mountainous areas with an altitude of 985-2153 m, where mountainous sub-type zoonotic VL occurs (see Figure 1 in the ref. [3]). The sampling sites had a representative geographic coverage of P. chinensis and P. sichuanensis in the high altitude areas.
The significant deviations from HWE due to the heterozygote deficits were detected in most samples, suggesting the presence of population subdivision within samples (Wahlund effect). Indeed, cluster analysis, implemented by using STRUCTURE, recognized three gene clusters in the six sandfly collections. Almost all individuals in SCD population were assigned to cluster I (Figure 2). The data identified SCD as a distinct genetic unit with an isolated gene pool. The other two genetic clusters were distributed in the rest of five collections. Consistent with the pattern inferred by microsatellite, the ITS2 sequences from the SCD (GenBank accession HM747279, HM747280) plus one sequence (GU385746) from the Gu and Zhang collection were clustered in one clade, while the other ITS2 sequences grouped into two sister clades (Figure 4). Therefore, the ITS2 analysis supported the genetic distinction between SCD and other P. chinensis collections. When six sample collections were compared, the differentiation between SCD and others was evident by high FST and reduced gene flow (FST = 0.15-0.20, N m = 0.998-1.419, Table 3). Apparently, SCD represents a genetic unit that has diverged from the other populations. The SCD specimens were sampled in a location at an elevation of 2035-2153 m, a typical habitat of P. sichuanensis[6]. The morphological characters of the SCD population specimens were consistent with the descriptions of P. sichuanensis[5, 6], although morphology only is often not conclusive for distinguishing the two species. Taken together, both ITS2 and microsatellites separated SCD from other populations. Therefore, we concluded that the molecular data supported the species status of P. sichuanensis. The ecological adaptation of P. sichuanensis in high altitude regions may have contributed to its separation from P. chinensis in the low altitude regions. The relative low level of divergence may represent adaptation-driven incipient speciation of P. sichuanensis by divergent environment at high altitudes. Such ongoing speciation has been exemplified in the mosquito Anopheles gambiae, in which two molecular forms, M and S, are experiencing a speciation-with-ongoing-gene-flow [43–45]. Further investigation with more genetic markers, eventually whole genomic sequencing, will clarify the issue ultimately.
The pairwise level of genetic variation was small in the remaining five populations (FST = -0.003-0.090, N m = 2.516-6.581, Table 3). The two gene clusters did not show any correlation to geographic origin. The AMOVA data suggested that most variations among populations may attribute to the individual variation, which may explain the source of the two clusters that were shared by the P. chinensis populations. The small genetic differentiation in P. chinensis populations and no evidence of isolation by distance suggested that no obvious barriers limit the dispersal of sandflies in the P. chinensis populations sampled in this study.
Leishmaniasis has shown an increasing trend in recent years in China [3]. Different Leishmania genotypes have been found in different geographic origins, such as hill, plain and desert [46]. The current study investigated the sandflies in the mountainous regions where the VL is epidemic. Both P. chinensis and P. sichuanensis are competent vectors [8, 9]. The different ecological habitats may be accompanied by different behavior and physiology that may affect disease transmission and compromise anti-vector measures. Particularly, P. sichuanensis occurs in regions of above 2000 m, where Leishmania has been cycling among wild reservoir animal hosts. It is conceivable that P. sichuanensis constitutes a necessary component connecting the zoonotic VL to the human community. The current characterization of sandfly population structure and ITS2 sequences provide molecular data to develop objective and reliable methods for molecular identification of sandfly specimens. Such tools will be particularly useful to further investigate the ecology, behavior and vector capacity of P. chinensis and P. sichuanensis in the mountainous regions. This type of data will facilitate the development of appropriate measures to control sandfly vectors.
Conclusion
The microsatellite data suggest that SCD represents a population of P. sichuanensis possessing a distinct gene pool, which was differentiated from the P. chinensis populations. The molecular data support the species status of P. sichuanensis. The five P. chinensis populations did not have a significant divergence from each other. The genetic distinction of P. sichuanensis from P. chinensis warrants further study to explore potential influence on physiology, behavior, and vector competence that may be associated with different ecological habitats between the two species.
References
Leng YJ, Zhang LM: Check list and geographical distribution of phlebotomine sandflies in China. Ann Trop Med Parasitol. 1993, 87 (1): 83-94.
Zhang LM, Leng YJ: Eighty-year research of phlebotomine sandflies (Diptera: Psychodidae) in China (1915-1995). II. Phlebotomine vectors of leishmaniasis in China. Parasite. 1997, 4 (4): 299-306.
Wang JY, Cui G, Chen HT, Zhou XN, Gao CH, Yang YT: Current epidemiological profile and features of visceral leishmaniasis in People’s Republic of China. Parasit Vectors. 2012, 5: 31-10.1186/1756-3305-5-31.
Guan LR, Shen WX: Recent advances in visceral leishmaniasis in China. Southeast Asian J Trop Med Public Health. 1991, 22 (3): 291-298.
Leng YJ, Yin ZC: The taxonomy of phlebotomine sandflies (Diptera: Psychodidae) of Sichuan Province, China, with descriptions of two species, Phlebotomus (Adlerius) sichuanensis sp. n. and Sergentomyia (Neophlebotomus) zhengjiani sp. n. Ann Trop Med Parasitol. 1983, 77 (4): 421-431.
Leng YJ, Zhang LM: Chinese phlebotomine sandflies of subgenus Adlerius nitzulescu,(Diptera: Psychodidae) and the identity of Phlebotomus sichuanensis Leng & Yin, 1983. Part I-Taxonomical study and geographical distribution. Parasite. 1931, 8 (1): 3-9.
Leng YJ, Zhang LM, Zhang BX: A study of phlebotomine sandflies (Diptera: Psychodidae) in Yunnan Province, China. I. Phlebotomine sandflies of northeastern and western Yunnan. Parassitologia. 1991, 33 (Suppl): 373-375.
Xiong GH, Jin CF, Hong YM: Further study on the type of sandfly Ph. chinensis in southern Gansu and Northern Sichuan. Endemic Dis Bull. 1990, 5: 53-60.
Xiong GH, Jin CF: Studies on the longitudinal distribution of sandfly Phlebotomus chinensis and its relation to Kala Azar in southern Gansu and northern Sichuan. Endemic Diseases Bulletin. 1989, 4 (4): 15-21.
Latrofa MS, Annoscia G, Dantas-Torres F, Traversa D, Otranto D: Towards a rapid molecular identification of the common phlebotomine sand flies in the Mediterranean region. Vet Parasitol. 2012, 184 (2–4): 267-270.
Kumar NP, Srinivasan R, Jambulingam P: DNA barcoding for identification of sand flies (Diptera: Psychodidae) in India. Mol Ecol Resour. 2012, 12 (3): 414-420. 10.1111/j.1755-0998.2012.03117.x.
Khalid N, Elnaiem D, Aboud M, Al Rabba F, Tripet F: Morphometric and molecular differentiation of Phlebotomus (Phlebotomus) sandflies. Med Vet Entomol. 2010, 4 (4): 352-360.
Depaquit J, Naucke TJ, Schmitt C, Ferte H, Leger N: A molecular analysis of the subgenus Transphlebotomus Artemiev, 1984 (Phlebotomus, Diptera, Psychodidae) inferred from ND4 mtDNA with new northern records of Phlebotomus mascittii Grassi, 1908. Parasitol Res. 2005, 95 (2): 113-116. 10.1007/s00436-004-1254-x.
Depaquit J, Ferte H, Leger N, Lefranc F, Alves-Pires C, Hanafi H, Maroli M, Morillas-Marquez F, Rioux JA, Svobodova M: ITS2 sequences heterogeneity in Phlebotomus sergenti and Phlebotomus similis (Diptera, Psychodidae): possible consequences in their ability to transmit Leishmania tropica. Int J Parasitol. 2002, 32 (9): 1123-1131. 10.1016/S0020-7519(02)00088-7.
Depaquit J, Ferte H, Leger N, Killick-Kendrick R, Rioux JA, Killick-Kendrick M, Hanafi HA, Gobert S: Molecular systematics of the phlebotomine sandflies of the subgenus Paraphlebotomus (Diptera, Psychodidae, Phlebotomus) based on ITS2 rDNA sequences. Hypotheses of dispersion and speciation. Insect Mol Biol. 2000, 9 (3): 293-300. 10.1046/j.1365-2583.2000.00179.x.
Di Muccio T, Marinucci M, Frusteri L, Maroli M, Pesson B, Gramiccia M: Phylogenetic analysis of Phlebotomus species belonging to the subgenus Larroussius (Diptera, psychodidae) by ITS2 rDNA sequences. Insect Biochem Mol Biol. 2000, 30 (5): 387-393. 10.1016/S0965-1748(00)00012-6.
Belen A, Kucukyildirim S, Alten B: Genetic structures of sand fly (Diptera: Psychodidae) populations in a leishmaniasis endemic region of Turkey. J Vector Ecol. 2011, 36 (Suppl 1): S32-S48.
Hamarsheh O, Presber W, Yaghoobi-Ershadi MR, Amro A, Al-Jawabreh A, Sawalha S, Al-Lahem A, Das ML, Guernaoui S, Seridi N: Population structure and geographical subdivision of the Leishmania major vector Phlebotomus papatasi as revealed by microsatellite variation. Med Vet Entomol. 2009, 23 (1): 69-77. 10.1111/j.1365-2915.2008.00784.x.
Hamarsheh O, Presber W, Al-Jawabreh A, Abdeen Z, Amro A, Schonian G: Molecular markers for Phlebotomus papatasi (Diptera: Psychodidae) and their usefulness for population genetic analysis. Trans R Soc Trop Med Hyg. 2009, 103 (11): 1085-1086. 10.1016/j.trstmh.2009.02.011.
Depaquit J, Lienard E, Verzeaux-Griffon A, Ferte H, Bounamous A, Gantier JC, Hanafi HA, Jacobson RL, Maroli M, Moin-Vaziri V: Molecular homogeneity in diverse geographical populations of Phlebotomus papatasi (Diptera, Psychodidae) inferred from ND4 mtDNA and ITS2 rDNA epidemiological consequences. Infect Genet Evol. 2008, 8 (2): 159-170. 10.1016/j.meegid.2007.12.001.
Hamarsheh O, Presber W, Abdeen Z, Sawalha S, Al-Lahem A, Schonian G: Genetic structure of mediterranean populations of the sandfly Phlebotomus papatasi by mitochondrial cytochrome b haplotype analysis. Med Vet Entomol. 2007, 21 (3): 270-277. 10.1111/j.1365-2915.2007.00695.x.
Hamarsheh O, Amro A: Characterization of simple sequence repeats (SSRs) from Phlebotomus papatasi (Diptera: Psychodidae) expressed sequence tags (ESTs). Parasit Vectors. 2011, 4: 189-10.1186/1756-3305-4-189.
Aransay AM, Ready PD, Morillas-Marquez F: Population differentiation of Phlebotomus perniciosus in Spain following postglacial dispersal. Heredity (Edinb). 2003, 90 (4): 316-325. 10.1038/sj.hdy.6800246.
Mahamdallie SS, Pesson B, Ready PD: Multiple genetic divergences and population expansions of a Mediterranean sandfly, Phlebotomus ariasi, in Europe during the Pleistocene glacial cycles. Heredity (Edinb). 2011, 106 (5): 714-726. 10.1038/hdy.2010.111.
Zhang L, Ma Y: Identification of Phlebotomus chinensis (Diptera: Psychodidae) inferred by morphological characters and molecular markers. Entomotaxonomia. 2012, 34 (1): 71-80.
Lu B, Wu H: Classification and identification of important medical insects of China. 2003, Henan: Henan Science and Technology Publishing House
Zhang L, Ma Y: Isolation of microsatellite DNA and the polymorphic locus screening from Phlebotomus chinensis. Chin J Parasitol Parasit Dis. 2009, 27 (6): 503-507.
Schuelke M: An economic method for the fluorescent labeling of PCR fragments. Nat Biotechnol. 2000, 18 (2): 233-234. 10.1038/72708.
Nei M: Molecular evolutionary genetics. 1987, New York: Colombia University Press
Goudet J: FSTAT (Version 1.2): A computer program to calculate F-statistics. J Hered. 1995, 86 (6): 485-486.
Brookfield JF: A simple new method for estimating null allele frequency from heterozygote deficiency. Mol Ecol. 1996, 5 (3): 453-455.
Van Oosterhout C, Hutchinson WF, Wills DPM, Shipley P: Micro-Checker: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes. 2004, 4 (3): 535-538. 10.1111/j.1471-8286.2004.00684.x.
Raymond M, Rousset F: GENEPOP (Version 1.2): Population genetics software for exact tests and ecumenicism. J Hered. 1995, 86 (3): 248-249.
Evanno G, Regnaut S, Goudet J: Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol. 2005, 14 (8): 2611-2620. 10.1111/j.1365-294X.2005.02553.x.
Pritchard JK, Stephens M, Donnelly P: Inference of population structure using multilocus genotype data. Genetics. 2000, 155 (2): 945-959.
Excoffier L, Laval G, Schneider S: Arlequin (version 3.0): an integrated software package for population genetics data analysis. Evol Bioinform Online. 2005, 1: 47-50.
Slatkin M: A measure of population subdivision based on microsatellite allele frequencies. Genetics. 1995, 139 (1): 457-462.
Gaggiotti OE, Lange O, Rassmann K, Gliddon C: A comparison of two indirect methods for estimating average levels of gene flow using microsatellite data. Mol Ecol. 1999, 8 (9): 1513-1520. 10.1046/j.1365-294x.1999.00730.x.
Mantel N: The detection of disease clustering and a generalized regression approach. Cancer Res. 1967, 27 (2): 209-220.
Ovenden JR, Peel D, Street R, Courtney AJ, Hoyle SD, Peel SL, Podlich H: The genetic effective and adult census size of an Australian population of tiger prawns (Penaeus esculentus). Mol Ecol. 2007, 16 (1): 127-138.
Wang ZJ, Xiong GH, Guan LR: The achievement of Leishmaniasis control in China. Chin J Epidem. 2000, 21 (1): 51-54.
Guan LR, Wang J, Liu PZ, Chang ZG: The Bionomics of Phleboomus chinensis in the nountainous regions of southern Kansu and the Loess Plateau of northern Shensi. Acta Entomol Sin. 1980, 23 (1): 25-31.
Della Torre A, Fanello C, Akogbeto M, Dossou-yovo J, Favia G, Petrarca V, Coluzzi M: Molecular evidence of incipient speciation within Anopheles gambiae s. s. in West Africa. Insect Mol Biol. 2001, 10 (1): 9-18. 10.1046/j.1365-2583.2001.00235.x.
Gentile G, Slotman M, Ketmaier V, Powell JR, Caccone A: Attempts to molecularly distinguish cryptic taxa in Anopheles gambiae s. s. Insect Mol Biol. 2001, 10 (1): 25-32. 10.1046/j.1365-2583.2001.00237.x.
Lawniczak MK, Emrich SJ, Holloway AK, Regier AP, Olson M, White B, Redmond S, Fulton L, Appelbaum E, Godfrey J: Widespread divergence between incipient Anopheles gambiae species revealed by whole genome sequences. Science. 2010, 330 (6003): 512-514. 10.1126/science.1195755.
Ma Y, Bu L, Hua X: [20-year search on molecular markers of Leishmania isolates from different Kala-azar foci in China to confirm whether genetic fingerprints of Kala-azar pathogens correlate with disease types]. J Biomed Engineer. 2011, 28 (5): 997-1000.
Acknowledgements
The authors thank Leyuan Shang, Yunpu Su, Qiuye Yan for providing Henan specimens, Xinyuan CDC in Xinjiang, Kashi CDC in Xinjiang, and Yin Ma and Lin Lin for assistance with specimen collection. We thank Changfa Jin for his help in the morphological identification and for collecting specimens specifically for this study. This work was supported by the National Natural Science Foundation of China (No. 30771869 for YM). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
YM conceived and designed the experiments. LZ performed the experiments. YM and JX analyzed the data, wrote the manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Zhang, L., Ma, Y. & Xu, J. Genetic differentiation between sandfly populations of Phlebotomus chinensis and Phlebotomus sichuanensis (Diptera: Psychodidae) in China inferred by microsatellites. Parasites Vectors 6, 115 (2013). https://doi.org/10.1186/1756-3305-6-115
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1756-3305-6-115