
Abstract
Background
Cutaneous Leishmaniasis (CL) is endemic in Sudan. It is caused by Leishmania major parasites and transmitted by Phlebotomus papatasi sandflies. Recently, uncommon clinical manifestations of CL have been reported. Moreover, L. donovani parasites that cause Visceral Leishmaniasis (VL) have been isolated from CL lesions of some patients who contracted the disease in Khartoum State, Central Sudan with no history of travelling to VL endemic sites on south-eastern Sudan. Because different clinical manifestations and the parasite behaviour could be related to genetic differentiation, or even sub-structuring within sandfly vector populations, a population genetic study was conducted on P. papatasi populations collected from different localities in Khartoum State known for their uncommon CL cases and characterized by contrasting environmental conditions.
Methods
A set of seven microsatellite loci was used to investigate the population structure of P. papatasi samples collected from different localities in Khartoum State, Central Sudan. Populations from Kassala State, Eastern Sudan and Egypt were also included in the analyses as outgroups. The level of genetic diversity and genetic differentiation among natural populations of P. papatasi was determined using F ST statistics and Bayesian assignments.
Results
Genetic analyses revealed significant genetic differentiation (FST) between the Sudanese and the Egyptian populations. Within the Sudanese P. papatasi populations, one population from Gerif West, Khartoum State, exhibited significant genetic differentiation from all other populations including those collected as near as 22 km.
Conclusion
The significant genetic differentiation of Gerif West P. papatasi population from other Sudanese populations may have important implication for the epidemiology of leishmaniasis in Khartoum State and needs to be further investigated. Primarily, it could be linked to the unique location of Gerif West which is confined by the River Nile and its tributaries that may act as a natural barrier for gene flow between this site and the other rural sites. The observed high migration rates and lack of genetic differentiation among the other P. papatasi populations could be attributed to the continuous human and cattle movement between these localities.
Similar content being viewed by others


Background
Female sandflies of the genus Phlebotomus in the Old World are vectors of Leishmania parasites, the causative agent of different clinical forms of leishmaniasis [1]. The disease is endemic in 88 countries in five continents with a total of 350 million people at risk [2].
The parasites within the Leishmania donovani complex usually invade the macrophages of the liver, spleen and bone marrow causing the severe symptoms of the fatal Visceral Leishmaniasis (VL) [3, 4]; while the parasites of L. major complex which cause Cutaneous Leishmaniasis (CL) invade the subcutaneous reticulo-endothelial system and cause self-healing lesions that leave life-long scars [5, 6].
Phlebotomus papatasi (Scopoli, 1786) is the principal vector of Leishmania major in the Old World [1]. It is a widely distributed species found in variable habitats and associated with a wide range of vertebrate hosts [7, 8]. Due to their poor dispersal capacity, populations of P. papatasi are expected to show some genetic structuring along their geographical range as a result of adaptation to local habitats and limited gene flow [9]. Such genetic variability could play an important role in the epidemiology and clinical manifestations of leishmaniasis since it affects the vectorial capacity of the vector [10, 11].
Previous studies focusing on potential population sub-structuring in P. papatasi used a number of molecular markers and provided contrasting results. For example, isoenzyme analyses clearly separated Western Mediterranean P. papatasi populations from those from eastern countries but, predictably, failed to reveal differences between urban and rural sandfly populations in Morocco [12]. Mitochondrial DNA sequences coding for cytochrome b (mtDNA cyt b) and sequences from the second internal transcribed spacer of the ribosomal DNA (rDNA ITS2) were both used to study the population structure of P. papatasi populations from North Africa and the Mediterranean sub-region countries [13, 14]. The analyses of mtDNA cyt b sequences suggested some level of genetic differentiation among widely separated populations and revealed a pattern of isolation-by distance between populations from Syria, Egypt, Israel/Palestine and Turkey [13]. The ND4 mtDNA region was also used in combination with the rDNA ITS2 to study the population structure of P. papatasi from 18 countries from North Africa, the Mediterranean sub-region, Saudi Arabia and India, but revealed no clear phylogeographic structure between those populations. However, signs of restricted gene flow were found among populations from Iran, Egypt, Syria, Yemen and Turkey [14].
Microsatellite markers combined with Bayesian statistic analysis were recently used to study the population structure of Phlebotomus papatasi populations in countries from the North-African and the Mediterranean sub-regions. This study confirmed the occurrence of highly significant genetic differentiation between some populations [15]. However, the geographical scale of the study did not allow for detecting possible genetic differentiation at the local level, which may be the most relevant for explaining the observed patterns of variation in epidemiologically relevant traits observed in some regions.
Thus, so far, genetic differentiation among P. papatasi populations could not be demonstrated at the local geographical level, despite evidence suggesting that it may occur. For example, Schmidt and Schmidt (1963) observed marked morphometric variations within the Egyptian populations of P. papatasi suggestive of sub-structuring [16]. Using isoenzyme analysis, Kassem et al. reported the presence of polymorphisms at many isoenzyme loci among populations of P. papatasi from Egypt but no significant genetic differentiation could be detected [17, 18]. It must be stressed that the possibility of genetic differentiation in P. papatasi populations at the local scale has not yet been explored using microsatellite markers which, with their higher mutation rates, should be comparatively much more informative than isoenzymes and other sequence loci [19].
In Sudan, CL is caused by L. major and transmitted by P. papatasi[20, 21]. Before the 1970s, the disease was confined to the western parts of the country. Thereafter, major epidemics occurred along the River Nile and the disease became endemic in many regions of the country [21]. The usual clinical forms of the disease usually heal spontaneously without the need for medical treatment, a matter which discourages patients from attending the health centres, and therefore obscures the incidence of the disease [22]. Visceral Leishmaniasis (VL) which is caused by L. donovani and transmitted by P. orientalis is known to be endemic in eastern and southern parts of Sudan [4, 23], with few scattered foci along the White Nile and Darfur [24]. Recently, uncommon clinical manifestations of CL that did not heal spontaneously nor responded to usual drugs have been reported [25]. Moreover, L. donovani parasites have been isolated from CL lesions of some patients who contracted the disease in Khartoum State, Central Sudan, with no history of travelling to VL endemic sites [26]. Recent studies have demonstrated the possibility of genetic exchanges between different strains and species of the Leishmania parasites [27–29] which may further complicate the epidemiology of the disease since hybrid parasites may adapt differently to the vector and reservoir hosts [30].
Because different clinical manifestations and the parasite behaviour may also be related to genetic differentiation or sub-structuring within sandfly vector populations [11, 31], we conducted a population genetic study of P. papatasi populations in Sudan from broad to local geographical scale. This was done using a set of 5 microsatellite markers, especially developed for P. papatasi[32], and given the paucity of such markers, by testing and using additional markers developed for P. perniciosus a related phlebotomine species [33].
P. papatasi populations were collected from different localities characterized by the distribution of the atypical CL cases. The level of genetic diversity and genetic differentiation among natural populations of P. papatasi was determined using F ST and Bayesian assignments. Identifying potential factors leading to genetic differentiation and structuring in P. papatasi populations might improve our understanding of the epidemiology of the disease and help develop appropriate control strategies.
Methods
Study sites
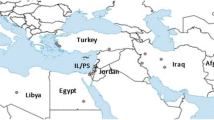
A total of 126 male specimens of P. papatasi, which are typically easier to identify morphologically than females in this and other phlebotomine species, were collected from five different localities in Sudan separated by 20 to 440 Km (Figure 1, Table 1). Individuals from a colonized population from Egypt (EGY) (~1600 Km from Khartoum State) were also included. The Sudan study sites were selected according to the distribution of the uncommon CL cases. They also represent different ecological zones separated by the River Nile and its tributaries. Four sites were located in Khartoum State; namely, Moyleih (MO), a semi-desert area on the west bank of the White Nile, El-Trais (TR), an irrigated area on the west bank of the White Nile; Sirougia village (SR) is a green rich irrigated area with many horticultural schemes on the east bank of the River Nile, northern Khartoum and Gerif West (GW) used to be green farms on the west bank of the Blue Nile, but recently changed into a residential area (also known as El-Manshya). Cutaneous leishmaniasis is not common in Moyleih and El-Trais areas; however some of the patients with CL lesions due to L. donovani were from these sites (Pers. Comm. Prof M. M. Mukhtar, Institute of Endemic Diseases, Sudan).Sirougia is a well-known endemic focus of CL where previous epidemic occurred beside some of the uncommon CL cases came from the nearby villages (Pers. Comm. Prof A. M. Musa, Institute of Endemic Diseases, Sudan). Gerif West is not known to be endemic for cutaneous leishmaniasis. However, some of the uncommon CL and CL due to L. donovani cases were from this site. The fifth Sudan study site was the village of Wad Shariefai (SHR) in Kassala State, Eastern Sudan which is located in the immediate vicinity of the dry savannah habitats of the Tajouj Forest. Wad Shariefai village is a site where three closely-related species belonging to the subgenus Phlebotomus phlebotomus (P. papatasi, bergeroti and duboscqi) were found sympatrically. Due to the far location of this Eastern site it is expected that P. papatasi from this site would be genetically different from P. papatasi populations from Khartoum Sate, Central Sudan.

Map of the Sudan showing leishmaniasis endemic sites and the main study sites from which Phlebotomus papatasi populations were collected. VL endemic area (grey area), CL endemic sites (●), Khartoum and Kassala States (■) (right). Map of the main Study sites in Khartoum State (left).
Microsatellite PCR amplification
Samples were processed and DNA was extracted as previously described [34]. Microsatellite loci were amplified using the PCR primers originally developed by Hamarsheh et al. and Aransay et al. [32, 33]. The PCR reactions were performed in a gradient thermocycler (PT200, MJ) for 30 cycles. The PCR mix contained 1.5 to 3μl of genomic DNA (5–8ng/μl) extracted from individual male sandfly, 5x PCR buffer (Promega, Madison, WI), 15pmol of each primer, 200mM of each dNTPs, 1.2μl of 25mm MgCl2 and 0.5U Taq ® DNA polymerase (Promega, Madison, WI) in 20μL total reaction volume. The thermal cycling conditions were an initial hold at 95°C for 2min, followed by 30 cycles of 94°C for 30s, 55°C for 30s and 72°C for 30s and a final extension at 72°C for 5min. For amplifications involving the primer AA13, a touchdown PCR programme was performed. The programme steps were as mentioned above except that the number of cycles was increased to 35 and the starting annealing temperature was 59°C, which was then decreased by 1°C for the first five cycles. For correct sizing of polymorphic PCR products, the forward primers were labelled with 5’-fluorescent tags. Details of the primers sequences; repeat motif, annealing temperature, and fluorescent labelling are given in Table 2.
PCR product multiplexing
PCR product multiplexing was done according to the amplified allele size and the fluorescent dye labelling of each product in order to create two primer sets. Set A contained a mix of Pap1, Pap2, AA13, and AA82, while set B was a mix of Pap4, AA18, and AA24. About 16 samples with different dilutions (1:10, 1:20, and 1:50) were initially genotyped at Eurofins MWG Operon Company, Ebersberg, Germany. According to the results from the first genotyped samples, a suitable dilution factor was chosen and the two sets of sample microplates were prepared, tightly packaged in dry ice and submitted for subsequent genotyping in the aforementioned company.
Microsatellite data analysis
Because the repeat arrays of all the microsatellite loci consist of trinucleotide repeats (except for Pap2 which consist of dinucleotide repeats), the allele sizes were expected to differ predominantly by multiples of three nucleotides. Therefore any exceptions were treated as a result of insertions or deletions (indels) of odd numbers of bases in the flanking region.
The data were screened for the presence of stutters, null alleles and large allele dropout using the software Microchecker version 2.2.3 available at http://www.microchecker.hull.ac.uk[35]. Then the data were analyzed using ARLEQUIN version 3.5 available at http://cmpg.unibe.ch/software/arlequin3[36]. The programme was used to calculate allele frequencies at each microsatellite locus and to test whether they conformed to Hardy-Weinberg equilibrium (HWE) in each population. The same programme was used to estimate genetic differentiation (FST) between populations and the level of gene flow (Nm) among them, as well as assessing whether patterns of genetic divergence among P. papatasi populations followed an isolation-by-distance pattern. The genotype assignment test implemented in ARLEQUIN programme was used to assign individual sandfly genotypes to their population of origin by computing the log-likelihood of each individual multi-locus genotype in each population sample, assuming that the individual belongs to that population [36]. The significance of all tests (P <0.05) was obtained with 1000 permutations and 100000 Markov chain steps.
Hidden population sub-structure within P. papatasi populations were further investigated using the Bayesian clustering algorithm implemented in the software STRUCTURE version 2, available at http://pritch.bsd.uchicago.edu[37]. The programme was run using an admixture model with a burn-in period of 10000 iterations, followed by 100000 iterations of Markov chain Monte Carlo (MCMC) repeats for each setting of K from 1 to 10 and no prior information about the population structure was provided. After calculations, the value of the ad hoc ΔK was plotted against the assumed number of populations (K = 10), the highest peak of the graph was considered as the number of possible subpopulations in the data set.
Results
Allelic richness and heterozygosity
The three markers Pap1, Pap2 and Pap4 showed varying degrees of heterozygosity among the different study populations; although the two markers Pap3 and Pap5 did not work well with the Sudanese P. papatasi populations even after several trials with different amplification conditions. Amongst the markers for P. perniciosus, AA13, AA18, AA24 and AA82, were successfully amplified in P. papatasi. However, these markers tended to be less polymorphic than in the original species. The total number of alleles per locus ranged from 1 to 5 with locus AA18 being monomorphic in all populations. In addition, each of the two loci AA13 and AA24 were found to amplify two different polymorphic loci with distinct allele size ranges (i.e. AA13a: 81-96bp and AA13b 129-132bp; AA24a: 141–147 and AA24b: 255–273) which were not found to be in linkage disequilibrium. This suggests that these two loci were the results of duplications to different unlinked regions of the genome; thus they were treated as two additional loci thereby increasing the total number of loci examined to nine (Table 3).
There was no evidence for scoring error due to stuttering, large allele dropout or null alleles after checking the dataset with Microchecker software. Except for locus AA18 which was monomorphic in all P. papatasi populations, the other eight studied microsatellite loci were found to be polymorphic. The total number of alleles per locus ranged from 2 to 5 with an average of 2.77 alleles per locus and an average gene diversity (mean expected heterozygosity) of 0.355 ± 0.226 (Table 3).
The six P. papatasi populations were found to be polymorphic at 88.9% of the amplified loci. Allele frequency distribution between the 6 P. papatasi populations varied from locus to locus, with some loci having similar distributions and others being highly variable. For example, allele 128 for locus Pap1 was the most common allele among all Sudanese populations while allele 125 was the most common among the Egyptian population. For locus Pap2, allele 85 was the most common among populations from Sudan while allele 89 was the most common in a population from Egypt. Private alleles were detected at locus Pap1 in the MO population, locus AA24a in the SR population, loci AA13a and AA24b in the SHR population, and locus Pap2 in population EGY. Locus AA18 was monomorphic and was excluded from subsequent analyses (Table 4).
There was a considerable variation in mean heterozygosity in P. papatasi populations, including the Egyptian laboratory colony, despite its long history of colonization. Surprisingly, this population was the most variable with approximately 0.493 heterozygosity (i.e. on average 49% of the individuals were heterozygous at any given locus). Meanwhile, MO population was the most heterogeneous among all the Sudanese populations (40% heterozygosity). The most uniform population in the study was TR with 31.5% heterozygosity (Table 5).
Linkage disequilibrium and Hardy–Weinberg equilibrium
As few as 14 out of 168 comparisons between pairs of loci were found to be in significant linkage disequilibrium. However, none of those comparisons remained significant after Bonferroni correction.
Each locus was tested for significant departure from Hardy-Weinberg equilibrium (HWE). Observed heterozygosity varied from 0.000 to 0.960 while expected heterozygosity ranged from 0.039 to 0.660. The observed heterozygosity was higher than the expected value in 25 cases and lower than expected in 13 cases of the comparisons made between P. papatasi populations (Table 6). Significant deviations from HWE due to heterozygotes deficiency were observed in locus Pap2 within the SR and EGY populations, locus Pap4 within the MO, TR, SR, and GW populations, and locus AA24a within SR population. This could be due to population subdivision rather than the existence of null alleles since amplification was successful for all individuals. Significant deviations from HWE due to homozygote deficiency were found in locus Pap1 within GW population and locus AA13b in all populations. The Egyptian P. papatasi population was monomorphic for two loci (Pap4, and AA13a) and the GW population was monomorphic for AA13a (Table 6). Because of their consistent deviation from HWE among all populations, the two loci Pap4 and AA13 b were excluded from subsequent analyses.
Estimates of genetic differentiation between populations
The genetic differentiation between pairs of populations, as measured by FST , ranged from 0.007 to 0.233. Significant FST estimates (FST =0.147-0.233) were observed between all P. papatasi populations from Sudan when compared to those from Egypt (Table 7). Significant genetic differentiation was also observed between P. papatasi populations from GW and SHR (FST =0.033), whereas moderate but non-significant differentiation (0.011-0.014) was observed between SHR and other populations from Khartoum State (MO, TR, and SR). Within Khartoum State, significant FST (0.100 to 0.125) estimates were also observed between GW and MO, TR, and SR populations. The populations from MO, TR, and SR showed little genetic differentiation amongst themselves as indicated by low FST values ranging from 0.007 to 0.046. A neighbour-joining tree based on all pair-wise FST estimates confirmed that the GW population, whilst being distinct from all other populations in Sudan is comparatively more similar to SHR than to nearer populations (Figure 2).

Neighbour-joining (NJ) dendrogram showing the genetic distance between Phlebotomus papatasi populations from Sudan and Egypt. Relationships based on Wright’s F ST statistics
As expected, FST estimates translated into high estimates of migration rate, Nm, among populations from MO and TR, Southwest Khartoum State. High gene flow estimates were also observed between populations from MO, TR and SR and SHR, despite the large geographical distance between them. However, very little gene flow was observed between those same populations and GW which is approximately geographically equidistant to the other three sites in Khartoum State (Table 7).
A Mantel test showed that there was a positive but non-significant correlation (r = 0.08, P > 0.05) between linearized estimates of genetic differentiation FST (FST/(1-FST)) and geographical distances (log km) (Figure 3).

Relationship between linearized F ST and log geographic distance among Phlebotomus papatasi populations from Sudan and Egypt. 1 = MO, 2 = TR, 3 = GW, 4 = SR, 5 = SHR, 6 = EGY (Mantel test: P > 0.05, non-significant).
Population structure
The STRUCTURE genotype assignment programme clustered our study populations into two groups (Figure 4). The dataset was further analyzed by sorting out individuals between the two suggested subpopulations as belonging to subpopulation A (grey colour) and subpopulation B (black colour) based on the likelihood of their genotype to belong to one or the other population (Figure 5). Most of the individuals in the Egyptian population (88%) were assigned to subpopulation B, while most of the individuals in the Sudanese populations belonged to subpopulation A. However, the observed heterogeneity in population assignment - e.g. 83% of TR and 73% of MO belong to the population A - suggested that the two clusters do not represent reproductively isolated populations (Table 8).

Relationship between the second order rates of change of the likelihood function (Delta K) averaged over 10 runs (K). The highest peak corresponds to the number of the possible subpopulations inferred by the software STRUCTURE (see text for details).

Estimated membership coefficients of individual sandflies from six P . papatasi populations to the two putative populations predicted by program STRUCTURE. Subpopulation (A) grey; subpopulation (B) black. Membership coefficients were estimated from microsatellite loci genotypic frequencies.
The same result was also observed when attempting to assign individual sandflies to the different collection sites by computing the log-likelihood of the genotype of each individual based on the allelic frequencies of each population. According to the population assignment test, about 80% of the Egyptian sandfly individuals were correctly assigned to their population of origin. However, none of the individual sandflies from the Sudanese populations of MO and TR were assigned to the Egyptian one (Table 9).
Discussion
This study further validates the use of microsatellites for studying subtle patterns of genetic differentiation between populations at the microgeographic scale. In such situations, microsatellite markers with their high-mutation rates and high allelic diversity are known to perform much better than slow evolving allozymes and other nuclear and mitochondrial markers [38, 39]. However, useful microsatellites are neither highly abundant nor easily isolated from arthropod species [40]. Homologous microsatellite loci have sometimes been amplified in species related to the one they were isolated from using the same primers, but amplification success strongly depends on the divergence time separating the species concerned [41].
Here, given the low number of microsatellite loci available for P. papatasi, additional loci originally designed for P. perniciosus were tested and successfully cross-amplified. As expected, some of these loci produced fewer alleles than in the original species (P. perniciosus). For example, the total number of alleles per locus for these loci ranged from 4 to 9 in 13 Spanish populations of P. perniciosus with a sample size ranging from 13 to 38 but here in our study allelic richness ranged from 1 to 5. Surprisingly, two primer pairs AA24 and AA13 amplified more than one locus in P. papatasi with distinct allele size-ranges and genetically unlinked, indicating a duplication of the genome in the region of those repeat sequences in P. papatasi. These loci were polymorphic in most P. papatasi populations, which contrasts with previous results in P. perniciosus where locus AA24 was not significantly polymorphic in 12 out of 13 studied populations, and locus AA13 was monomorphic in 5 of them [42].These variations suggest strikingly different evolutionary trajectories. Future comparative genomic studies might help elucidate the phylogenetic relationships and genome rearrangements characterizing these sandfly vector species.
Interestingly, no significant pattern of isolation-by-distance was observed amongst the 6 P. papatasi populations sampled from Central and Eastern Sudan, and Egypt. This result is due to the unexpected significant genetic differentiation found at the local geographical scale between the Sudanese populations that interfered with broader geographical patterns of genetic differentiation. Importantly, the P. papatasi population from GW was significantly genetically differentiated from that of SHR, located 400km away in Eastern Sudan. But the same population (GW) was also significantly genetically differentiated from the other 3 populations from Khartoum State (MO, TR and SR) despite these populations being only 22-27km apart. It is noteworthy that GW locality is isolated by the River Nile and its tributaries from the other collection sites in 3 directions (North, West and East). Furthermore there are no direct routes of transportations between this and other locations in Khartoum State (see map in Figure 1), a matter that may constitute a further barrier to gene flow for the GW population. It is noteworthy that uncommon clinical manifestations of CL were also reported from SR which used to be an endemic site for common CL cases. However, P. papatasi population from this site was significantly genetically differentiated from GW than from the other two sites from Khartoum State (MO and TR). Previous studies demonstrated the presence of different genetic groups among L. major strains from different locations in the same country which could be related to different P. papatasi subpopulations [43, 44]. Whether the parasite strains causing these complications in SR and GW belong to the same genetic groups or not, remains to be further explored.
It is well known that populations in close proximity are genetically more similar than distant populations and that high gene flow usually precludes local adaptation [45]. Therefore, the lack of interpopulation genetic differentiation among Khartoum State populations (MO, TR and SR) could be attributed to the flies’ own dispersal abilities, to human transportation and potentially to wind dispersal. P. papatasi is known to be capable of flying up to 2km per night in open desert where they can be also be carried by air currents. Moreover, sandflies usually swarm over their host during feeding and usually mating takes place on the host; thus they may be capable of movement along with animal herds [46, 47]. It should be stated that the Moyleih area (MO) hosts a famous cattle market where animals are brought from Western and Southern Sudan either for slaughter in the nearby “Ghunawa” abattoir or to be herded across the Nile bridge towards the “Kadaro” abattoir on the Eastern bank of the Nile very near the village of Sirougia (SR). In addition, the seasonal movement of the camel- herding nomads northward during the wet season and southward at the end of the wet season [48] may contribute to the dispersal of sandfly over a wide range.
The close genetic similarity among sandfly populations from Khartoum State (MO, TR and SR) and Wad Shariefai (SHR), Eastern Sudan, despite the large geographical distance, could be attributed to the prevailing environmental conditions such as wind speed, humidity and temperatures that may play an important role in sandfly dispersal [1]. Although each of the studied sites has their own peculiar environmental conditions, they all lie within the dry savannah belt [49]. Moreover MO and TR, although they are apparently desert areas, used to be covered by thick savannah forests that may have had similar environmental conditions as SHR [50]. Since the mid-1970s the whole Western Sudan, including the area west of the White Nile, suffered from continuous drought periods that lead to deterioration of the plant cover, soil, water and biodiversity [50–52]. Previous studies on the distribution and phenology of P. papatasi indicate that this species can withstand extreme environmental conditions and survive even in areas with extreme temperature and aridity [53–55]. The prevailing conditions within deep wall- cracks and small muddy huts provide high humidity and cooler temperatures suitable for sandfly proliferation in these sites. All these factors may explain the genetic similarity observed between distant sandfly populations since flies can disperse largely freely between vast expenses of largely similar habitats.
Low genetic differentiation between P. papatasi populations separated by large geographical distances has also been reported in previous studies based on different molecular markers [14, 56, 57]. In addition, population genetic studies of other important sandfly species demonstrated the homogeneity of these populations in a radius of 20km while a certain degree of structuring was observed in a zone of approximately 1km [58]. It is worth mentioning that little genetic differentiation was also shown in populations of P. orientalis in Sudan using 30 RAPD markers. In addition, populations from Dinder National Park, South-Eastern Sudan were found to be genetically very similar to population from SR, Khartoum State [59]. It has been recently postulated that latitude is the main factor determining the degree of genetic diversity among P. papatasi populations [13].
This study also revealed significant genetic differentiation and restricted gene flow between the Egyptian EGY population and all the Sudanese populations, which suggests some degree of isolation-by-distance despite the overall lack of a significant correlation between the geographical distance and estimates of genetic differentiation. The Egyptian P. papatasi colony also showed unexpected high degree of heterozygosity compared to the Sudanese field populations, although long colonization and adaptation to laboratory conditions are usually thought to minimize the genetic variability in laboratory populations [60, 61]. Unfortunately, no detailed information was available about the EGY colony, although it may be speculated that this diversity may be a result of its augmentation with new collections, which is a common practice in sand fly colonization. It must be mentioned that in previous studies, high levels of polymorphism and heterozygosity were also observed in P. papatasi colony from Sinai, Egypt even after the 33 generations [62].
Conclusion
The significant differentiation of the GW Phlebotomus papatasi population from the other Sudanese populations may have important consequences on the epidemiology of leishmaniases in Khartoum State and warrants further investigation. Such genetic variations could potentially correlate with differences in the vectorial capacity of the sandfly vector to transmit different strains of the parasites or even hybrids. Future studies using a larger number of microsatellite loci, more populations and more specimens per population are also needed to further elucidate patterns of genetic differentiation of P. papatasi in Sudan. This knowledge would improve our understanding of the epidemiology of the disease in Sudan, and ultimately would result in improved disease and vector control programs.
Abbreviations
- CL:
-
Cutaneous leishmaniasis
- VL:
-
Visceral leishmaniasis
- MO:
-
Moyleih, a semi-desert area on the west bank of the White Nile
- TR:
-
El-Trais, an irrigated area on the west bank of the White Nile
- SR:
-
Sirougia village, a green rich irrigated area on the east bank of the River Nile
- GW:
-
Gerif West, a residential area on the west bank of the Blue Nile
- SHR:
-
Wad Shariefai village, in Kassala State, Eastern Sudan where the three closely-related species belonging to the subgenus Phlebotomus Phlebotomus were found sympatrically
- EGY:
-
Colony samples from Egypt
- HWE:
-
Hardy-Weinberg equilibrium
- He:
-
Expected heterozygosity
- Ho:
-
Observed heterozygosity
- SD:
-
Standard deviation
- Wright’s FST:
-
Measure of population differentiation
- FIS:
-
Inbreeding coefficient
- (Nm):
-
Rate of gene flow among populations.
References
Killick-Kendrick R: The biology and control of phlebotomine sand flies. Clin Dermatol. 1999, 17: 279-289. 10.1016/S0738-081X(99)00046-2.
World Health Organization (WHO): Disease watch focus: leishmaniasis. TDR | Nature Reviews Microbiology. 2004, 2: 692-693. 10.1016/S0140-6736(98)10178-2.
Herwaldt BL: Leishmaniasis. Lancet. 1999, 354: 1191-1199.
El-Hassan AM, Zijlstra EE: Leishmaniasis in sudan. Visceral leishmaniasis. Trans R Soc Trop Med Hyg. 2001, 95 (suppl 1): 27-58. 10.1016/S0738-081X(99)00041-3.
Oumeish OY: Cutaneous leishmaniasis: a historical perspective. Clin Dermatol. 1999, 17: 249-254. 10.1016/S0738-081X(99)00051-6.
Ashford RW: Cutaneous leishmaniasis: strategies for prevention. Clin Dermatol. 1999, 17: 327-332.
Lewis DJ: A taxonomic review of the genus phlebotomus (diptera: psychodidae). Bull Br Mus (Nat Hist) (Entomol). 1982, 45: 121-209. 10.1046/j.1365-2915.1999.00140.x.
Ghosh KN, Mukhopadhyay JM, Guzman H, Tesh RB, Munstermann LE: Interspecific hybridization and genetic variability of phlebotomus sandflies. Med Vet Entomol. 1999, 13: 78-88.
Ready PD: Should sand fly taxonomy predict vectorial and ecological traits?. J Vec Ecol. 2011, 36: S17-S22. 10.1080/00034989860175.
Hanafi HA, El Sawaf BM, Fryauff DJ, Beavers GM, Tetreault GE: Susceptibility to leishmania major of different populations of phlebotomus papatasi (diptera:psychodidae:) from endemic and non-endemic regions of Egypt. Ann Trop Med Parasitol. 1998, 92: 57-64. 10.1016/0169-4758(95)80137-5.
Lanzaro GC, Warburg A: Genetic variability in phlebotomine sandflies: possible implications for leishmaniasis epidemiology. Parasitol Today. 1995, 11: 151-154.
Boussaa S, Perrotey S, Boumezzough A, Harrak R, Hilali S, Pesson B: Isoenzymatic characterization of phlebotomus papatasi (diptera: psychodidae) of the Marrakech area, Morocco. J Med Entomol. 2009, 45: 370-374. 10.1111/j.1365-2915.2007.00695.x.
Hamarsheh O, Presber W, Abdeen Z, Sawalha S, Al-Lahem A, Schönian G: Genetic structure of mediterranean populations of the sandfly Phlebotomus papatasi by mitochondrial cytochrome b haplotype analysis. Med Vet Entomol. 2007, 21: 270-277. 10.1016/j.meegid.2007.12.001.
Depaquit J, Lienard E, Verzeaux-Griffon A, Ferté H, Bounamous A, Gantier J-C, Hanafi HA, Jacobson RL, Maroli M, Moin-Vaziri V: Molecular homogeneity in diverse geographical populations of Phlebotomus papatasi (diptera: psychodidae) inferred from ND4 mtDNA and ITS2 rDNA: epidemiological consequences. Infect Genet Evol. 2008, 8: 159-170. 10.1111/j.1365-2915.2008.00784.x.
Hamarsheh O, Presber W, Yaghoobi-Ershadi MR, Amro A, Al-Jawabreh A, Sawalha S, Al-Lahem A, Das ML, Guernaoui S, Seridi N: Population structure and geographical subdivision of the Leishmania major vector Phlebotomus papatasi as revealed by microsatellite variation. Med Vet Entomol. 2009, 23: 69-77.
Schmidt ML, Schmidt JR: A morphological study of Phlebotomus papatasi from Egypt (diptera: psychodidae). Ann Entomol Soc Am. 1963, 56: 567-573.
Kassem H, Fryauff DJ, El Sawaf BM, Shehata MG, Shoumar NF: Electrophoretic comparison of the leishmania vectors Phlebotomus papatasi and P. Langeroni (diptera: psychodidae). J Med Entomol. 1990, 4: 592-601.
Kassem H, Fryauff DJ, Shehata MG: Enzyme polymorphism and genetic variability of one colonized and several field populations of Phlebotomus papatasi (diptera: psychodidae). J Med Entomol. 1993, 30: 407-413. 10.1146/annurev.ento.48.091801.112647.
Krzywinski J, Besansky NJ: Molecular systematics of anopheles: from subgenera to subpopulations. Annu Rev Entomol. 2003, 48: 111-139. 10.1016/0035-9203(55)90066-3.
Kirk R, Lewis DJ: Studies in leishmaniasis in the Anglo-Egyptian Sudan. XI. Phlebotomus in relation to leishmaniasis in the Sudan. Trans R Soc Trop Med Hyg. 1955, 49: 229-240.
El-Safi SH, Peters W: Studies on leishmaniasis in Sudan. Epidemic of cutaneous leishmaniasis in Khartoum. Trans R Soc Trop Med Hyg. 1991, 85: 44-47. 10.1016/S0035-9203(01)90314-1.
El-Hassan AM, Zijlstra EE: Leishmaniasis in Sudan. Cutaneous leishmaniasis. Trans R Soc Trop Med Hyg. 2001, 95: 1-17. 10.1093/ije/25.4.862.
Seaman J, Mercer AJ, Sondorp E: The epidemic of visceral leishmaniasis in western Upper Nile, southern Sudan: course and impact from 1984 to 1994. Int J Epidemiol. 1996, 25: 862-871. 10.3201/eid1404.071099.
Kolaczinski JH, Hope A, Ruiz JA, Rumunu J, Richer M, Seaman J: Kala-azar epidemiology and control, southern Sudan. Emerg Infect Dis. 2008, 14: 664-666. 10.1016/j.trstmh.2005.04.009.
Elamin EM, Guerbouj S, Musa AM, Guizani I, Khalil EAG, Mukhtar MM, Elkadaro AMY, Mohamed HS, Ibrahim ME, Abdel Hamid MM: Uncommon clinical presentations of cutaneous leishmaniasis in Sudan. Trans R Soc Trop Med Hyg. 2005, 99: 803-808. 10.1016/j.trstmh.2007.10.005.
Elamin EM, Guizani I, Guerbouj S, Gramiccia M, El Hassan AM, Di Muccio T, Taha MA, Mukhtar MM: Identification of Leishmania donovani as a cause of cutaneous leishmaniasis in Sudan. Trans R Soc Trop Med Hyg. 2008, 102: 54-57. 10.1016/j.ijpara.2006.06.019.
Ravel C, Cortes S, Pratlong F, Morio F, Dedet J-P, Campino L: First report of genetic hybrids between two very divergent leishmania species: Leishmania infantum and Leishmania major. Int J Parasitol. 2006, 36: 1383-1388. 10.1073/pnas.0703678104.
Lukes J, Mauricio IL, Schönian G, Dujardin J-C, Soteriadou K, Dedet J-P, Kuhls K, Tintaya KWQ, Jirku M, Chocholova E: Evolutionary and geographical history of the Leishmania donovani complex with a revision of current taxonomy. PNAS. 2007, 104: 9375-9380.
Hamad SH, Musa AM, Khalil EAG, Abebe T, Younis BM, Elthair MEE, EL-Hassan AM, Hailu A, Bart A: Leishmania: probable genetic hybrids between species in Sudanese isolates. J Microbiol Antimicrob. 2011, 3: 142-145. 10.1126/science.1169464.
Akopyants NS, Kimblin N, Secundino N, Patrick R, Peters N, Lawyer P, Dobson DE, Beverley SM, Sacks DL: Demonstration of genetic exchange during cyclical development of Leishmania in the sand fly vector. Science. 2009, 324: 265-268. 10.1186/1471-2148-8-183.
Al-Jawabreh A, Diezmann S, Müller M, Wirth T, Schnur LF, Strelkova MV, Kovalenko DA, Razakov SA, Schwenkenbecher J, Kuhls K, Schönian G: Identification of geographically distributed sub-populations of Leishmania (Leishmania) major by microsatellite analysis. BMC Evol Biol. 2008, 8: 183-10.1111/j.1471-8286.2006.01359.x.
Hamarsheh O, Presber W, Abdeen Z, Sawalha S, AL-Lahem A, Schönian G: Isolation and characterization of microsatellite loci in the sand fly Phlebotomus papatasi (diptera: psychodidae). Mol Ecol Notes. 2006, 6: 826-828.
Aransay AM, Malarky G, Ready PD: Isolation (with enrichment) and characterization of trinucleotide microsatellites from Phlebotomus perniciosus, a vector of Leishmania infantum. Mol Ecol Notes. 2001, 1: 176-178. 10.1111/j.1365-2915.2010.00893.x.
Khalid N, Elnaiem D, Aboud M, Al Rabba F, Tripet F: Morphometric and molecular differentiation of Phlebotomus (Phlebotomus) sandflies. Med Vet Entomol. 2010, 24: 352-360. 10.1111/j.1471-8286.2004.00684.x.
Van Oosterhout C, Hutchinson WF, Wills DPM, Shipley P: Micro-checker: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes. 2004, 4: 535-538.
Excoffier L, Laval G, Schneider S: ARLEQUIN ver. 3.0: an integrated software package for population genetics data analysis. Evol Bioinform. 2005, 1: 47-50.
Pritchard JK, Stephens M, Donnelly P: Inference of population structure using multilocus genotype data. Genetics. 2000, 155: 945-959. 10.1038/sj.hdy.6801002.
Hedrick PW: Genetics of populations. 2005, Jones and Bartlett, 3
Conn JE, Mirabello L: The biogeography and population genetics of neotropical vector species. Heredity. 2007, 99: 245-256. 10.1046/j.1365-2583.1999.00142.x.
Day JC, Ready PD: Relative abundance, isolation and structure of phlebotomine microsatellites. Insect Mol Biol. 1999, 8: 575-580. 10.1111/j.1461-0248.2006.00889.x.
Selkoe KA, Toonen RJ: Microsatellites for ecologists: a practical guide to using and evaluating microsatellite markers. Ecol Lett. 2006, 9: 615-629. 10.1038/sj.hdy.6800246.
Aransay AM, Ready PD, Morillas-Marquez F: Population differentiation of Phlebotomus perniciosus in Spain following postglacial dispersal. Heredity. 2003, 90: 316-325. 10.1016/j.actatropica.2006.01.010.
Mahnaz T, Katrin K, Amer A-J, Isabel M, Gabriele S, Safar F, Hossein AM: Leishmania major: genetic heterogeneity of Iranian isolates by single-strand conformation polymorphism and sequence analysis of ribosomal DNA internal transcribed spacer. Acta Trop. 2006, 98: 52-58. 10.1186/1756-3305-4-9.
Hamarsheh O: Distribution of Leishmania major zymodemes in relation to populations of Phlebotomus papatasi sand flies. Parasit Vectors. 2011, 4: 9-10.1046/j.0962-1083.2001.01436.x.
François B, Nicolas L-M: The estimation of population differentiation with microsatellite markers. Mol Ecol. 2002, 11: 155-165. 10.1111/j.1365-2915.1988.tb00213.x.
Yuval B, Warburg A, Sleinch Y: Leishmaniasis in the Jordan valley. Dispersal characteristics of the sandfly Phlebotomus papatasi. Med Vet Entomol. 1988, 2: 391-395.
Tesh RB, Guzman H: Sand flies and the agents they transmit. The Biology of Disease Vectors. Edited by: Beaty BJ, Marquardt WC. 1996, Niwot, Colorado, USA: University Press of Colorado, 117-127.
Teklu T, Braun JV, Zaki E, Ali A: Research reports No. 88. Drought and famine relationships in Sudan: policy implications. 1991, Washington, D.C, USA: International Food Policy Research Institute (IFPRI), 140-
Abu Sin A, Abbakar AM: Kassala: The bridging state of eastern Sudan. Peace in eastern Sudan. 2009, Norway: BRIC
Alhassan SA: MA dissertation. Transformation in rural economic system in dry land areas of central Sudan. 2003, University of Khartoum, Faculty of Art, Department of Geography
Seid-Ahmed SA: Agriculture and globalization challenges. Strategic Studies Series. 1999, Khartoum, Sudan: Centre of Strategic Studies, in Arabic
Ibrahim AA, Dawoud HA: National perspective and strategic interventions for the impact of desertification on gender immigration from western states of sudan. Strategic and National Security. 2008, 1: 1-28. 10.1111/j.1365-2915.2003.00461.x.
Elnaiem DA: M.Sc dissertation. Studies on sandflies in active focus of cutaneous leishmaniasis, at Wad Elzaki and Elhashaba,White Nile province, Sudan. 1986, Khartoum, Sudan: University of Khartoum, Faculty of Science, Department of Zoology
Wasserberg G, Yarom I, Warburgy A: Seasonal abundance patterns of the sandfly Phlebotomus papatasi in climatically distinct foci of cutaneous leishmaniasis in Israeli deserts. Med Vet Entomol. 2003, 17: 452-456. 10.1603/033.046.0229.
Chelbi I, Kaabi B, Bijaoui M, Derbali M, Zhioua E: Spatial correlation between Phlebotomus papatasi scopoli (dipteral: psyehodidae) and incidence of zoonotie cutaneous leishmaniasis in Tunisia. J Med Entomol. 2009, 46: 400-402. 10.1046/j.1365-2583.1997.00175.x.
Esseghir S, Ready PD, Killick-Kendrick R, Ben-Ismail R: Mitochondrial haplotypes and phylogeography of Phlebotomus vectors of Leishmania major. Insect Mol Biol. 1997, 6: 211-225.
Parvizi P, Ready PD: Molecular investigation of the population differentiation of Phlebotomus papatasi, important vector of Leishmania major, in different habitats and regions of Iran. Iran Biomed J. 2006, 10: 69-77.
Rocha LDS, Falqueto A, Santos CBD, Grimaldi G, Cupolillo E: Genetic structure of Lutzomyia (nyssomyia) intermedia populations from two ecologic regions in Brazil where transmission of Leishmania (viannia) braziliensis reflects distinct eco-epidemiologic features. AmJTrop Med Hyg. 2007, 76: 559-565.
Numairy MS: Genetic diversity and population structure of Phlebotomus oreintalis (diptera: psychodidae) from selected regions in Sudan. M.Sc thesis. 2009, Khartoum, Sudan: University of Khartoum, Factulty of Science, Department of Zoology
Mukhopadhyay JEF, Rangel K, Ghosh K, Munstermann LE: Patterns of genetic variability in colonized strains of Lutzomyia longipalpis (diptera: psychodidae) and its consequences. AmJTrop Med Hyg. 1997, 57: 216-221.
Volf P, Volfova V: Establishment and maintenance of sandfly colonies. J Vec Ecol. 2011, 36: S1-S9.
Kassem HA, Fryauff DJ, Hanafi HA: Enzyme polymorphism and genetic variability of laboratory populations of Phlebotomus papatasi, P. bergeroti, P. langeroni, and P. perniciosus. J Egypt Soc Parasitol. 1999, 29: 459-472.
Acknowledgements
Many thanks are due to Dr Rania Baleela (Dept. of Zoology, University of Khartoum) for helping with microsatellite data analyses. Special thanks are due to Dr A. Rafie Mekhawi (Dept of Zoology, University of Buhri) for his help during the field trips. Many thanks for Prof. Hala Kassem (Ain shams university, Egypt) for providing P. papatasi samples from Egypt. This research received financial support from UNDP/ World Bank/WHO Special Programme for Research in Tropical Diseases (TDR) ID No (A60120), and Gordon Memorial trust, UK.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
NK carried out the field and laboratory work, microsatellite analysis and drafted the manuscript. MA, FA and DE helped in designing the field study, interpretation of the results and editing the manuscript. FT helped with the data analyses, interpretation of the results and editing the manuscript. All authors read and approved the final version of the manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Khalid, N.M., Aboud, M.A., Alrabba, F.M. et al. Evidence for genetic differentiation at the microgeographic scale in Phlebotomus papatasi populations from Sudan. Parasites Vectors 5, 249 (2012). https://doi.org/10.1186/1756-3305-5-249
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1756-3305-5-249