
Abstract
Background
Glossina fuscipes, a riverine species of tsetse, is the major vector of human African trypanosomiasis (HAT) in sub-Saharan Africa. Understanding the population dynamics, and specifically the temporal stability, of G. fuscipes will be important for informing vector control activities. We evaluated genetic changes over time in seven populations of the subspecies G. f. fuscipes distributed across southeastern Uganda, including a zone of contact between two historically isolated lineages. A total of 667 tsetse flies were genotyped at 16 microsatellite loci and at one mitochondrial locus.
Results
Results of an AMOVA indicated that time of sampling did not explain a significant proportion of the variance in allele frequencies observed across all samples. Estimates of differentiation between samples from a single population ranged from approximately 0 to 0.019, using Jost's DEST. Effective population size estimates using momentum-based and likelihood methods were generally large. We observed significant change in mitochondrial haplotype frequencies in just one population, located along the zone of contact. The change in haplotypes was not accompanied by changes in microsatellite frequencies, raising the possibility of asymmetric mating compatibility in this zone.
Conclusion
Our results suggest that populations of G. f. fuscipes were stable over the 8-12 generations studied. Future studies should aim to reconcile these data with observed seasonal fluctuations in the apparent density of tsetse.
Similar content being viewed by others

Introduction
Tsetse flies, Glossina spp (Diptera: Glossinidae) transmit several species of pathogenic trypanosomes causing Human African Trypanosomiasis (HAT) and African Animal Trypanosomiasis (AAT). HAT affects human welfare directly through the chronic and acute forms of the disease caused by Trypanosoma brucei gambiense and T. b. rhodesiense respectively. AAT, on the other hand, stands as a major obstacle to the development of more efficient and sustainable livestock production systems in tsetse-infested areas [1]. A major challenge to controlling HAT is lack of suitable prophylactic drugs and vaccines against trypanosomiasis. Furthermore, chemotherapeutic agents for treatment of HAT are expensive, difficult to administer in remote areas and exhibit poor safety profiles. Consequently, vector control remains a viable alternative for large-scale control of trypanosomiasis.
Understanding tsetse population dynamics is critical for determining which control strategy is most appropriate (e.g., suppression, eradication), for choosing the best method for enacting that strategy (e.g., traps, insecticide-treated cattle, sterile insect technique), and for determining the scale at which vector control activities must be implemented [2]. Determinants of population dynamics include both life history and ecological correlates such as mating system, dispersal ability and population size, which influence the extent to which tsetse populations can recover from refugia following intervention, or re-colonize a cleared zone from neighboring sources. Recently, the use of population genetics has provided insights into tsetse ecology [3], with important ramifications for the implementation of control programs [4]. For example, studies of tsetse in Guinea and Senegal have identified populations that are sufficiently isolated to warrant attempts at complete elimination [5–7]. Elsewhere though, studies have documented relatively high levels of gene flow, necessitating integration of barriers into elimination schemes [8] or warranting an area-wide control effort that encompasses the dispersal-linked populations [9, 10].
Across Africa, Glossina fuscipes is one of the most important vectors of HAT, transmitting an estimated 90% of all disease cases [11]. Glossina fuscipes is a member of the palpalis group of tsetse, which inhabit low bushes or forests at the margins of rivers, lakes or temporarily-flooded scrub land. In eastern Africa, populations of the subspecies G. f. fuscipes appear to respond to seasonal weather patterns, often disappearing during the bi-annual dry season from sites where they were previously abundant [12]. If populations in refugia are small, then seasonal bottlenecks could result in large temporal changes in gene frequencies. In order to investigate the impact of seasonal climate changes on population size and to gain further insight into the population dynamics of G. f. fuscipes, we evaluated temporal changes in gene frequencies at one mitochondrial locus and 16 microsatellite loci in multiple Ugandan populations. Our sampling scheme included three populations situated at a zone of contact between two divergent lineages of G. f. fuscipes. These two lineages exhibit distinct mitochondrial DNA (mtDNA) haplotypes and strong differentiation at microsatellite loci, suggesting a long history of isolation, and providing a unique opportunity to monitor their interaction over time [9, 10].
Materials and methods
Tsetse collection and study area
Tsetse flies were collected using biconical traps [13] during the period from March 2008 to January 2010. All sites were sampled in 2008 [10] and then at least one year later in 2009 or 2010. Four sites were also sampled a third time (Table 1). Each fly was stored individually in 80% ethanol.
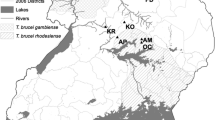
Tsetse collections were conducted at seven sites spanning central and southeastern Uganda (Figure 1). These sites generally reflected the riverine/woodland habitat preferred by G. fuscipes, but varied somewhat in regard to the immediate environment. Sites at Busime (BU) and Junda (JN) were located in a transition zone between marsh and woodland on the edge of Lake Victoria and Lake Kyoga, respectively. Sites at Bunghazi (BN), Dokolo/Otuboi (OT) and Okame (OK) were situated along permanent streams in a region of mixed agriculture and pastureland. Sampling at Mukongoro (MK) was conducted at the margin of ephemeral wetlands associated with rice cultivation. Sampling at Masindi (MS) was conducted within a region of banana and sugar cane plantations.

Map of sites at which populations of G. f. fuscipes were sampled in Uganda. Location codes are shown in Table 1. The dotted line indicates the approximate extent of a zone of contact between two historically isolated groups of tsetse.
The study sites spanned a zone of contact between two divergent groups of G. fuscipes co-occuring in Uganda [9, 10]. Sites MK and OT were situated north of the zone of contact and flies here were expected to possess solely northern mtDNA haplotypes. Sites BN, JN, and MS were located at the zone of contact and flies here were expected to possess both northern and southern haplotypes. Sites BU and OK were located south of the zone of contact and flies at these sites were expected to possess exclusively southern mtDNA haplotypes.
DNA Extraction
DNA was extracted from tsetse legs using NucleoSpin 96 Tissue Kits (Clontech, Mountain View, CA) or DNeasy kits (Qiagen, Valencia, CA) following the manufacturer's protocols.
Mitochondrial DNA sequencing
PCR was used to amplify a 570 bp fragment of mtDNA from a random subset of flies from each population using the primers COIF1 (CCT CAA CAC TTT TTA GGT TTA G) and COIIR1 (GGT TCT CTA ATT TCA TCA AGT A) as described by [10]. We amplified COIF1/COIIR1 in a 25 μl PCR reaction containing 1 × buffer (GoTaq colorless, Promega), 0.8 mM each dNTP, 0.4 mM primers, 1.5 mM MgCl2 and 0.5 U Go Taq polymerase. The amplification involved a denaturation step at 95°C for 8 min, followed by 50 cycles each at 94°C for 30 s, 51°C for 30 s, 72°C for 45 s, with a final extension step at 72°C for 7 min. PCR products were sequenced using an ABI Model 3730 automated sequencer (Applied Biosystems, Foster City, CA, USA). Electropherograms were visually inspected and sequences were trimmed to remove poor quality data. The resulting sequences (530 bp) were aligned by eye using the computer program Sequencher 4.2.2 (Gene Codes Corporation).
Microsatellite genotyping
We genotyped individual flies at 16 loci. We used 11 of the 13 loci described by [10], excluding D05 and Pgp17 due to possible null allele problems. We also employed five new dinucleotide loci identified in the G. morsitans genome and optimized for use in G. fuscipes: GmmA06, GmmB20, GmmD15, GmmL03, GmmL11 [14]. Amplifications were performed with fluorescently labeled forward primers (6-FAM, HEX and NED) using a touchdown PCR (10 cycles of annealing at progressively lower temperatures from 60°C to 51°C followed by 35 cycles at 50°C) in 12.5 μl reaction volumes employing 1 × buffer, 0.8 mM dNTPs, 2.0 mM MgCl2 and 0.5 U Go Taq polymerase. PCR products were multiplexed in groups of two or three and then genotyped on the ABI 3730 automated sequencer. Alleles were scored using the program Genemarker v 5.0 (SoftGenetics) with manual editing of the automatically scored peaks.
Marker validation and genetic diversity
Microsatellite loci were evaluated for Hardy Weinberg equilibrium (HWE) and linkage disequilibrium (LD) using Genepop version 4.0 [15]. Markov chain parameters were set at 10,000 dememorizations, 1000 batches, 10,000 iterations per batch for HWE and 100,000 dememorizations,1000 batches,10,000 iterations per batch for LD. We used the method of [16] as implemented in MultiTest v.1.2 to correct for multiple tests. Locus- and population-specific estimates of microsatellite allele frequencies were generated using the program Genalex version 6.3 [17]. We used the program FSTAT version 3.1 [18] to calculate allelic richness and the program Arlequin v. 3.5 [19] to calculate observed (Ho) and expected (He) heterozygosity for each population. DnaSP version 5.0 [20] was used to calculate mtDNA haplotype diversity (Hd) and nucleotide diversity (π).
Temporal genetic differentiation and population stability
For microsatellite data, we used Jost's DEST[21] to quantify genetic differentiation between populations and between temporal samples from the same population. DEST provides a less-biased estimate of differentiation than FST and related statistics, especially when estimated using highly polymorphic microsatellite loci [22]. Locus-specific calculations of DEST were performed using the web-based program SMOGD [23] and then averaged across loci. For mtDNA data, we used Fisher's exact test and the statistical software SAS version 9.1 to test for differences in haplotype frequencies among temporal samples from the same population. For both microsatellite and mtDNA data, we performed an analysis of molecular variance (AMOVA) as implemented in Arlequin v. 3.5 [19] to characterize the proportion of the variance in microsatellite allele frequencies or haplotype frequencies that was attributable to differences in date of sampling. For this analysis, we used only the two samples from each population that were separated by the longest time interval.
We estimated current effective population sizes based on temporal changes in microsatellite allele at all seven sites. The effective size of a population (Ne) is defined as the size of an ideal population (i.e., one of constant size, discrete generations, and negligible selection and gene flow), which would exhibit the same genetic characteristics as the population at hand [24]. Ne, therefore, reflects the rate of change in gene frequencies due to random drift alone [25]. We used two methods: the moment-based approach [26] and a likelihood approach implemented in TM3 [27]. Estimates were generated using the software NeEstimator [28]. For TM3, we employed 100,000 updates and a maximum Ne of 20,000.
For all analyses, we assumed that G. fuscipes undergoes approximately 8 generations per year using observations from colony flies (~7.3 generations per year, [29] ~8.5 generations per year at 25°C, [30] and those reported in other studies of the palpalis group (G. palpalis gambiensis and G. palpalis palpalis) in Guinea and Equatorial Guinea [5, 31]. All populations were evaluated at an interval of at least one year (~8 generations apart). For four populations (BU, OK, BN, MK), we generated estimates at two different sampling intervals (0 to 8 generations, and 0 to 12 generations).
For each temporal sample in all seven populations, we also tested for an excess of heterozygosity relative to observed allelic diversity, which may be indicative of a recent bottleneck [32]. For each temporal sample, tests of heterozygosity excess were performed separately for each microsatellite locus. Significance was assessed across loci using Wilcoxon's test, which is the most appropriate test given the number of microsatellite loci evaluated. All tests were performed using the program BOTTLENECK [33].
Results
Marker validation and diversity
We genotyped a total of 667 tsetse flies at 16 microsatellite loci. We detected 17 values of FIS (out of 288) that exhibited significant departures from HWE at p < 0.05 (Additional file 1: Table S1). Assessed by locus, the number of significant FIS values observed was consistent with chance at an overall value of p < 0.05. Assessed by population, the number of significant FIS values observed was consistent with chance for all populations except the sample representing generation 12 from BN. Following sequential Bonferroni correction, only one locus pair exhibited significant linkage, and only in one population, confirming previous work showing that these loci were unlinked [10, 14].
Microsatellite diversity was lowest in the samples from Mukongoro (MK) and highest in the samples from Bunghazi (BN). Allelic richness ranged from 3.0 to 4.4 and expected heterozygosity (HE) ranged from 0.418 to 0.609, (Table 1). MtDNA haplotype diversity was relatively low across samples with the number of haplotypes ranging from 1 to 4. As expected, nucleotide diversity was generally higher in populations from the zone of contact which were composed of flies with both northern and southern ancestry. We detected only two haplotypes that had not been previously reported [10]. Both of these haplotypes were recovered in population OT and differed by just one substitution from Hap26 or Hap27 [10].
Temporal variation in genetic diversity
Variation in allele frequencies by population and locus are depicted in Figure 2. Genetic differentiation between samples taken from the same population but at different times was extremely low, and uniformly lower than the differentiation observed between populations. DEST averaged 0.001 for temporally-spaced samples within populations, compared to 0.308 between populations (Additional file 2: Table S2). The largest values of DEST among temporally-spaced samples were observed in Masindi (MS generation 0 vs.13, DEST = 0.019 ± 0.022) and Otuboi (OT generation 0 vs. 11, DEST = 0.013 ± 0.007).

Microsatellite allele frequencies observed in seven populations of G. f. fuscipes sampled at either two or three different times. Numbers after location code indicate the time interval (in generations) since the first sampling.
Mitochondrial haplotype frequencies also exhibited little change over time (Figure 3). We observed a significant change in haplotype frequencies only between the two temporally spaced samples from Junda (JN, p = 0.046). This was attributable to the loss of the two least common haplotypes in the sample representing generation 13.

Mitochondrial haplotype frequencies observed in seven populations of G. f. fuscipes sampled at two different time periods. Numbers after location codes indicate the time interval (in generations) since the first sampling. Only temporal samples from Junda (JN) differed significantly.
An analysis of molecular variance using both microsatellite allele frequencies and haplotype frequencies suggested that differences between temporally-spaced samples explained an insignificant amount of the overall genetic variation (Table 2). Differences among sites, on the other hand, contributed significantly to the overall variation in genetic diversity. The percent variation explained was greater in the case of mtDNA data, compared to microsatellite data.
Effective size
Estimates of Ne were generated for the seven populations based on microsatellite allele frequency changes observed among samples collected at different times from the same population. Momentum-based estimates ranged from 216 to infinity, but only the estimate from OT was bounded by a 95% confidence interval that did not include infinity (Table 3). Likelihood estimates ranged from 152 to 19,550 and all estimates were bounded by 95% confidence intervals that included 20,000, the maximum value of Ne considered (Table 3).
For populations MK and OK, estimates of Ne were similar regardless of whether the calculations were performed using data for generations 0 and 8 or generations 0 and 12. In populations BN and BU, however, estimates of Ne derived from the moment method differed by an order of magnitude depending on whether the sample representing 8 generations or 12 generations was included. In population BN, the estimate of Ne generated by the Likelihood method was similarly unstable.
Population bottlenecks
Following Bonferonni correction, seven samples (taken from Bunghazi (BN), Masindi (MS), Okame (OK) and Otuboi (OT)) exhibited significant signatures of a recent population bottleneck under the infinite allele model (IAM) model. Only one of these samples (MS generation 0) also tested positive for a bottleneck under the two phase model (TPM; Table 4). Samples from Busime (BU) and Mukongoro (MK) exhibited the least evidence for past bottlenecks (all but one p-value >> 0.05), however power to detect a bottleneck may have been low in MK on account of relatively low genetic diversity (Table 1).
Discussion
We assessed changes in genetic composition of seven tsetse populations in southeast Uganda in order to gain insight into the population dynamics of G. f. fuscipes. In general, our results provide evidence for temporal stability of G. f. fuscipes populations over the one to two year period that we examined. With the exception of just one or two populations discussed below, mitochondrial haplotype frequencies and microsatellite allele frequencies exhibited little change over time and effective population sizes were generally large.
Compared to other riverine species of tsetse, estimates of Ne for G. f. fuscipes were similar to or larger than estimates for G. palpalis palpalis in Equatorial Guinea [31] and 2 to 3 orders of magnitudes larger than estimates for G. p. gambiensis on islands off the coast of Guinea [5]. Values of Ne for G. f. fuscipes populations were also generally larger than estimates for a savannah species, G. pallidipes, in Kenya [34]. The large effective population sizes and overall stability of G. f. fuscipes populations support the hypothesis [35] that seasonal variation in tsetse numbers, in which larva develop in utero, should be relatively small, since they do not depend on surface water or moist media for breeding. Nonetheless, the lack of variation in genetic structure over time is surprising given the reduced abundance of G. f. fuscipes observed during the dry season in Uganda [12]. To reconcile our results with this observation, which may reflect the low efficiency of traps used for monitoring [36, 37], we suggest that populations of G. f. fuscipes in dry season refugia remain large, and that seasonal invasion of marginal wet-season habitat (e.g., at Mukongoro, Bunghazi) must occur in waves of tsetse that are large enough to be representative of the refugia population. Large populations of pupa, which develop in the ground over a period of weeks, may also help to ensure the continuity of tsetse populations and would contribute to reducing the variance in genetic changes over time.
In contrast to the other populations, estimates of Ne were low for populations MS and especially OT, where both moment and likelihood methods produced values of only about 200. These values could be indicative of small populations. Ne may also be influenced by overlapping generations and temporal variance in reproductive success as well as the forces of selection, mutation and migration. In this study, however, the low values of Ne observed in these populations probably reflected small differences in the location of trapping sites used for the two temporal samples. Generation 13 from MS was sampled at a distance of about 4 km from the original site at which generation 0 was sampled. Likewise, generation 11 from OT was sampled at a single site that was 11-20 km from the relatively widely dispersed sites from which generation 0 was sampled. Thus, for these sites, which were the only two sites sampled at different locations across years, fine-scale spatial genetic variation could be responsible for the apparent temporal variation in gene frequencies, thus depressing estimates of Ne.
Given that genetic variation in MS and OT samples can probably be attributed to microgeographic variation, the change in genetic composition of the population at JN likely reflects the only significant temporal change observed in this study. Although microsatellite allele frequencies were largely invariate, mtDNA haplotype frequencies here differed significantly between generation 0 and generation 13. Junda (JN), along with sites BN and MS, lies along a narrow zone of contact between two long-diverged and historically-isolated groups of G. f. fuscipes[9, 10]. In 2008, populations at all three of these sites harbored both "southern" and "northern" mtDNA haplotypes. Interestingly, in Junda, individuals with the "southern" haplotypes disappeared from the sample after 13 generations. This could be due to a particularly small population of females and stochastic variation in female reproductive success, although in tsetse, the latter is more likely to be true among males than females [5]. Mating success can also be influenced by Wolbachia, a symbiont that may impose mating barriers due to cytoplasmic incompatibility between infected and uninfected tsetse individuals [38], thereby biasing mating in favor of infected females and potentially producing mitochondrial sweeps [39]. Given the change in mtDNA observed at Junda, flies here should be examined for Wolbachia. If present, the zone of contact in Uganda may provide a unique opportunity to monitor symbiont-induced population changes over time.
References
Itard J, Cuisance D, Tacher G: Trypanosomoses: historique répartition géographique. Principales maladies infectieuses et parasitaires du bétail. Europe et Régions Chaudes. Editions Tec et Doc and Editions Médicales Internationales. 2003, Lavoisier, Paris, 2: 1607-1615.
Torr S, Hargrove J, Vale G: Towards a rational policy for dealing with tsetse. Trends Parasitol. 2005, 21: 537-541. 10.1016/j.pt.2005.08.021.
Krafsur ES: Tsetse flies: genetics, evolution, and role as vectors. Infect Genet Evol. 2009, 9: 124-141. 10.1016/j.meegid.2008.09.010.
Solano P, Ravel S, De Meeus T: How can tsetse population genetics contribute to African trypanosomiasis control?. Trends Parasitol. 2010, 26 (5): 255-263. 10.1016/j.pt.2010.02.006.
Solano P, Ravel S, Bouyer J, Camara M, Kagbadouno MS, Dyer N, Bardes L, Herault D, Donnelly MJ, De Meeus T: The Population Structure of Glossina palpalis gambiensis from Island and Continental Locations in Coastal Guinea. PLoS Negl Trop Dis. 2009, 3 (3): e392-10.1371/journal.pntd.0000392.
Kagbadouno M, Camara M, Bouyer J, Hervouet JP, Courtin F, Jamonneau V, Morifaso O, Kaba D, Solano P: Tsetse elimination: its interest and feasibility in the historical sleeping sickness focus of Loos islands, Guinea. Parasite. 2009, 16: 29-35.
Solano P, Kaba D, Ravel S, Dyer NA, Sall B, Vreysen MJ, Seck MT, Darbyshir H, Gardes L, Donnelly MJ, De Meeûs T, Bouyer J: Population Genetics as a Tool to Select Tsetse Control Strategies: Suppression or Eradication of Glossina palpalis gambiensis in the Niayes of Senegal. PLoS Negl Trop Dis. 2010, 4 (5): e692-10.1371/journal.pntd.0000692.
Bouyer J, Balenghien T, Ravel S, Vial L, Sidibé I, Venon ST, Solano P, Demeeus T: Population sizes and dispersal pattern of tsetse flies: rolling on the river. Molecular Ecology. 2009, 18: 2787-2797. 10.1111/j.1365-294X.2009.04233.x.
Abila PP, Slotman MA, Parmakelis A, Dion KB, Robinson AS, Muwanika VB, Enyaru JCK, Okedi LM, Aksoy S, Caccone A: High levels of genetic differentiation between Ugandan Glossina fuscipes fuscipes populations separated by Lake Kyoga. PLoS Negl Trop Dis. 2008, 2 (5): e242-10.1371/journal.pntd.0000242.
Beadell JS, Hyseni C, Abila PP, Azabo R, Enyaru JCK, Ouma JO, Mohammend YO, Okedi LM, Aksoy S, Caccone A: Phylogeography and population structure of Glossina fuscipes fuscipes in Uganda: Implications for control of tsetse. PLoS Negl Trop Dis. 2010, 4 (3): e636-10.1371/journal.pntd.0000636.
Omolo MO, Hassanali A, Mpiana S, Esterhuizen J, Lindh J, Lehane MJ, Solano P, Rayaisse JB, Vale GA, Torr J, Tirado I: Prospects for Developing Odour Baits To Control Glossina fuscipes spp., the Major Vector of Human African Trypanosomiasis. PLoS Negl Trop Dis. 2009, 3 (5): e435-10.1371/journal.pntd.0000435.
Katunguka-Rwakishaya E, Kabagambe EK: Tsetse survey in Mukono district, south-east Uganda: population structure, distribution and blood meal status. Trop Anim Hlth Prod. 1996, 28: 151-157.
Challier A, Laveissiere C: Un nouveau pie'ge pour la capture des glossines (Glossina: Diptera, Muscidae): description et essais sur le terrain. Cah ORSTOM sér Ent Méd Parasitol. 1973, 11: 251-262.
Hyseni C, Beadell J, Gomez Ocampo Z, Ouma J, Okedi L, Gaunt M, Caccone A: The Glossina morsitans morsitans (Diptera: Glossinidae) genome as a source of microsatellite markers for other tsetse fly (Glossina) species. Molecular Ecology Resources. 2011, 11 (3):
Rousset F: GENEPOP'007: a complete re-implementation of the Genepop software for Windows and Linux. Mol Ecol Res. 2008, 8: 103-106. 10.1111/j.1471-8286.2007.01931.x.
De Meeûs T, Guegan JF, Teriokhin A: MultiTest V.1.2, a program to binomially combine independent tests with a comparison to other related methods on proportional data. BMC Bioinformatics. 2009, 10: 443-
Peakall R, Smouse PE: GENALEX 6: genetic analysis in Excel. Population genetic software for teaching and research. Molecular Ecology Notes. 2006, 6: 288-295. 10.1111/j.1471-8286.2005.01155.x.
Goudet J: FSTAT (version 1.2): a computer program to calculate Fstatistics. J Heredity. 1995, 86: 485-486.
Excoffier L, Laval G, Schneider S: Arlequin (version 3.0): an integrated software package for population genetics data analysis. Evolutionary Bioinformatics. 2005, 1: 47-50.
Rozas JJC, Sanchez-Delbarrio X, Messeguer , Rozas R: DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics. 2003, 19: 2496-2497. 10.1093/bioinformatics/btg359.
Jost L: G(ST) and its relatives do not measure differentiation. Molecular Ecology. 2008, 17: 4015-4026. 10.1111/j.1365-294X.2008.03887.x.
Heller R, Siegismund HR: Relationship between three measures of genetic differentiation G(ST), D-EST and G'(ST): how wrong have we been?. Molecular Ecology. 2009, 18: 2080-2083. 10.1111/j.1365-294X.2009.04185.x.
Crawford NG: smogd: software for the measurement of genetic diversity. Molecular Ecology Resources. 2010, 10: 556-557. 10.1111/j.1755-0998.2009.02801.x.
Wright S: Evolution in Mendelian populations. Genetics. 1931, 16: 97-159.
Charlesworth D: Plant sex determination and sex chromosomes. Heredity. 2002, 88: 94-101. 10.1038/sj.hdy.6800016.
Waples RS: A generalized approach for estimating effective population size from temporal changes in allele frequency. Genetics. 1989, 121: 379-391.
Berthier P, Beaumont MA, Cornuet JM, Luikart G: Likelihood-based estimation of the effective population size using temporal changes in allele frequencies: a genealogical approach. Genetics. 2002, 160: 741-751.
Jorde PE, Ryman N: Unbiased estimator for genetic drift and effective population size. Genetics. 2007, 177: 927-935. 10.1534/genetics.107.075481.
Krafsur ES, Marquez JG, Ouma JO: Structure of some East African Glossina fuscipes fuscipes (Diptera: Glossinidae) populations. Med Vet Entomol. 2008, 22: 222-227. 10.1111/j.1365-2915.2008.00739.x.
Hargrove JW: Extinction probabilities and times to extinction for populations of tsetse flies Glossina spp. (Diptera: Glossinidae) subjected to various control measures. Bulletin of Entomological Research. 2005, 95: 13-21. 10.1079/BER2004335.
Dyer NA, Furtado A, Cano J, Ferreira F, Afonso MO, Mabale NN, Asumu PN, Lima SC, Benito A, Weetman D, Donnelly MJ, Pinto J: Evidence for a discrete evolutionary lineage within Equatorial Guinea suggests that the tsetse fly Glossina palpalis palpalis exists as a species complex. Molecular Ecology. 2009, 18: 3268-3282. 10.1111/j.1365-294X.2009.04265.x.
Cornuet JM, Luikart G: Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics. 1996, 144: 2001-2014.
Piry S, Luikart G, Cornuet JM: BOTTLENECK: A computer program for detecting recent reductions in the effective population size using allele frequency data. J Heredity. 1999, 90: 502-503. 10.1093/jhered/90.4.502.
Ouma JO, Marquez JG, Krafsur ES: Macrogeographic population structure of the tsetse fly, Glossina pallidipes (Diptera: Glossinidae). Bull Entomol Res. 2005, 95: 437-447. 10.1079/BER2005376.
Hargrove JW: Tsetse population dynamics. The Trypanosomiases. Edited by: Maudlin I, Holmes PH, Miles MA. 2004, CABI Publishing, Wallingford, 113-138. full_text.
Bouyer J, Seck MT, Sall B, Ndiaye EY, Guerrini L, Vreysen MJB: Stratified entomological sampling in preparation for an area-wide integrated pest management program: the example of Glossina palpalis gambiensis (Diptera: Glossinidae) in the Niayes of Senegal. J Med Entomol. 2010, 47: 543-552. 10.1603/ME09149.
Rayaisse JB, Tirados I, Kaba D, Dewhirst SY, Logan JG, Diarrassouba A, Salou E, Omolo MO, Solano P, Lehane MJ, Pickett JA, Vale GA, Torr SJ, Esterhuizen J: Prospects for the development of odour baits to control the tsetse flies Glossina tachinoides and G. palpalis s.l. PLOS Negl Trop Dis. 2010, 4: e632-10.1371/journal.pntd.0000632.
O'Neill SL, Gooding RH, Aksoy S: Phylogenetically distant symbiotic microorganisms reside in Glossina midgut and ovary tissues. Medical and Veterinary Entomology. 1993, 7: 377-383.
Hurst GDD, Jiggins FM: Problems with mitochondrial DNA as a marker in population, phylogeographic and phylogenetic studies: the effects of inherited symbionts. Proc R Soc. 2005, B 272: 1525-1534.
Acknowledgements
We are thankful for the continued support of Drs. Elizabeth Opiyo (Gulu University, Uganda) and John Charles Enyaru (Makerere University, Uganda), who are part of Mr. Echodu's thesis committee. This study was supported by grants from the NIH (AI068932) to SA, AC and LMO, the Fogarty Center (D43TW 007391) to SA and AC. Our thanks go to Dr. Ssewannyana Edward and his staff at the National Livestock Resources Research Institute, Tororo, Uganda for facilitating field collections.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
RE and JSB contributed to field collections, performed lab work, analyzed the data, and drafted an initial version of the manuscript. CH also performed lab work and data analysis. LMO, SA and AC helped design the study, coordinated fieldwork and provided guidance on the manuscript. All authors read and approved the final manuscript.
Electronic supplementary material
13071_2010_263_MOESM1_ESM.DOC
Additional File 1: Table S1. FIS values for the 16 microsatellite loci. Significance was assessed at p < 0.05 (*) and a Bonferroni-corrected value of p < 0.0028 (bold). Low variability precluded calculation of FIS in some populations (n/a). (DOC 62 KB)
13071_2010_263_MOESM2_ESM.DOC
Additional File 2: Table S2. Pairwise estimates of genetic differentiation (Jost's DEST) between samples taken from seven populations of G. f. fuscipes. Estimates of differentiation (below diagonal) and associated standard error (above diagonal) between populations of flies sampled at the same site but different times are shaded in grey. (DOC 66 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Echodu, R., Beadell, J.S., Okedi, L.M. et al. Temporal stability of Glossina fuscipes fuscipes populations in Uganda. Parasites Vectors 4, 19 (2011). https://doi.org/10.1186/1756-3305-4-19
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1756-3305-4-19