
Abstract
Background
Rickets can occur due to Vitamin D deficiency or defects in its metabolism. Three rare genetic types of rickets with different alterations of genes have been reported, including: Vitamin D dependent rickets type 1, Vitamin D dependent rickets type 2 or also known as Vitamin D resistant rickets and 25 hydroxylase deficiency rickets. Vitamin D dependent rickets type 1 is inherited in an autosomal recessive pattern, and is caused by mutations in the CYP27B1 gene encoding the 1α-hydroxylase enzyme. We report here a new mutation in CYP27B1, which lead to Vitamin D dependent rickets type 1.
Case presentation
We report on a 13-month-old Arabic Saudi girl with Vitamin D dependent rickets type 1 presented with multiple fractures and classic features of rickets. A whole exome sequencing identified a novel pathogenic missense mutation (CYP27B1:Homozygous c.1510C > T(p.Q504X)) which results in a protein truncating alteration. Both parents are heterozygous carriers of the mutation. Based on data search in Human Gene Mutation Database, 63 CYP27B1 alterations were reported: only 28.6% are protein truncating (5 nonsense, 13 frameshift insertions/deletions, 0 gross deletions), while 61.9% are non-truncating (38 missense, 1 small in-frame insertions/deletion), and 9.5% are possible protein-truncating (5 splice, 1 regulatory).
Conclusion
The deleterious effect of this alteration, which was the only mutation detected in the CYP27B1 common gene of Vitamin D dependent rickets type 1 in the proband, and its autosomal recessive inheritance fashion, both support a pathogenic nature of this mutation as the cause of Vitamin D dependent rickets type 1.
Similar content being viewed by others

Background
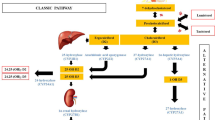
Vitamin D, whether synthesized in the skin or obtained from the diet, is biologically inert and requires activation via two selective metabolic steps; the second of them is rate-limiting and hormonally regulated (Figure 1). Both of the steps are catalyzed by cytochrome P450 enzymes to form the active secosteroid hormone 1,25 dihydroxyvitamin D3 (1,25(OH)2D3) [1, 2].

Bio-activation pathway of vitamin D.
Vitamin D deficiency remains the commonest cause of rickets worldwide [3, 4]. So far, apart from liver or renal diseases that may affect vitamin D metabolism, three rare genetic errors of vitamin D metabolism that can cause rickets have been described [3–6]. The first involves 1α-hydroxylase deficiency, also described as vitamin D dependent rickets type 1 (VDDR-1). The second type involves defective vitamin D receptor (VDR), resulting in vitamin D resistant rickets (VDRR), also known as vitamin D dependent rickets type II (VDDR II) [3]. More rare, 25 hydroxy vitamin D3 (25OHD3) deficiency have been reported and linked to a selective mutation in CYP2R1 gene that leads to 25-hydroxylase deficiency [4, 5].
Children with VDDR-1 may present with hypotonia, muscle weakness, joint pain and deformity, growth failure, seizures, or fractures in early infancy [3]. We report a 13-month-old girl with VDDR-1 presented with multiple fractures, classic biochemical and severe radiological findings of rickets. An initial CYP27B1 gene analysis for common alterations was performed and showed no disease-associated mutations in this patient. However, the whole exome sequencing identified a novel pathogenic mutation (CYP27B1: Homozygous c.1510C > T(p.Q504X)) in our patient.
Case presentation
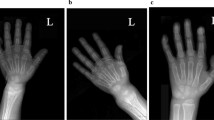
A 13-month-old Arabic Saudi girl, previously well, presented with multiple fractures as a result of minor traumas. She was adequately exposed to sunlight and had normal perinatal, developmental, and nutritional history. She is the only child for first-cousins parents, with no family history of similar problems or metabolic bone disease. Examination revealed normal growth for age, mildly reduced muscle tone, normal teeth, rachitic rosary, Harrison’s sulcus, widening of the wrist and bowing deformity of distal arms and legs. Her head circumference was on the 50th centile for age with a frontal bossing and wide anterior fontanel. She had a swelling, secondary to fracture, over her right clavicle. However, the rest of her skin examination was normal and no other signs, which might suggest physical abuse. Initial investigations showed: low serum calcium and phosphorus, high serum parathyroid hormone (PTH), high serum alkaline phosphatase, a normal serum 25OHD3 which was repeated two times, and inappropriately low normal serum 1,25(OH)2D3 suggesting 1α-hydroxylation defect (Table 1). Radiological workup showed generalized osteopenia with multiple fractures involving the right clavicle, right ulna, left proximal ulna and tibia bones. Disorganization of the growth plate was apparent with classic features of rickets (Figure 2).

X-ray images at time of presentation showing generalized osteopenia with altered texture (a & b). Humeral metaphysis shows frying with more pronounced lucency (a), and ulnar/radial metaphysis shows frying and cupping of their outline (b) - (white arrows). The cortices are indistinct with coarse fuzzy trabecuale (a & b). Fracture of right clavicle (a), and proximal shaft of the ulna (b) - (black arrows).
In the light of the above biochemical results and severe radiological findings, genetic types of rickets were suspected. Initially, CYP27B1 gene analysis was performed only for the common mutations in exon 2 and 8 and it was negative. Unfortunately, the genetic laboratory, where the common mutations of CYP27B1 gene tests were tested, did not offer further analysis of the same common gene. Alternatively, It was suggested to perform mutations analysis for hypophosphataemic rickets but this was clinically and biochemically irrelevant in this patient. With a high clinical suspicion of 1α-hydroxylation defect, a whole exome sequencing test was performed in another genetic lab, with a report of a likely diagnosis of VDDR-1 (MIM-609506). The exome sequencing revealed a novel pathogenic nonsense deleterious mutation in the proband’s CYP27B1 gene (CYP27B1: Homozygous c.1510C > T (p.Q504X)), with both parents found to be heterozygous carriers of the mutation (Table 2).
Awaiting the genetic results, the patient required high doses of 1α-hydroxyvitamin D and calcium supplements to heal her bones and to return serum chemistries to normal (Table 1).
Discussion
CYP27B1 gene is located on chromosome 12q13.3. It encodes a member of the cytochrome P450 superfamily of enzymes, which are monooxygenases that catalyze many reactions involved in drug metabolism and synthesis of cholesterol, steroids and other lipids. CYP27B1 gene encodes 1α-hydroxylase enzyme that is localized to the inner mitochondrial membrane of renal cells; in which it activates 25OHD3 to synthesise 1,25(OH)2D3, which binds to vitamin D receptor and regulates calcium metabolism. Homozygous or compound heterozygous alterations in this gene are inherited in autosomal recessive pattern, and are in association with VDDR-1 [7].
In our patient, the clinical, biochemical and radiological findings were all consistent with those of previously reported patients with CYP27B1 gene alterations [3, 8]. The first reported negative result of genetic mutations suggestive of VDDR-1 was based on inadequate examination, only for the common alterations in exons 2 and 8 of CYP27B1. This, beside the suggestion of further testing for hypophosphataemic rickets by the genetic lab, which was clinically irrelevant in our patient, both had mislead the directions of choice for further genetic testing. It would have been reasonable to perform a full analysis of the CYP27B1 gene alone for any other mutations in the first place, which could have detect the novel mutation in our patient, before proceeding for a whole exome sequencing in this patient. Nonetheless, genetic analysis with full exome sequencing, bioinformatics analysis and filtering based on autosomal and X-linked dominant and recessive inheritance models of the proband, mother, and father revealed 53 genes (70 unique alterations) (Table 3). Manual review to rule out sequencing artefacts and polymorphisms along with the medical interpretation to rule out genes lacking clinical overlap with the patient’s evaluated phenotype resulted in 25 genes (36 unique alterations) (Table 3).
Alterations with likely clinical relevance “candidate genes” underwent co-segregation analysis, in which 1 candidate gene (1 alteration): (CYP27B1:Homozygous c.1510C > T(p.Q504X)) was selected for further investigations. The familial co-segregation analysis sustained the significance of the identified gene alteration, and revealed an inheritance fashion of the mutation as both parents were found to be heterozygous carriers of the alteration (Table 2).
This identified novel CYP27B1 alteration displayed a deleterious nature and it is the likely cause of our patient’s clinical findings. This is implemented by: Firstly, the premature protein truncation effect of the alteration. The c.1510C > T(p.Q504X) alteration results from a C to T nucleotide substitution at position 1510. This changes an amino acid from a glutamine to a stop codon; and premature stop codons are typically deleterious in nature [9–11]. Secondly, the inconsistency of the alteration type with the previously reported predominant CYP27B1 alterations. Based on data search in the Human Gene Mutation Database (HGMD), 63 CYP27B1 alterations were reported: only 28.6% are protein truncating (5 nonsense, 13 frameshift insertions/deletions, 0 gross deletions), while 61.9% are non-truncating (38 missense, 1 small in-frame insertions/deletion), and 9.5% are possible protein-truncating (5 splice, 1 regulatory) [12]. Thirdly, the inheritance pattern of the detected alteration in our patient is consistent with an autosomal recessive fashion.
Conclusion
The deleterious nature of the reported novel CYP27B1: c.1510C > T(p.Q504X) alteration and its inheritance fashion in our patient supports its classification as a pathogenic mutation for VDDR-1.
Consent
Ethical approval for this study was obtained from the ethics committee at King Khalid University Hospital, and parents consented to genetic testing and consented to genetic testing in their child. Copies of the written consents are available for review by the Editor-in-Chief of this journal.
Authors’ information
All of the authors in this article are currently employed by King Khalid University Hospital and King Saud University, Saudi Arabia. Amir Babiker (AB) is a Fellow of the Royal College of Paediatrics and Child Health. He is a Consultant Paediatric endocrinologist and Assistant Professor of Paediatrics. Iman Al Gadi (AG) is a Paediatric resident and demonstrator in Endocrine division (Paediatrics). Nasir A. Al Jurayyan (NAA), MD, is Professor of Paediatrics, Consultant Paediatric endocrinologist and Head of Endocrine division. Abdulrahman Al-Nemri (AA), MD, is Chairman of the Paediatric Department and Associate Professor of Paediatrics. Ali Abdu Haboob (AAA), MD, is a consultant Paediatric Intensivist and Assistant Professor of Paediatrics. Ahmed Amer Al Boukai (ABO), MD, FRKSU is an Associate Professor and Consultant Radiologist. Ali Al Zahrani (AA), a Technician in the Department of Pathology. Hanan Habib (HH), MD, is Professor of Pathology and a Consultant Pathologist.
Abbreviations
- 1:
-
25(OH)2D3: 1,25 dihydroxyvitamin D3
- 25OHD3:
-
25 hydroxy vitamin D3
- HGMD:
-
Human Gene Mutation Database
- PTH:
-
Parathyroid hormone
- VDDR-1:
-
Vitamin D dependent rickets type 1
- VDR:
-
Vitamin D receptor
- VDRR:
-
Vitamin D resistant rickets
- VDDR II:
-
Vitamin D dependent rickets type II.
References
Hewison M, Zehnder D, Bland R, Stewart PM: 1 alpha-Hydroxylase and the action of vitamin D. J Mol Endocrinol. 2000, 25: 141-148. 10.1677/jme.0.0250141.
Portale AA, Miller WL: Human 25-hydroxyvitamin D- 1 alpha-hydroxylase: cloning, mutations, and gene expression. Pediatr Nephrol. 2000, 14: 620-625. 10.1007/PL00009639.
Malloy PJ, Feldman D: Genetic disorders and defects in vitamin D action. Rheum Dis Clin North Am. 2012, 38: 93-106. 10.1016/j.rdc.2012.03.009.
Allgrove J, Shaw N: Calcium and bone disorders in children and adolescents. Endocrine Development. Volume 16. 2009, Basel: Karger AG
Cheng JB, Levine MA, Bell NH, Mangelsdorf DJ, Russell DW: Genetic evidence that the human CYP2R1 enzyme is a key vitamin D 25-hydroxylase. Proc Natl Acad Sci. 2004, 101 (20): 7711-7715. 10.1073/pnas.0402490101.
Misra M, Pacaud D, Petryk A, Collett-Solberg PF, Kappy M, and on behalf of the Drug and Therapeutics Committee of the Lawson Wilkins Pediatric Endocrine Society: Vitamin D deficiency in children and its management: review of current knowledge and recommendations. Pediatrics. 2008, 122 (2): 398-417. 10.1542/peds.2007-1894.
RefSeq: The NCBI Handbook [homepage on the Internet]. Chapter 18, The Reference Sequence (RefSeq) Project. c2013. Bethesda (MD): National Library of Medicine (US), National Center for Biotechnology Information; 2002 [updated 2013 July 19; cited 2014 August 20] Available from: [http://www.ncbi.nlm.nih.gov/books/NBK21091/]
Wang JT, Lin CJ, Burridge SM, Fu GK, Labuda M, Portale AA, Miller WL: Genetics of vitamin D 1 alpha-hydroxylase deficiency in 17 families. Am J Hum Genet. 1998, 63 (6): 1694-1702. 10.1086/302156.
Griffiths AJF, Miller JH, Suzuki DT, Lewontin RC, Gelbart WM: An Introduction to Genetic Analysis. 2000, New York: W. H. Freeman, 7
Culbertson MR: RNA surveillance. Unforeseen consequences for gene expression, inherited genetic disorders and cancer. Trends Genet. 1999, 15 (2): 74-80. 10.1016/S0168-9525(98)01658-8.
Wilkinson MF, Shyu AB: RNA surveillance by nuclear scanning?. Nat Cell Biol. 2002, 4 (6): E144-E147. 10.1038/ncb0602-e144.
Human Gene Mutation Database [homepage in the Internet]. [cited 2014 August 20] Available from: [http://www.hgmd.cf.ac.uk/]
Acknowledgement
The authors would like to thank and extend their appreciation to the College of Medicine Research Centre, Deanship of Scientific Research, King Saud University, Riyadh, Saudi Arabia for funding this work.
The authors would also like to acknowledge Professor Fowzan Al Kuraya, Head of Developmental Genetics Unit, King Faisal Specialist Hospital and Research Centre, College of Medicine, Alfaisal University, Riyadh, Saudi Arabia, for his kind efforts of reviewing the genetic results from the two labs, confirming the interpretation of the genetic results and helping us in searching the HGMD to confirm the novelty of this mutation.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
Idea conceived by AB - The main author and treating consultant of the patient in the case report. AB, IG and NJ - Contributed in patient’s management as well as writing and drafting the manuscript, IG - also designed Figure 1. ABO, AN, AAA, HH and AA - Contributed in patient’s management as well as arranging and interpreting the biochemical, genetic and radiological findings in this patient, ABO - also reported the findings in Figure 2 and put ligands for it. All authors approved the final format of the submitted manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Babiker, A.M., Al Gadi, I., Al-Jurayyan, N.A. et al. A novel pathogenic mutation of the CYP27B1 gene in a patient with vitamin D-dependent rickets type 1: a case report. BMC Res Notes 7, 783 (2014). https://doi.org/10.1186/1756-0500-7-783
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1756-0500-7-783