
Abstract
Background
Loeys–Dietz syndrome, also known as Marfan syndrome type II, is a rare connective tissue disorder caused by dominant mutations in transforming growth factor-beta receptors (TGFBR1 and 2).
Case presentation
We report a 7-year-old Japanese boy with Loeys–Dietz syndrome who carried a novel, de novo missense mutation in TGFBR2 (c.1142g > c, R381P). He showed dysmorphic faces and skeletal malformations that were typical in previous cases with Loeys-Dietz syndrome. The cardiac studies disclosed the presence of markedly dilated aortic root and patent ductus aorteriosus. The cranial magnetic resonance imaging (MRI) and angiography (MRA) detected the tortuous appearances of the bilateral middle cerebral and carotid arteries.
Conclusion
This study depicts the systemic vascular phenotypes of a child with Loeys–Dietz syndrome that were caused by a novel heterozygous mutation of TGFR2. A large cohort with serial imaging studies for vascular phenotypes will be useful for delineating the genotype-phenotype correlations of Loeys–Dietz syndrome.
Similar content being viewed by others


Background
Loeys–Dietz syndrome is a rare congenital disorder characterized by loose connective tissue [1, 2]. As a variant form of Marfan syndrome, affected individuals exhibit progressive vascular diseases such as dilation of the aortic root, aortic dissection and valvular insufficiency [1–3]. In addition to these common phenotypes, patients with Loeys–Dietz syndrome manifest distinct anomalies, such as ocular hypertelorism, high-arched palate, bifid uvula, scoliosis and clubfoot [1, 2].
Vascular complications in Loeys–Dietz syndrome are known to progress more rapidly than those in Marfan syndrome [2, 3]. Hence the precise diagnosis of Loeys–Dietz syndrome is a critical issue in the long-term management of affected individuals. In this report, we present a Japanese boy with Loeys–Dietz syndrome who carried a novel mutation in the TGFBR2 gene. This study highlights the moderate to severe vascular phenotypes that resulted from an R381P mutation in TGFBR2.
Methods
MR imaging was performed using a 3.0T system (Intera Achieva; Philips Medical Systems, Best, Netherlands). Polymerase chain reaction (PCR) amplifying the coding exons of TGFBR2 (chr3:30647994–30735633) was performed as previously described [1, 2, 4]. Sequencing reactions were performed with the BigDye® Terminator v3.1 cycle sequencing kit (Life Technologies, Grand Island, NY, U.S.A.) according to the manufacturer’s protocol.
Case Presentation
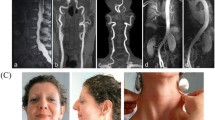
A 7-year-old Japanese boy born to healthy Japanese parents was referred to our department for assessment of systemic anomalies. He had undergone surgical repairs for right undescended testis, inguinal hernia and right clubfoot. Genetic disorders were suspected from his dysmorphic facial appearance consisting of hypertelorism, ectopic hair formation on the right lower mandible, and bifid uvula (Figure 1A-C). Physical examination also revealed hypermobility of joints (not shown), talipes equinovarus and scoliosis (Figure 1D-F). His intelligence was normal. He had no history of stroke, heart attack or arrhythmia. Ocular examination excluded ectopia lentis. Ehlers-Danlos syndrome or related connective disorders were suspected from these clinical features. Computed tomography (CT) scan revealed protrusion of the aortic arch (Figure 2A) and the presence of a patent ductus arteriosus (Figure 2B). Echocardiography revealed aortic root dilatation to 26.7 mm in diameter (Figure 2C). The dysmorphic features and cardio-vasuclar phenotypes of the present case allowed us to diagnose him as having Loeys–Dietz syndrome. To confirm the genetic diagnosis of Loeys–Dietz syndrome, we sequenced the coding exons of the TGFBR2 gene. A novel heterozygous missense mutation (c.1142g > c, R381P) was detected in TGFBR2 exon 5 of the proband (Figure 3A) but not his parents (Figure 3B).

Dysmorphic features of the present case. The patient shows the typical facial appearance for Loeys-Dietz syndrome with ocular hypertelorism and mild strabismus (A). Other symptoms include ectopic hair growth on the right mandible (B), high-arched cleft palate with bifid uvula (C), bilateral clubfoot (D), and kyphoscoliosis (E, F).

Cardiovascular phenotypes of the present case. Imaging studies from a 3D-reconstructed CT scan (A) and echocardiographies (B and C) are shown. The case had abnormally dilated the aortic root (A, B) and the patent ductus arteriosus (arrows in A and C). Note that, in panel C, the shunt flows are present across the ductus.

The de novo mutation at TGFBR2 exon 5. The sequence chromatograms show the heterozygous mutation in TGFBR2 (c.1142g > c) in the proband’s DNA (A). The arrow indicates the position of the missense mutation replacing Arg381 (cgg) with Pro (ccg). The parents of the case (B) show the normal TGFBR2 genotype (left, father; right, mother).
To assess systemic vascular malformation, we performed cranial and cervical magnetic resonance imaging (MRI) studies. The MR angiographies revealed the prominently tortuous appearance of the vertebral, carotid and middle cerebral arteries (Figure 4A and B). These findings were consistent with those in previous reports [5, 6]. No vascular aneurysm was identified in the cervical and cranial MRI. Of note, we found in the cranial MRI that unusually enlarged perivascular spaces at the bilateral posterior white matter without ischemic damage, hemorrhagic lesions or metabolic degeneration (Figure 5A-C). Spinal MRI detected enlarged sacral foramina as typically observed in Marfan syndrome and Loeys–Dietz syndrome (data not shown).

Neurovascular phenotypes in magnetic resonance imaging. The cranial and cervical magnetic resonance angiography displays the tortuous appearances of the carotid, middle cerebral (A) and vertebral arteries (B).

Enlarged Virchow–Robin spaces in the present case. The sagittal (A) and axial (B-D) views of T1- (A and B), T2- (C) and diffusion-weighted images (D) show the dilated perivascular “Virchow–Robin” spaces (arrows). Note that the dilated perivascular regions exhibit iso-intensity to the fluidal spaces in the lateral ventricle (A-D). Diffusion-weighted images were acquired with the b-factor of 1,000 s/mm2. Asterisk (A) denotes the presence of subarachnoid cyst at the right middle cranial fossa.
Conclusions
This report presents the systemic phenotypes of Loeys–Dietz syndrome that were caused by a novel TGFBR2 mutation (R381P) in a Japanese case. The systemic vascular phenotypes of this case indicate the strong penetrance as well as the dominant effects of the heterozygous missense mutation.
The TGFBR2 gene encodes transforming growth factor-beta receptor II (70/80 kDa), a membrane-bound, serine/threonine kinase domain-containing protein. This protein forms a heterodimeric complex with another receptor protein, thereby activating the downstream signaling molecules, such as SMAD proteins, upon stimulation by TGF-beta ligands. Previous studies showed that the majority of pathogenic TGFBR2 mutations were identified in the exons encoding the protein kinase domain [1–3]. In vitro studies suggested that such mutations disturbed not only the kinase activity of TGFBR2, but also internalization process of the receptor, suggesting the dominant effects of heterozygous mutations. Given that the R381P mutation in this case (Figure 3A) was also located at the kinase domain of TGFBR2, we speculated that the mutation caused deleterious effects on both its kinase activity and the receptor internalization. An angiotensin II receptor antagonist, losartan, has been recently shown to down-regulate the expression of TGFBRs, and thus proved to be effective in preventing the progressive aortic dilation and development of aneurysm in Marfan and Loeys-Dietz syndromes. A large cohort, including the present case, is thus needed to monitor its efficacy and side effects, thereby elucidating prognostic factors and contraindications of the therapies.
It is well known that prevention of aortic dilation is a vital issue for the long-term survival of patients with Loeys-Dietz syndrome. On the other hand, there are no reports clearly demonstrating the association of Marfan or Loeys-Dietz syndrome with neurovascular complications [7, 8]. Nonetheless, recent reports showed sporadic cases with intracranial aneurysm as well as tortuosity of carotid arteries in this disorder [7, 9, 10]. Given the progressive nature of the cardiovascular phenotypes of Loeys–Dietz syndrome, one could argue that only severely affected cases may experience such late-onset neurovascular complications. It also remains to be determined whether the enlarged perivascular lumens in this case (Figure 5A-C) are associated with the neurovascular phenotypes of Loeys-Dietz syndrome. Future studies will address the issues of their genotype-phenotype correlations, particularly on their comorbidity with neurovascular complications. Large cohorts with serial imaging studies can be considered to determine whether certain genotypes may correlate to their specific phenotypes.
Consent
This study was conducted in compliance with the institutional review board at Kyushu University Hospital. Written informed consent was obtained from the patient’s parents for publication of this Case Report and any accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
References
Loeys BL, Chen J, Neptune ER, Judge DP, Podowski M, Holm T, Meyers J, Leitch CC, Katsanis N, Sharifi N, et al: A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat Genet. 2005, 37: 275-281. 10.1038/ng1511.
Loeys BL, Schwarze U, Holm T, Callewaert BL, Thomas GH, Pannu H, De Backer JF, Oswald GL, Symoens S, Manouvrier S, et al: Aneurysm syndromes caused by mutations in the TGF-beta receptor. N Engl J Med. 2006, 355: 788-798. 10.1056/NEJMoa055695.
Judge DP, Dietz HC: Marfan’s syndrome. Lancet. 2005, 366: 1965-1976. 10.1016/S0140-6736(05)67789-6.
Mizuguchi T, Collod-Beroud G, Akiyama T, Abifadel M, Harada N, Morisaki T, Allard D, Varret M, Claustres M, Morisaki H, et al: Heterozygous TGFBR2 mutations in Marfan syndrome. Nat Genet. 2004, 36: 855-860. 10.1038/ng1392.
Rodrigues VJ, Elsayed S, Loeys BL, Dietz HC, Yousem DM: Neuroradiologic manifestations of Loeys-Dietz syndrome type 1. Am J Neuroradiol. 2009, 30: 1614-1619. 10.3174/ajnr.A1651.
Togashi Y, Sakoda H, Nishimura A, Matsumoto N, Hiraoka H, Matsuzawa Y: A Japanese family of typical Loeys-Dietz syndrome with a TGFBR2 mutation. Intern Med. 2007, 46: 1995-2000. 10.2169/internalmedicine.46.0467.
Maski KP, Sengupta S, Silvera M, Rivkin MJ: Intracranial artery dissection in an adolescent with Marfan syndrome. Pediatr Neurol. 2011, 45: 39-41. 10.1016/j.pediatrneurol.2010.12.011.
van den Berg JS, Limburg M, Hennekam RC: Is Marfan syndrome associated with symptomatic intracranial aneurysms?. Stroke. 1996, 27: 10-12. 10.1161/01.STR.27.1.10.
Hughes BD, Powers CJ, Zomorodi AR: Clipping of a cerebral aneurysm in a patient with Loeys-Dietz syndrome: case report. Neurosurgery. 2011, 69: E746-E755. 10.1227/NEU.0b013e31821964a3.
Rahme RJ, Adel JG, Bendok BR, Bebawy JF, Gupta DK, Batjer HH: Association of intracranial aneurysm and Loeys-Dietz syndrome: case illustration, management, and literature review. Neurosurgery. 2011, 69: E488-E492. 10.1227/NEU.0b013e318218cf55.
Acknowledgments
We thank Dr. Steve Maricich (Case Western University) for critical reading of this manuscript. This work was supported by a Grant-in-Aid from the Ministry of Education, Culture, Sports, Science and Technology of Japan (KAKEN #23810025 to Y.S., 24791072 to K.Y., 23591503 to H.T., 23591502 to Y.I., and 22249043 to T.H.), Life Science Foundation of Japan (Y.S.) and the Takeda Science Foundation (Y.S.).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
KU and YM wrote the first draft of the manuscript. KU, YM, and YS conducted the experiments. YS and TH directed the study, supervised experiments and edited the manuscript. OT and MN supervised and confirmed the radiological findings. KU, HN, and KY assessed the cardiovascular complications. KU, KU, YS, YI, HN, KY, and HT managed the patient. All authors read and approved the final manuscript.
Kiyoshi Uike, Yuki Matsushita contributed equally to this work.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Uike, K., Matsushita, Y., Sakai, Y. et al. Systemic vascular phenotypes of Loeys-Dietz syndrome in a child carrying a de novo R381P mutation in TGFBR2: a case report. BMC Res Notes 6, 456 (2013). https://doi.org/10.1186/1756-0500-6-456
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1756-0500-6-456