
Abstract
Background
Some organisms can survive extreme desiccation by entering into a state of suspended animation known as anhydrobiosis. Panagrolaimus superbus is a free-living anhydrobiotic nematode that can survive rapid environmental desiccation. The mechanisms that P. superbus uses to combat the potentially lethal effects of cellular dehydration may include the constitutive and inducible expression of protective molecules, along with behavioural and/or morphological adaptations that slow the rate of cellular water loss. In addition, inducible repair and revival programmes may also be required for successful rehydration and recovery from anhydrobiosis.
Results
To identify constitutively expressed candidate anhydrobiotic genes we obtained 9,216 ESTs from an unstressed mixed stage population of P. superbus. We derived 4,009 unigenes from these ESTs. These unigene annotations and sequences can be accessed at http://www.nematodes.org/nembase4/species_info.php?species=PSC. We manually annotated a set of 187 constitutively expressed candidate anhydrobiotic genes from P. superbus. Notable among those is a putative lineage expansion of the lea (late embryogenesis abundant) gene family. The most abundantly expressed sequence was a member of the nematode specific sxp/ral-2 family that is highly expressed in parasitic nematodes and secreted onto the surface of the nematodes' cuticles. There were 2,059 novel unigenes (51.7% of the total), 149 of which are predicted to encode intrinsically disordered proteins lacking a fixed tertiary structure. One unigene may encode an exo-β-1,3-glucanase (GHF5 family), most similar to a sequence from Phytophthora infestans. GHF5 enzymes have been reported from several species of plant parasitic nematodes, with horizontal gene transfer (HGT) from bacteria proposed to explain their evolutionary origin. This P. superbus sequence represents another possible HGT event within the Nematoda. The expression of five of the 19 putative stress response genes tested was upregulated in response to desiccation. These were the antioxidants glutathione peroxidase, dj-1 and 1-Cys peroxiredoxin, an shsp sequence and an lea gene.
Conclusions
P. superbus appears to utilise a strategy of combined constitutive and inducible gene expression in preparation for entry into anhydrobiosis. The apparent lineage expansion of lea genes, together with their constitutive and inducible expression, suggests that LEA3 proteins are important components of the anhydrobiotic protection repertoire of P. superbus.
Similar content being viewed by others


Background
Dehydration is a severe stress for organisms--most animals die if they lose more than 15-20% of their body water [1], while loss of more than 20-50% of their water content is lethal to most higher plants [2]. Some organisms have the capacity to survive extreme desiccation by entering into a state of suspended animation known as anhydrobiosis [3]. When rehydrated, anhydrobiotes revive and resume active metabolism. For example, a viable culture of the nematode Panagrolaimus sp. PS443 was isolated from dry soil that had been stored for 8 years [4]. An understanding of the molecular mechanisms responsible for anhydrobiotic survival will provide insights which may ultimately lead to the ability to confer desiccation tolerance on desiccation sensitive organisms by utilizing the strategies of anhydrobiosis, a development termed anhydrobiotic engineering [5]. Anhydrobiotic taxa have a wide distribution in nature, being found in bacteria, archaea, fungi, invertebrates, terrestrial microalgae, mosses, lichens, plant seeds and pollen, and there are approximately 350 species of angiosperm "resurrection plants" [6]. This distribution demonstrates that anhydrobitoic phenotypes are likely to have evolved independently on multiple occasions and provides support for the concept of anhydrobiotic engineering. Invertebrate anhydrobiotes include members of the Nematoda, Rotifera, Tardigrada, Crustacea and Insecta. These anhydrobiotes typically occupy aquatic or terrestrial habitats that are prone to temporary water loss. Free-living nematodes, rotifers and tardigrades contain representatives which are capable of entering anhydrobiosis at all stages of their life cycle. Crustacean anhydrobiotic stages are confined to the embryonic cysts of aquatic brine shrimps and other microcrustaceans [7]. The chironomid Polypedilum vanderplanki is the only anhydrobiotic insect described to date, but an anhydrobiotic capacity is restricted to the aquatic larval stages of this insect [8].
Most anhydrobiotic organisms are slow dehydration strategists [9], being unable to survive exposure to extreme desiccation unless they have first experienced a period of gradual water loss at high relative humidity (RH). During this period of slow dehydration the biochemical and molecular changes necessary for anhydrobiotic survival are induced. Slow dehydration strategists are found in aquatic and terrestrial habitats that lose water slowly. Many lichens, algae and bryophytes can survive rapid water loss; their tissues can tolerate the passage from the fully hydrated state to air dryness within an hour [10]. The bryophyte Tortula ruralis can survive rapid cellular dehydration and this vegetative desiccation tolerance is characterised by two components: constitutive expression of protective molecules and an inducible repair and recovery programme that is activated upon rehydration [10, 11]. Some nematodes which live in exposed environments such as moss cushions, or the aerial parts of plants can also survive rapid desiccation [9, 12]. Perry and Moens have recently proposed that, for nematode anhydrobiotes, the terms slow- and fast-dehydration strategists be replaced by external dehydration strategists and innate dehydration strategists, respectively [13]. External dehydration strategists, having little independent ability to control water loss, occur in environments that experience slow rates of water loss; whereas innate dehydration strategists have intrinsic adaptations to control the rate of water loss. These intrinsic adaptations may include behavioural (coiling/clumping) responses or morphological adaptations (e.g. surface lipids [14]) that slow the rate of water loss and allow time for inducible molecular protection mechanisms to be put in place. It may also be possible that, like the bryophyte T. ruralis, some innate dehydration strategist nematodes may also be constitutively adapted at a cellular level to survive desiccation [15].
Nematodes are a species-rich phylum with members occurring in marine and freshwater sediments, in soil and in moist terrestrial habitats. The phylum also contains economically important parasites of plants and animals. Free living anhydrobiotic nematodes occur in habitats susceptible to desiccation, but a capacity to undergo anhydrobiosis has also been important in the evolution of parasitic nematodes since many parasitic nematodes have anhydrobiotic infective stages or cysts [16] (Figure 1). Anhydrobiotic nematodes are abundant in the sub-order Tylenchina, a diverse group of nematodes that contains free living microvores and predators, as well as parasites of invertebrates, vertebrates and plants. The Tylenchina are particularly noted for the repeated evolution of plant parasitic clades within this sub-order [17] and phylogenetic reconstructions show that anhydrobiotic Tylinchina also have had multiple independent evolutionary origins. The genus Panagrolaimus, a member of the Tylenchina, contains both external dehydration and innate dehydration strategist nematodes [15, 18, 19]. Panagrolaimids feed on bacteria and occupy a variety of niches ranging from Antarctic, temperate and semi-arid soils to terrestrial mosses. Panagrolaimus species have a short generation time (~ 10 days at 20°C); they can be cultured with Escherichia coli as a food source using protocols developed for the model nematode Caenorhabditis elegans and an RNA interference protocol has been described for P. superbus[20]. In addition to its anhydrobiotic capacity, P. davidi from maritime Antarctica can survive freezing when fully hydrated [21]. We have found that a similar cryotolerant capacity also exists in other hydrated Panagrolaimus species isolated in temperate and continental regions (McGill et al., unpublished). Phylogenetic analysis shows that the anhydrobiotic species and strains of Panagrolaimus described to date belong to a single clade [15], which will facilitate comparative transcriptome analyses of the molecular basis of dehydration within a single genus. Here we present an analysis of 4,009 unique sequences (unigenes) derived from 9,216 Sanger-sequenced ESTs from an unstressed mixed population of the innate/fast desiccation strategist Panagrolaimus superbus and we identify putative anhydrobiosis related genes which may have application in future anhydrobiotic engineering experiments. P. superbus was originally isolated in 1981 from a gull's nest in Surtsey [22], an Icelandic island formed during 1963-1967 from volcanic eruptions [23]. The objective of the 959 Nematode Genomes Initiative is to encourage genome sequencing across the diversity of the phylum Nematoda [24]. Because of its provenance and its anhydrobiotic and cryotolerant phenotypes the genome of P. superbus is currently being sequenced as part of the Nematode Genomes Initiative (http://www.nematodes.org/nematodegenomes/index.php?title=About#tab=Welcome). In addition to providing cDNA clones and sequence data for candidate anhydrobiotic genes, the dataset presented here will also provide anchor sequences important for the assembly of the genome and transcriptome of P. superbus.

A phylogenetic tree of the Phylum Nematoda based on that of Blaxter et al. [25]showing the distribution of anhydrobiotic and desiccation tolerant taxa, along with their trophic ecology, across the Phylum. (¶Erkut et al. have recently shown that C. elegans dauer larvae can survive exposure to low relative humidity following preconditioning at 98% RH for 4 days [26]).
Results and discussion
EST assembly
A total of 9,216 ESTs were obtained by Sanger sequencing from a directionally cloned cDNA library prepared from an unstressed mixed stage population of P. superbus. Processing these ESTs through the PartiGene pipeline [27] resulted in 7,606 high quality ESTs with an average length of 425 bp (Table 1). These EST sequences have been deposited in dbEST with the accession numbers GW405912-GW413517. The Partigene pipeline then clustered these ESTs into 1,079 consensus sequences (contigs) and 2,958 singletons. Removal of putative bacterial sequences and rRNA genes yielded a total of 3,982 putative protein-coding transcripts (unigenes). BLASTX analysis showed that 1,923 of these unigenes had significant hits to a combined NemPep+, WormPep and the NCBI non-redundant (nr) NCBI database (see Methods for details). Among these were 100 unigenes which had unique hits to the NCBI nr database, leaving 2,059 (51.7%) novel P. superbus unigenes. These P. superbus unigene annotations can be subjected to keyword queries and the sequences can be downloaded from the NEMBASE4 database at http://www.nematodes.org/nembase4/species_info.php?species=PSC.
Of the 100 unigenes with unique hits to the NCBI nr database, 38 returned BLAST hits only to bacterial taxa. Bacterial sequences had been screened and removed from the dataset during the assembly process using stringent matching criteria (BLASTN with an e-value cut-off of 1e-50), thus some of these 38 sequences may correspond to residual contaminant sequences. Others may represent sequences from bacterial associates of P. superbus, or horizontally transferred sequences, e.g. one unigene sequence (PSC01785) had highest similarity to an ankyrin gene from a Wolbachia endosymbiont from Culex quinquefasciatus[28]. The remaining 62 sequences returned hits to eukaryote taxa and these included several hits to plant LEA (late embryogenesis abundant) sequences, important in plant desiccation tolerance. PSC01785 had best similarity to an exo-β-1,3-glucanase, a member of the glycosyl hydrolase family 5 (GHF5), from the Stramenopile Oomycete Phythophthora infestans[29]. β-1,3-glucans are a major structural component of fungal cell walls and, as a microbivore, P. superbus may also use GH5 glucanases to incorporate soil fungi into its diet. Endo-β-1,3-glucanase GHF5 genes have been reported from the fungal feeding nematodes Bursaphelenchus xylophilus, B. mucronatus[30] and Aphelenchus avenae[31], with horizontal gene transfer (HGT) from bacteria being proposed to explain their evolutionary origin. Endo-β-1,4-glucanase (cellulase) genes, also belonging to GHF5, have been reported from several species of plant parasitic nematodes of the order Tylenchida [32], with HGT from bacteria also proposed as their likely source [33]. The GHF5 cellulases recently reported from the free living nematode Pristionchus[34] are most similar to sequences from two slime mould species (Amoebozoa). Thus the P. superbus sequence reported here represents another possible HGT event (this time from the eukaryote Stramenopile lineage) for nematode GHF5 genes.
Large EST datasets have been generated for nematode parasites, however with the exception of the model nematodes C. elegans, other Caenorhabditis species and Pristionchus pacificus, there are relatively few EST datasets for free-living nematodes. Metagenomic analyses of nematode ESTs [35, 36] show that nematode gene-space is substantially under-sampled. BLAST analyses of the EST cluster consensus sequences from 37 species represented in NemPep3 showed that 34-70% of the sequences from each nematode species were unique to that species [35]. Data from Nembase4 (derived from 54 parasites and 8 free living species) show that 61.8% of the predicted proteins had no GO, EC or KEGG annotations [37]. Thus the finding that 51.7% of the P. superbus unigenes correspond to novel sequences is consistent with previous studies and is a reflection of the diversity of nematode gene space.
Abundantly expressed transcripts
The 44 most abundantly expressed P. superbus protein-coding sequences comprise 1,200 ESTs and represent 15.7% of the total EST dataset (Table 2). The most abundant sequence, containing 79 ESTs, encodes a member of the nematode specific family of SXP/RAL-2 proteins [38, 39]. These proteins have been detected in the pharyngeal glands and as secreted and surface associated antigens in diverse animal parasitic nematodes. Immunization with recombinant antigens derived from SXP/RAL-2 has been effective in protecting treated animal hosts against filarial worm [40], roundworm [41] and hookworm [42] infections. SXP/RAL-2 proteins have also been described in plant parasitic nematodes [43, 44] and SXP/RAL-2 sequences from 11 species of plant parasitic nematodes are represented in NEMBASE4. GenBank searches show that SXP/RAL-2 homologs also occur in other free living nematodes in addition to P. superbus viz.: C. elegans (Accession Numbers: NP_496220 and NP_495640); C. briggsae (XP_002630517) and Pristionchus pacificus (5 ESTs). Nematode SXP/RAL-2 sequences are likely to be encoded by a small multigene family [43, 44]. We detected five SXP/RAL-2 unigenes in P. superbus, comprising 95 ESTs and representing 2.2% of the total EST dataset. With the exception of Ascaris lumbricoides, the level of SXP/RAL-2 expression in P. superbus was higher than that observed for any of the 19 species of parasites with SXP/RAL-2 homologs in NEMBASE4. SXP/RAL-2 are small (16-21 kDa) basic proteins which share a common domain of unknown function (DUF148, PF02520 http://pfam.sanger.ac.uk/). No RNAi phenotypes have been detected for SXP/RAL-2 homologs in C. elegans, but one homolog (NP_496220/ZK970.7) was among 14 genes which were upregulated in C. elegans on response to fungal infection [45]. All SXP/RAL-2 sequences characterised to date, including PSC00077, have a signal peptide indicative of a secreted protein. Parasitic nematode studies suggest that these nematode-specific proteins are most likely secreted from the pharyngeal glands onto the surface of the cuticle where they appear to carry out a structural or protective function. The very high level of expression of SXP/RAL-2 sequences in P. superbus suggests that this cuticular protein may have an important role in anhydrobiotic protection in this nematode.
Ten abundantly expressed sequences were associated with reproductive function, eight corresponding to major sperm protein genes (MSPs) and two to vitellogenin genes. In total we detected 32 P. superbus MSP unigenes and 7 vitellogenin unigenes. In C. elegans MSPs are encoded by a multigene family comprising more than 50 genes [47, 48]. Nematode MSPs are small, basic proteins required for the amoeboid movement of sperm. A family of six genes vit-1 to vit-6 encode C. elegans vitellogenin [49, 50], a major yolk component which is expressed exclusively in the adult hermaphrodite intestine from which it is secreted into the pseudocoelomic space and taken up by oocytes [51]. Four structural genes were abundantly expressed in P. superbus: an actin family member (homolog of C. elegans act-2); a gene encoding a core histone protein required for chromatin assembly and chromosome function [52] and genes encoding two proteins associated with the 60S ribosomal subunit. Two abundantly expressed contigs were associated with lipid metabolism. PSC00187 encodes a homolog of C. elegans HEH-1 and human NPC2/He1, a cholesterol-binding protein whose deficiency in humans causes Niemann-Pick type C2 disease involving retention of cholesterol in lysosome [53, 54]. Transcripts for the mitochondrially encoded cytochrome c oxidase subunit 1, essential for oxidative phosphorylation and ATP synthesis, were also highly expressed in this mixed stage P. superbus library.
Twenty one of the abundant sequences listed in Table 2 are novel. These novel unigenes correspond to 641 ESTs, representing 8.4% of the total EST dataset. Data on the predicted physico-chemical parameters of the putative proteins encoded by these 21 unigenes are presented in Additional file 1. Thirteen (65%) of these novel unigenes encode a signal peptide indicative of a secreted protein. The association between sequence novelty and likely secretion has been noted previously in the parasitic nemataode Nippostrongylus brasiliensis[55]. Three of the putative novel proteins are predicted to be natively unfolded over 80-100% of their primary sequence. The P. superbus dataset contains a total of 2,059 novel unigenes. Further analysis of these novel sequences is presented in a later section.
Functional annotation
In order to identify candidate stress-related genes which may have a role in anhydrobiosis the annot8r program [56] was used to assign KEGG (Kyoto Encyclopaedia of Genes and Genomes) pathway annotations [57] and Gene Ontology (GO) terms [58] to the P. superbus unigenes.
Assignments to metabolic pathways using KEGG
One thousand six hundred and eighty four KEGG orthology assignments were inferred by searching for P. superbus unigenes that have homologs among the default set of manually curated eukaryotic genes in the KEGG database (which contains 26 genomes); similarly 1,412 KEGG assignments specific to the C. elegans genome were also inferred (Table 3). KEGG pathways associated with metabolism had the highest representation, with a large number of the P. superbus sequences associated with the 'carbohydrate metabolism', 'energy metabolism', 'lipid metabolism' and 'amino acid metabolism' pathways. In the environmental information processing category, 'signal transduction' was highly represented. Other highly represented pathways were found in the genetic information processing category including 'translation' and 'folding, sorting and degradation' and a large number of sequences had KEGG assignments to the human neurodegenerative disease sub-category. Many neurodegenerative diseases are associated with the dysfunction or overload of the protection systems responsible for repairing or degrading damaged proteins and macromolecules [59–61]. Cells exposed to severe water stress experience serious damage to their macromolecules and membranes; proteins lose their structures, become unfolded and aggregate. Thus anhydrobiotic organisms are adapted to survive cellular dehydration by deploying efficient cellular protection and repair systems (Figure 2) and it is likely that some gene products that have roles in anhydrobiotic protection in nematodes may also have human homologs which are required for neural survival. For example the molecular chaperone DJ-1, which is associated with familial Parkinson's disease [62, 63], is also upregulated in response to desiccation stress in the anhydrobiotic nematode Aphelenchus avenae[64]; and AAvLEA1, a natively unfolded late embryogeneis (LEA) protein which is upregulated in response to desiccation stress in A. avenae[65], has been shown in vitro to protect complex mixtures of proteins from aggregation [66].

Possible steps involved in the detection and expression of anhydrobiotic protection mechanisms in nematodes (sHSP = small heat shock protein; LEA = late embryogenesis abundant protein; IUP = intrinsically unfolded protein).
Gene ontology assignments
The Gene Ontology consortium has developed a vocabulary of defined terms that describe gene products in the context of three domains--Biological Process, Molecular Function and Cellular Component in a species-independent manner [58]. The representation of GO terms as found by BLAST searches of the P. superbus unigenes against genes in the GO database are presented in Additional file 2. In summary, these representations consist of: Biological Process--3,148 BLAST hits; Molecular Function--1,671 hits; and Cellular Component--1,245 hits. Several of these hits were to gene products whose descriptions indicate roles in anhydrobiotic protection, as discussed in the following section.
Putative anhdyrobiotic and stress response genes constitutively expressed in unstressed P. superbus
When cells suffer severe dehydration metabolism ceases, macromolecules denature, membranes undergo phase changes and fuse with other normally separate membranes. Unlike desiccation sensitive taxa, anhydrobiotes have evolved mechanisms which maintain the structure and integrity of macromolecules and membranes in the absence of water and also during rehydration and revival. Comparative studies of the desiccation tolerance phenotypes of anhydrobiotes show lineage specific differences in the response patterns and biochemical adaptations, which implies that anhydrobiotic phenotypes can be achieved in different taxa by the expression of functionally equivalent molecules. Based on currently available data from nematodes and other anhydrobiotic animals we present a model showing the possible steps involved in the detection and expression of anhydrobiotic protection mechanisms in nematodes (Figure 2). Using GO, KEGG and BLAST description data we have identified components of this model in the P. superbus unigene dataset and we have assembled and manually annotated a set of 187 candidate genes whose products may be involved in the anhydrobiotic response of P. superbus. This dataset is summarized in Table 4 and presented in full in Additional file 3.
Signal transduction, protein kinases and transcription factors
Transduction of environmental stress signals is achieved in eukaryotes through a conserved cascade of sequentially acting stress activated protein kinases (SAPKs) which form a branch of the mitogen-activated kinase (MAP-kinase) system [67–70]. In Saccharomyces cerevisiae the SAPK pathway is activated by osmostress and the terminal kinase Hog1 [71], when phosphorylated, translocates to the nucleus [72]. Here it phosphorylates several transcription factors, and associates at stress-responsive promoters through such transcription factors [73], resulting in the expression of osmotic response genes. Our P. superbus EST dataset contains 35 protein kinases. Among these were three contigs encoding MAP kinases. One of these PSC00478, is a homolog of MAPKAP kinase-2 which is responsible for the phosphorylation of the small heat-shock proteins Hsp27 [74] and α-B-crystallin [75]. The phosphorylation of these small HSPs in response to stresses such as heat shock and oxidation is proposed to regulate actin filament dynamics and to stabilize microfilaments [76]. Two unigenes encode members of the STE20 family of serine/threonine kinases [77]. One of these, PSC03670, has high BLAST identity (3e-51) to the gck-3 gene whose function is required by C. elegans for volume recovery and survival after exposure to extreme hypertonic stress [78]. Nine unigenes encode putative casein kinases, important regulatory molecules in cell division and differentiation and in DNA damage repair [79, 80]. Casein kinase 2 (CK2) was found to be upregulated in response to desiccation stress in the nematode Steinernema feltiae[81]. Other P. superbus stress-related kinases include a homolog of akt-1, a regulatory component within the insulin/IGF-1 signaling pathway [82, 83], which plays an important role in regulating nematode life span, dauer formation and stress tolerance and diacetylglycerol (DAG) kinase, which modulates DAG levels in the cell membrane, regulating intracellular signalling proteins that have evolved the ability to bind this lipid [84]. DAG kinase is also activated in plants during cold and osmotic stress [85].
Several P. superbus unigenes are predicted to encode transcription factors. Among these are a forkhead protein, a member of a conserved family of transcriptional regulators of cellular processes including metabolism, ageing, apoptosis, cell cycle progression and stress resistance [86, 87]. Two unigenes encode putative jumonji (JmjC) domain-containing proteins. The C. elegans jmjc-1 gene functions as a transcriptional activator of stress related genes in response to multiple stimuli including heat-shock and osmotic and oxidative stress [88]. Two unigenes encode putative high mobility group (HMG) proteins, which are important in modulating chromatin structure and gene expression. HMG transcripts are upregulated in response to desiccation osmotic heat and cold stresses in the anhydrobiotic nematode Aphelenchus avenae[64] and HMG function is required for the transcription of stress-responsive genes in Arabidopsis thaliana[89].
Anti-oxidant activity
Reactive oxygen species (ROS) accumulate in cells as a result of cellular dehydration [90, 91]. ROS cause oxidative damage to proteins, lipids, DNA and other macromolecules. Therefore proteins with antioxidant properties are required to rapidly neutralise ROS immediately they are formed. We have characterized 21 unigenes encoding proteins that fall into this category: PSC00113 encodes a manganese superoxide dismutase responsible for converting O2•- radicals into H2O2; three unigenes encode glutathione peroxidases which function to reduce H2O2; and sequences encoding both the 1-Cys and 2-Cys class of peroxiredoxin enzymes (whose main function is the reduction of peroxides [92]) were also identified.
The tripeptide glutathione (GSH) functions as a co-factor for the antioxidant enzymes glutaredoxin (Grx) and glutathione S-transferase (GST). GSTs catalyse the conjugation of glutathione to reactive electrophilic compounds from endogenous and xenobiotic sources and are thus capable of detoxifying a large variety of cytotoxic molecules [93]. The P. superbus dataset contains seven GST unigenes, four from the sigma class of cytosolic GSTs and two from the kappa class of mitochondrial GSTs. The GST sigma and kappa classes are considered to be involved in protection against endogenously produced ROS [94]. PSC02300 encodes a Grx enzyme. Protein deglutathionylation is carried out by Grx [95], and this enzyme can also reduce the disulphide bridges of oxidised proteins [96]. Such disulphide bridges can also be reduced by thioredoxin (PSC00712), which shares a similar structure and overlapping function with Grx [97]. Other P. superbus unigenes whose gene products are likely to be involved in antioxidant activity and redox regulation include: aldehyde dehydrogenase which deactivates malondialdehyde, an important end product of lipid peroxidation; two aldo-keto reductase sequences [98, 99] and a putative cytosolic NADP-isocitrate dehydrogenase (NADP-ICDH). NADP-ICDH catalyzes the production of NADPH and, by supplying NADPH to the antioxidant systems, NADP-ICDH is an important component in the control of redox balance and the modulation of oxidative damage in the cytosol [100, 101].
Late embryogenesis abundant proteins
Thirteen P. superbus unigenes encode predicted late embryogenesis abundant (LEA) proteins (Table 5). Although LEA proteins have been shown to accumulate during the onset of desiccation in anhydrobiotic animals including nematodes [65, 81, 102], genes encoding LEA proteins are particularly numerous and heterogeneous in plant genomes. For example the Arabidopsis thaliana genome contains 51 lea genes placed into 9 different Pfam groups [103, 104]. In animal genomes lea genes are less abundant and predominantly belong to the Group 3 LEA family (Pfam F02987) [105], with members of LEA Group 1 (PF00477) being described to date only in the brine shrimp Artemia franciscana[106] and in an unspecified tardigrade species [107]. Group 3 LEA proteins are highly hydrophilic and largely lacking in secondary structure when fully hydrated [105]. Group 3 proteins also contain blocks of tandemly repeated 11-mer amino acid motifs [108], the number of repeats per LEA protein typically ranging in number from 5 to 24 [109].
A database of LEA proteins has been established recently [112]. Although 89% of the sequences in this database are from land plants, the LEAPdb database includes LEA sequences from animal taxa. Among BLAST searches against the LEAPdb (Table 5), ten P. superbus unigenes had best hits to LEA3 proteins from plant species, one unigene was most similar to a putative LEA protein from Haemophilus influenzae[113] and two to an LEA3 protein from C. elegans, the abundance of P. superbus sequences which had hits to plant genes may be a consequence of the large number of plant LEA sequences represented in the LEAP database. The LEA sequences encoded by six of the P. superbus unigenes are predicted to be 100% natively unfolded. Three of the 13 P. superbus LEA sequences are predicted to lack substantial regions of unfolded structure, however all 13 LEA sequences had negative GRAVY (Grand Average of Hydropathy) indices characteristic of hydrophilic proteins (Table 5). All the predicted sequences showed evidence of tandemly repeated 11-mer amino acid motifs (See Additional file 4). The C. elegans genome has been reported to contain three lea genes [109] and four lea genes have been detected in the C. briggsae genome [109]. The best characterized nematode LEA protein is AAv1 which is upregulated in the nematode A. avenae in response to desiccation. AAv1 has been shown to protect complex mixtures of proteins from aggregation in vitro and in vivo[66]. It is possible that some of the P. superbus unigenes reported here may represent alternatively spliced forms of a single lea gene. However, the relative abundance of lea genes in P. superbus as compared to C. elegans, along with their constitutive expression (we detected 34 LEA-encoding ESTs), suggest that LEA group 3 proteins are an important component of the anhydrobioitc protection repertoire of P. superbus.
Molecular chaperones and unfolded protein response
Heat shock proteins (HSPs) are essential for the correct folding and maturation of a great diversity of client proteins and for protecting proteins from stress induced unfolding and aggregation [114, 115]. Eukaryotic HSP families contain multiple genes, which may be either constitutively expressed or stress inducible and targeted to specific cellular compartments [116, 117]. The HSP expression repertoire of an anhydrobiotic organism may thus be very important in maintaining the integrity of the proteome during the dehydration and recovery phases of anhydrobiosis [118–121]. The P. superbus dataset contains representatives of all the heat shock protein (HSP) classes characteristic of nematodes, including four distinct small heat shock proteins (sHSP). sHSP are the major "holding" chaperones, retaining unfolding proteins in a conformation suitable for subsequent refolding thus preventing their irreversible aggregation [122, 123]. Anhydrobiotic encysted larvae of the brine shrimp Artemia franciscana accumulate large quantities of a sHSP known as p26 which constitutes ~15% of the non-yolk protein in these larvae [124]. A. franciscana cysts are resistant to desiccation, high temperature, γ-irrradiation and anoxia and the chaperoning activity of p26 is likely to be a very significant component of this remarkable stress resistance [125].
The accumulation of unfolded proteins in the endoplasmic reticulum (ER) arising from physiological or abiotic stress leads to the expression of several protein folding chaperones, including members of the HSP90 and HSP70 families and their co-chaperones [126]. Unfolded protein response (UPR) chaperones from the P. superbus dataset include three protein disulfide isomerases (PDI), which catalyse the formation and isomerization (rearrangement) of cysteine bonds during protein folding [127, 128]; five cyclophilin-type peptidyl-prolyl cis-trans isomerases which catalyse the isomerisation of the peptide bonds preceding proline residues; and a homolog of Derlin-2 which is required for the degradation of misfolded glycoproteins in the ER [129]. Five P. superbus unigenes encode proteins required for the facilitated folding of actin and tubulin to form microtubules: two prefoldin subunits, two subunits of cytosolic T-complex protein 1 and α-tubulin folding cofactor B (Additional file 3). Changes in microtubule dynamics have been shown to occur during osmotic stress in Zea mays[130] and during desiccation in Brassica napus[131] and it is possible that adjustments to the stability of the microtubule cytoskeleton are also required by P. superbus for successful entry into anhydrobiosis.
Removal of damaged proteins--the ubiquitin-proteasome (UPS) and autophagy systems
When the HSP chaperone system fails to correctly fold a denatured protein, the misfolded protein is polyubiquitinated. The 26S proteasome, a large multiprotein complex, then translocates polyubiquitinated proteins into the inner proteolytic chamber where they are hydrolysed [132]. If the generation of misfolded proteins exceeds the proteolytic capacity of the ubiquitin proteasomal system (UPS), misfolded proteins accumulate into aggregates which are degraded by autophagy [133, 134]. A whole genome RNA interference (RNAi) screen in C. elegans identified 40 genes that are essential for survival during acute hypertonic stress [135]. Half of these genes encode proteins that function to detect, transport, and degrade damaged proteins. The importance of the proteasomal system to unstressed nematodes is also apparent from its abundant representation in the P. superbus EST dataset, which contains 44 UPS unigenes comprising 68 ESTs (Additional file 3). In contrast, autophagy genes are not well represented in unstressed P. superbus. We detected only two P. superbus homologs of the 19 core C. elegans autophagy genes [136].
DNA damage response proteins
DNA extracted from anhydrobiotic stages of the plant parasitic nematode Ditylenchus dipsaci was intact, showing no increase in the frequency of double strand DNA breaks (DSBs) as compared with hydrated worms [137]. Data from the anhydrobiotic chironomid P. vanderplanki[138] and anhydrobiotic tardigrades [139, 140] show that DSBs accumulate with time in the dry state in these organisms. DSBs also accumulate during desiccation in the anhydrobiotic and radiation resistant bacterium Deinococcus radiodurans. Similar to P. vanderplanki[138] and anhydrobiotic tardigrades [139, 140], D. radiodurans has acquired the ability to rapidly repair DNA damage when rehydrated [141]. The P. superbus dataset contains 12 unigenes encoding proteins involved in DNA repair (Additional file 3). However these DNA repair proteins appear to be constitutively expressed at low levels in P. superbus, as each is represented by only a single EST.
Other putative anhydrobiotic genes
Other transcripts whose products may play a role in the anhydrobiotic response of P. superbus include two putative aquaporins; an erm (ezrin, radixin, and moesin) family member; an an1-like Zinc finger sequence; a thaumatin-like transcript; two copies of lon-1 which encodes a protease that selectively degrades oxidized mitochondrial proteins [142] and a homolog of the Ric1 family [143] which encodes plasma membrane proteins that are expressed in response to high salt or low temperature conditions in plants [144]. ERM proteins are activated by osmotic shrinkage [145] and they are thought to function as cross-linkers between plasma membranes and actin-based cytoskeletons [146]. Thaumatin-like proteins are induced in plants in response to pathogens, cold, drought and osmotic stress [147]. The an1-like multigene family is involved in plant abiotic stress responses and in inflammation responses in mammals [148].
A comparison the P. superbus EST unigene dataset with EST datasets from other anhydrobiotic nematodes
The 3,982 P. superbus unigenes were compared to EST unigene datasets from three other species of anhydrobiotic nematodes [31, 102, 149] to identify putative homologous protein families which may reveal some of the core anhydrobiotic processes shared by these nematodes. Plectus murrayi is an Antarctic soil nematode adapted to survive desiccation and freezing [102]. Aphelenchus avenae is a slow desiccation strategist soil dwelling fungiverous nematode. Ditylenchus africanus is an endoparasite of plants with peanut as its primary host. It migrates to the pods and seeds of the ground nut and can survive in an anhydrobiotic state in the seeds [149]. The phylogenetic relationships of these nematodes are indicated in Figure 1.
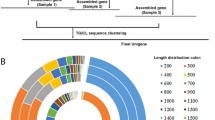
The combined dataset from the four anhydrobiotic nematodes comprised 10,791 unigenes. All against all BLASTP analyses of the predicted peptide sequences for these unigenes, followed by their classification into putative homologous groups using the TRIBE Markov clustering algorithm as implemented in the MCL software package [150], has identified 7,063 unigene families, where 6,308 consist of singletons. The distribution of these unigene families across the four nematode species is summarized in Figure 3(a). A total of 67 unigene families contain transcripts from all four anhydrobiotic nematodes. While our analysis is based on an incomplete coverage of the transcriptomes of all four nematodes, these 67 families provide a first indication of subsets of genes common to the four species, some of which may be involved in anhydrobiotic processes. The annotation of the 67 unigene families (inferred by BLAST), together with a list of their component P. superbus contigs is presented in Additional file 5. These families include representatives of several of the anhydrobiotic and stress response proteins discussed in the previous section. Among these are protein kinases and HMG proteins; glutathione S-transferase; sHSP, HSP70; HSP90; peptidyl-prolyl cis-trans isomerase; several components of the UPS system and RIC1, a poorly characterized family which encodes plasma membrane proteins that are expressed in response to high salt or low temperature conditions in plants [144]. Members of the nematode specific transthyretin-related (ttr) family [151] are also included among the 67 unigenes. The function of ttr genes remains elusive; so far the function of just one nematode ttr gene product has been discovered (TTR-52 mediates the recognition and engulfment of apoptotic cells in C. elegans[152]). Figure 3(b) shows the distribution of homologs of the P. superbus putative stress response genes from Table 4 across the four anhydrobiotic nematode species. These BLAST annotations are presented in full in Additional file 3. Since this analysis is based on partial transcriptomes of the four nematodes the results need to be interpreted conservatively, however the data show that these four anhydrobiotic nematodes express a great diversity of stress responsive genes. Surprisingly none of the 13 LEA unigenes were common to all four nematode datsets, and 8 LEA sequences were found only in P. superbus. This may indicate that constitutive expression of LEA transcripts is higher in P. superbus than in the three other anhydrobiotic nematodes. When more complete coverage of the transcriptomes of anhydrobiotic nematodes and other anhydrobiotic animals becomes available comparative transcriptomic analyses will be a powerful tool for the identification of candidate genes and processes required for successful anhydrobiotic survival.

(a)-Venn diagram indicating the number of unigene families that contain representatives from one or more anhydrobiotic nematodes: Panagrolaimus superbus, Plectus murrayi, Ditylenchus africanus, Aphelenchus avenae. The total dataset comprises 7,063 families 67 of which have representative ESTs from all four nematode species. The values in parentheses represent the percentage of the total number of families represented in each section. (b)-Venn diagram showing the distribution of homologs of the P. superbus putative stress response genes from Table 4 across four anhydrobiotic nematode species. The values represent the number of P. superbus unigenes that have BLAST hits to sequences from one or more of the nematodes Plectus murrayi, Ditylenchus africanus, Aphelenchus avenae. The numbers in parentheses correspond to the number of families (see Methods) these unigenes constitute.
Analysis of novel ESTs
Of the 3,982 unigenes in our dataset 2,059 (51.7%) have no significant similarity to any sequences in the Genbank or NemPep databases. The Prot4EST algorithm [46] was used to translate these novel unigenes into putative peptides. Analysis of the physical properties of these putative peptides reveals that 149 of them are predicted to lack a fixed tertiary structure (100% intrinsically disordered), while an additional 296 peptides are predicted to be 50-99% disordered. Intrinsically disordered proteins (IDPs) are hydrophilic, being characterized by a high proportion of polar and charged amino acids and low sequence complexity; they also have a low content of the hydrophobic amino acids which would normally form the core of a folded globular protein [153]. These physical features also occur in LEA proteins. A plot the hydropathy [111] of P. superbus putative novel peptides and the 13 predicted P. superbus LEA proteins against their predicted degree of disorder (determined using the IUPred program [110]) shows that there are 225 P. superbus peptides with a GRAVY (Grand Average of Hydropathy) value of ≤ -1 and > 50% disordered (Figure 4).

A plot of the hydropathy value (GRAVY Index) [111]of P. superbus putative novel peptides and the 13 predicted P. superbus LEA proteins against their predicted degree of disorder, as determined by the IUPred program [110]. (GRAVY = Grand average of hydropathicity); hydrophilic proteins typically have hydropathy values < -1 [154]. Green lines represent the boundaries that delimit a group of novel hydrophilic peptides that are predominantly disordered.
Garay-Arroyo et al.[154] proposed that LEA proteins are contained within a larger group of proteins called 'hydrophilins' that accumulate in response to osmotic stress in prokaryotes and eukaryotes. The characteristics that define this group are a glycine content of greater than 6% and hydropathy index of less than -1. Our dataset contains 170 novel putative peptides that meet these criteria (Figure 5). The P. superbus unigenes predicted to encode LEA proteins (Table 5) were identified on the basis of BLAST searches. Analysis of their physical properties reveals that all of these putative LEA proteins are hydrophilic, having GRAVY values ranging from -0.62 to -1.42; six are predicted to be 100% unstructured, a further three are largely (77-99%) unstructured, but three putative LEA unigenes: PSC01853, PSC00514 and PSC00416 are predicted to be only partly disordered (9, 10 and 18% disorder, respectively). Eleven of the 13 putative LEA sequences also have a glycine content of greater than 6%.

Plot of the putative glycine content and the hydropathy of the protein sequences encoded by the novel ESTs and putative LEA proteins in the P. superbus dataset. Green lines represent the boundaries of the properties that define hydrophilins; glycine content > 6% and a hydropathicity index of < -1 [154].
Many IDPs proteins function by molecular recognition: either by transient, or permanent binding to a structured partner molecule [155]. However the functions of some IDPs depend directly on the extended random coil conformation of the disordered state--the so-called entropic chain effect [156]. Entropic chain effects are likely to be central to many of the functions of LEA proteins. The elongated, natively unfolded conformation of LEA proteins may help to form a "molecular shield" [66], preventing protein aggregation and denaturation. These hydrophilic, proteins also have the capacity to bind and retain water molecules and, at later stages of dehydration process, an abundance of charged amino acids may enable some LEA proteins to replace water at the hydrogen bonding sites of dehydrated proteins. Although LEA proteins are natively unfolded when fully hydrated, some LEA proteins, including AavLEA1 [157], have been shown to develop secondary structure as they become desiccated [158], leading to the suggestion that some LEA proteins might function as intracellular space-filler molecules which prevent the collapse of cells as they become desiccated [105]. The combined group of putative hydrophilic proteins identified in Figures 3 and 4 contains 294 individual novel sequences. These sequences represent an important group of candidate anhydrobiotic genes that merit further investigation.
Expression of putative stress related genes upon desiccation
P. superbus is capable of surviving exposure to a dry atmosphere in desiccation chambers containing silica gel without the need for prior preincubation to mild desiccation stress [15]. However it likely that in its natural habitat P. superbus would experience more gradual change from a condition in which its cells and tissues are fully hydrated to one of extreme dehydration. In addition, intrinsic behavioural (coiling/clumping) responses or morphological adaptations (such as surface lipids [14] or possibly SXP/RAL-2 cuticular proteins) may slow the rate of water loss in P. superbus and allow time for inducible molecular protection mechanisms to be put in place. We used qPCR to investigate the inducible expression of several unigenes that represent homologues of stress related genes in other organisms. The expression of five of the 19 genes tested was upregulated in P. superbus following exposure to 98% RH for 12 h (Figure 6).

Real-Time Relative qPCR analysis of the expression of some putative stress-response genes in Panagrolaimus superbus following 12 h of desiccation at 98% RH. The following transcripts were tested: PSC03116: small heat shock protein (sHSP) family member; PSC01018 sHSP family member; PSC02842 HSP40/DNaJ protein family member; PSC00673 HSP70 family member; PSC02695 cyclophilin family member; PSC00740 protein disulfide isomerase; PSC00782 LEA3 protein; PSC01853 putative LEA3 protein; PSC00514 LEA3 protein; PSC02304 DJ-1; PSC03895 1-Cys peroxiredoxin; PSC02494 gluthatione peroxidase; PSC04819 gluthatione peroxidase; PSC02624 glutathione S-transferase (sigma class); PSC04040 glutathione S-transferase (kappa class); PSC01063 aldehyde dehydrogenase; PSC01095 aldehyde dehydrogenase; PSC01029 aquaporin; PSC01468; RIC1 putative stress responsive protein. The reference genes were the P. superbus 60S ribosomal protein L32 and ama-1 genes. Statistically significant differences (Student's t test) are indicated, **p < 0.001.
Three antioxidant genes gpx (glutathione peroxidase), dj-1 and prx (which encodes a 1-Cys peroxiredoxin) were upregulated in response to desiccation stress in P. superbus. ROS accumulation is triggered by cellular dehydration and our qPCR data show the importance of enzymatic antioxidant defense systems during the induction of anhydrobiosis. Glutathione peroxidases (GPx) catalyse the reduction of H2O2 and GPx have been previously found to be upregulated in A. avenae and in P. murrayi in response to desiccation [64, 102]. DJ-1 is a multifunctional protein associated with familial Parkinson's disease [62, 63]. One of its proposed functions is a redox-dependent molecular chaperone activity [63] and a role for DJ-1 as an atypical peroxiredoxin-like peroxidase in inactivating mitochondrial H2O2 has also been proposed [159]. We show that the expression of dj-1 is upregulated 9-fold in P. superbus in response to desiccation stress. This gene is also upregulated in response to desiccation stress in the anhydrobiotic nematode Aphelenchus avenae[64]. Peroxiredoxins (Prx) comprise two classes: 1-Cys Prx and 2-Cys Prx, based on the number of cysteinyl residues directly involved in catalysis [92]. Animal Prx sequences comprise 3 clades [160]: clades A and B contain 2-Cys Prx, while 1-Cys Prx occur in clade C which also contains plant 1-Cys Prx sequences [160]. In plants 1-Cys Prx are seed-specific [161]: they accumulate during seed maturation and their expression declines during germination, an expression pattern is also characteristic of many lea genes. A seed-specific 1-Cys Prx, was found to be abundantly expressed during desiccation in the leaves of the resurrection plant Xerophyta viscosa[162] and transcripts encoding a 1-Cys Prx are also upregulated during rehydration of the anhydrobiotic moss Tortula ruralis[163]. Here we show that a 1-Cys Prx is upregulated in response to desiccation in P. superbus, revealing a further parallel between the desiccation tolerance mechanisms of anhydrobiotic nematodes and plants.
Only one of the three lea sequences tested was upregulated, but this sequence (PSC01853) was upregulated 9.8 fold in response to desiccation stress. Of the four P. superbus hsp sequences assayed only one, an shsp sequence, was upregulated in response to desiccation. The genes encoding HSP70 and HSP90 are constitutively expressed in the Antarctic nematode Plectus murrayi and are not upregulated further by desiccation [164]. Similarly only 2 of 6 hsp70 paralogues show higher expression levels in diapausing eggs of the rotifer Brachionus plicatilis than in other metabolically active life stages [165]. sHSP are the major holding chaperones which prevent the irreversible aggregation of unfolding proteins [122, 123]. They have been shown to accumulate in anhydrobiotic encysted larvae of the brine shrimp Artemia franciscana[124] and they are also abundantly expressed in diapausing eggs of B. plicatilis[165]. These expression profiles for representatives of different HSP classes in anhydrobiotic animals from three different phyla suggest that sHSP proteins, in particular, have an important role in maintaining the integrity of the proteome during anhydrobiosis.
Conclusions
We obtained 9,216 ESTs from an unstressed mixed population of the anhydrobiotic nematode P. superbus and we derived 4,009 putative P. superbus unigene sequences from these ESTs. The finding that 51% of these unigenes correspond to novel sequences is consistent with previous metagenomic analyses of nematode EST datasets, and is a reflection of the diversity of nematode gene space. Functional annotation of the P. superbus unigenes has identified 187 constitutively expressed consensus sequences encoding putative stress-related genes that may have a role in anhydrobiosis. Among these were: MAP-kinases; members of the jumonji family of transcription activators; antioxidant enzymes; molecular chaperones; components of the ubiquitin-proteasome system; DNA damage response proteins and late embryogenesis abundant (LEA) proteins. Thirteen P. superbus unigenes encode predicted LEA proteins, all members of Group 3 as is typical of animal LEA proteins. The relative abundance of lea genes in P. superbus as compared to C. elegans, along with their constitutive expression (we detected 34 LEA-encoding ESTs), suggest that LEA proteins are an important component of the anhydrobiotic protection repertoire of P. superbus and that the lea gene family may have undergone lineage-specific expansion in this species. P. superbus appears to utilize a strategy of combined constitutive and inducible gene expression in preparation for entry into anhydrobiosis. Five of the 19 putative P. superbus stress response genes tested were upregulated in response to desiccation. Three of the upregulated genes encoded antioxidant enzymes, an indication of the importance of enzymatic antioxidant defense systems during the induction of anhydrobiosis. One of the upregulated genes encoded a 1-Cys Prx, revealing a parallel between the desiccation tolerance mechanisms of plant seeds and resurrection plants with those of anhydrobiotic nematodes. Of the four P. superbus hsp sequences assayed only one, an shsp sequence, was upregulated in response to desiccation. This is consistent with the expression profiles for representatives of different HSP classes in anhydrobiotic animals from other phyla and suggests that sHSP proteins have an important role in maintaining the integrity of the proteome during the dehydration phases of anhydrobiosis.
A large number of P. superbus unigenes are homologous to human disease genes, particularly those implicated in neurodegenerative diseases. Many neurodegenerative diseases are associated with the dysfunction of the protection systems responsible for repairing or degrading damaged proteins and macromolecules, thus some gene products that have roles in anhydrobiotic protection in P. superbus may have human homologs that are required for neural survival. So, in addition to providing candidate genes for use in anhydrobiotic engineering experiments, knowledge of the molecular mechanisms responsible for anhydrobiotic protection of macromolecules may also provide insights into some of the gene products required for the integrity of neural tissues.
Analysis of the physical properties of the putative peptides encoded by the 2,059 novel P. superbus unigenes reveals that 149 of them are predicted to be 100% intrinsically disordered (IDPs) and that 170 novel sequences meet the criteria used to define 'hydrophilin' molecules which accumulate in response to osmotic stress in prokaryotes and eukaryotes. These IDPs and putative hydrophilins represent a key group of potential stress-related genes. The most highly expressed P. superbus sequence belongs to the nematode specific family of SXP/RAL-2 proteins, which had previously been identified as a class of secreted and surface associated antigens in diverse animal parasitic nematodes. The abundant representation of the SXP/RAL-2 in P. superbus may indicative of a role for this protein in stabilizing the nematodes' integument and slowing the rate of water loss during evaporative desiccation.
Panagrolaimus is an excellent model system for the study of anhydrobiosis and cryobiosis. Panagrolaimid nematodes can be readily cultured in the laboratory and have a short generation time. The anhydrobiotic and cryobiotic species and strains of Panagrolaimus described to date belong to a single clade, which will facilitate comparative transcriptomic analyses of the molecular basis of anhydrobiosis in a single genus. This study is the first investigation of the putative molecular mechanisms involved in anhydrobiosis in Panagrolaimus. Because of its provenance and its anhydrobiotic and cryotolerant phenotypes the genome of P. superbus is currently being sequenced as part of the 959 Nematode Genomes Initative [24]. In addition to providing cDNA clones and sequence data for candidate anhydrobiotic genes, the dataset presented here will provide anchor sequences important for the assembly of the genome and transcriptome of P. superbus from high-throughput sequence data.
Methods
Nematode culture
Panagrolaimus superbus (strain DF5050) was obtained from Prof. Bjorn Sohlenius, Swedish Museum of Natural History, Stockholm. The nematodes were cultured at 20°C in the dark on nematode growth medium (NGM) plates containing a lawn streptomycin resistant E. coli strain HB101 obtained from the Caenorhabditis Genetics Center, University of Minnesota, USA. The NGM was supplemented with streptomycin sulfate (30 μg ml-1).
cDNA library construction and EST generation
Total RNA was extracted from mixed stage unstressed worms using the TRIzol® reagent (Invitrogen, Carlsbad, USA). The cDNA library was prepared using the SMART™ cDNA Library Construction Kit Long-Distance (LD) PCR protocol (Clontech, Mountain View, CA 94043 USA). Fifty ng of total RNA was used for the SMART cDNA synthesis and there were 25 PCR cycles in the LD PCR amplification step. The cDNAs were cloned into the pDNR-Lib vector (Clontech) and transformed into E. coli DH10B cells. A total of 15,360 recombinant E. coli were picked using a Q-Bot™ robot (Genetix, Hampshire BH25 5NN, UK) and transferred to 384 well microtitre plates containing freezing media [166] and chloramphenicol (30 μg ml-1) and the plates were stored at -80°C. The cDNA inserts from individual transformants (n = 9,216) from the cDNA library were sequenced by the Sanger method at the Scottish Crop Research Institute, Dundee (4,224 clones) and at The GenePool, University of Edinburgh (4,992 clones).
Clustering and sequence analysis
The raw EST sequences were processed through the PartiGene pipeline [27], first using trace2dbEST which removes vector-derived sequences, poor quality sequences and ESTs shorter than 150 bp, followed by CLOBB [167], an iterative program which groups the sequences on the basis of BLAST similarity into clusters that are putatively derived from the same gene. Clusters containing more than one sequence were then assembled into consensus sequences using phrap (http://www.phrap.org). The partial transcriptome consists of these consensus sequences, along with those clusters that contain only one sequence (singletons). Potential bacterial contaminant sequences (28 contigs) and nematode rRNA genes (27 contigs) were identified using a BLASTN search of the P. superbus consensus sequences against the GenBank nucleotide database (nt), with an e-value cut-off of 1e-50 to identify significant matches. Genes encoded on the mitochondrial genome were identified by BLASTN using 27 nematode mtDNA genomes from GenBank as queries against the P. superbus consensus sequences with an e-value cut-off of 1e-10.
The consensus sequence for each unigene was translated using prot4EST [46]. Each unigene was then subjected to a BLASTP search (e-value cut-off of 1e-4) against a non-redundant custom database containing sequences from a variety of sources: the GenBank NR database, Wormpep (version 224) (http://www.sanger.ac.uk/Projects/C_elegans/WORMBASE/current/wormpep.shtml) and an extended version of the Nempep4 database (http://www.nematodes.org/nembase4/), which we have named Nempep4+. Nempep4+ has been supplemented with sequences from the following nematodes: Plectus murrayi, Ditylenchus africanus, Aphelenchus avenae, Trihinella spiralis, Wuchereria bancrofti, Loa loa and Pristionchus pacificus. Putative genes were annotated using the annot8r algorithm [56]. This software tool assigns Gene Ontology (GO) terms, Enzyme Commission (EC) numbers [168] and Kyoto Encyclopaedia of Genes and Genomes (KEGG) pathway data [57] to EST sequences based on BLAST searches against annotated subsets of the EMBL UniProt database [169]. All BLAST results were parsed and the corresponding annotations were saved in a relational postgreSQL database (http://www.postgresql.org). A web interface where the annot8r annotations can be subjected to keyword queries and where output clusters can retrieved is available at http://www.nematodes.org/nembase4/species_info.php?species=PSC. Supplemental ortholog assignment and pathway mapping were carried out using the KAAS-KEGG Automatic Annotation Server [170].
To identify putative unigene families among four anhydrobiotic nematodes, EST consensus sequences were kindly provided by various groups: 1,387 Plectus murrayi sequences [102] from Dr. B. Adhikari; 2,596 Ditylenchus africanus sequences [149] from Dr. A. Haegeman and 2,700 A. avenae sequences [31] from Dr. N. Karim. All ESTs were translated into peptide sequences using prot4EST [46] and they were subjected to an all-vs-all BLASTP analysis to identify pairwise similarities. A graph representation of the homologous relationships among the unigenes was constructed, where each node is a unigene and an edge is drawn between any two nodes that have a BLASTP match. Each edge is weighted by -logE where E is the e-value of the alignment between two similar unigenes. E-values of 0 are transformed into 1e-200, i.e. an edge weight of 200. This graph is then used by the MCL algorithm [150] as input, with an inflation parameter of 2.1, to classify the unigenes into putative families.
Translation and primary structure analysis of novel ESTs
Novel ESTs were translated into putative peptide sequences using prot4EST which incorporates the ESTScan2.0 [171] and DECODER [172] programs which use 'de novo' prediction methods for predicting the amino acid sequence of cDNA sequences with putative sequencing errors (particularly insertions and deletions). The glycine content and the Grand Average of Hydropathy (GRAVY) values of these putative peptides were determined using the ProtParam tool available at http://www.expasy.ch/tools/protparam.html[173]. The GRAVY value is calculated as the sum of hydropathy values of all the amino acids, divided by the number of residues in the sequence [111]. Predictions of the extent of intrinsically disordered regions within each putative peptide were determined using the IUPred program (using the long disorder prediction algorithm) [110] kindly provided by Dr. Zsuzsanna Dosztányi.
Real-time relative qPCR analysis of gene expression
A mixed population of nematodes was vacuum filtered onto 25 mm Supor® Membrane Disc Filters at a concentration of 2,000 nematodes per filter. Five replicate filters were prepared for each treatment. The filters were placed in an 8 L glass desiccation chamber over a saturated solution of potassium dichromate (to generate an RH of 98% [174]) for 12 h at 20°C in the dark. Nematodes were then washed off the filters with distilled water and the nematodes from the five filters were pooled together. RNA was extracted using the TRIsure™ (BIO-38033, Bioline) method followed by treatment with DNAse I (Promega, M6101). Control nematodes were placed directly in to TRIsure and flash frozen with liquid N2 without vacuum filtration.
Total RNA (1 μg per reaction) was converted to cDNA using the Roche Transcriptor First Strand cDNA Synthesis Kit (04 379 012 001). One μl of cDNA from the above reaction was used for each real time qPCR reaction. These reactions were carried out on a Roche LightCycler 480 thermocycler using Roche SYBR I Master 1 kit (04 707 516 001). Each qPCR reaction also contained 5 μl SYBR Master Mix, 0.002 pmole of each primer and 2 μl H20. Primers were designed to produce an amplicon of approximately 125 bp for each gene tested. (These primer sequences are presented in Additional file 6). Relative expression data were calculated with the LightCycler 480 Efficiency Method analysis software using the second derivative maximum option. The P. superbus ama-1 and rpl32 genes were used as a reference. Having established that the crossing point data were normally distributed and that the variance of the controls and treatment data were equal, two sample Student's t-tests were carried out to identify statistically significant differences in expression levels between the controls and the experimental treatments.
Availability of supporting data
These P. superbus EST sequences have been deposited in dbEST with the accession numbers GW405912-GW413517. The unigene sequences and annotations along with their constituent ESTs can be downloaded from the NEMBASE4 database [37] at http://www.nematodes.org/nembase4/species_info.php?species=PSC and the unigene annotations can also be subjected to keyword queries at the NEMBASE4 database.
References
Barrett J: Anhydrobiotic nematodes. Agric Zool Rev. 1982, 4: 161-176.
Kranner I, Beckett RP, Wornik S, Zorn M, Pfeifhofer HW: Revival of a resurrection plant correlates with its antioxidant status. Plant J. 2002, 31: 13-24.
Keilin D: The Leeuwenhoek lecture--the problem of anabiosis or latent life--history and current concepts. Proc R Soc Lond B Biol Sci. 1959, 150: 149-191.
Aroian RV, Carta L, Kaloshian I, Sternberg PW: A free-living Panagrolaimus sp. from Armenia can survive in anhydrobiosis for 8.7 years. J Nematol. 1993, 25: 500-502.
de Castro AG, Lapinski J, Tunnacliffe A: Anhydrobiotic engineering. Nat Biotechnol. 2000, 18: 473-473.
Leprince O, Buitink J: Desiccation tolerance: from genomics to the field. Plant Sci. 2010, 179: 554-564.
Clegg JS: Origin of trehalose and its significance during formation of encysted dormant embryos of Artemia salina. Comp Biochem Physiol. 1965, 14: 135-143.
Watanabe M, Kikawada T, Okuda T: Increase of internal ion concentration triggers trehalose synthesis associated with cryptobiosis in larvae of Polypedilum vanderplanki. J Exp Biol. 2003, 206: 2281-2286.
Womersley C: A reevaluation of strategies employed by nematode anhydrobiotes in relation to their natural environment. Vistas on Nematology. Edited by: Veech DA, Dickson DW. 1987, Hyattsville, MD: Society of Nematologists Inc., 165-173.
Oliver MJ, Velten J, Wood AJ: Bryophytes as experimental models for the study of environmental stress tolerance: Tortula ruralis and desiccation-tolerance in mosses. Plant Ecol. 2000, 151: 73-84.
Oliver MJ, Hudgeons J, Dowd SE, Payton PR: A combined subtractive suppression hybridization and expression profiling strategy to identify novel desiccation response transcripts from Tortula ruralis gametophytes. Physiol Plant. 2009, 136: 437-460.
Womersley CZ, Wharton DA, Higa LM: Survival biology. The physiology and Biochemistry of Free-Living and Plant-Parasitic Nematodes. Edited by: Perry RN, Wright DJ. 1998, Wallingford, Oxon., UK: CABI Publishing, 271-302.
Perry RN, Moens M: Survival of parasitic nematodes outside the host. Molecular and Physiolgical Basis of Nematode Survival. Edited by: Perry RN, Wharton DA. 2011, Wallingford, UK: CAB International, 1-27.
Wharton DA, Petrone L, Duncan A, McQuillan AJ: A surface lipid may control the permeability slump associated with entry into anhydrobiosis in the plant parasitic nematode Ditylenchus dipsaci. J Exp Biol. 2008, 211: 2901-2908.
Shannon AJ, Browne JA, Boyd J, Fitzpatrick DA, Burnell AM: The anhydrobiotic potential and molecular phylogenetics of species and strains of Panagrolaimus (Nematoda, Panagrolaimidae). J Exp Biol. 2005, 208: 2433-2445.
Perry RN: Desiccation survival of parasitic nematodes. Parasitology. 1999, 119: S19-S30.
Bert W, Leliaert F, Vierstraete AR, Vanfleteren JR, Borgonie G: Molecular phylogeny of the Tylenchina and evolution of the female gonoduct (Nematoda: Rhabditida). Mol Phylogenet Evol. 2008, 48: 728-744.
Wharton DA, Barclay S: Anhydrobiosis in the free-living Antarctic nematode Panagrolaimus davidi (Nematoda, Rhabditida). Fundam Appl Nematol. 1993, 16: 17-22.
Ricci C, Pagani M: Desiccation of Panagrolaimus rigidus (Nematoda): survival, reproduction and the influence on the internal clock. Hydrobiologia. 1997, 347: 1-13.
Shannon AJ, Tyson T, Dix I, Boyd J, Burnell AM: Systemic RNAi mediated gene silencing in the anhydrobiotic nematode Pangrolaimus superbus. BMC Mol Biol. 2008, 9: 58-
Wharton DA, Ferns DJ: Survival of intracellular freezing by the Antarctic nematode Panagrolaimus davidi. J Exp Biol. 1995, 198: 1381-1387.
Boström S: Descriptions and morphological variability of three populations of Panagrolaimus Fuchs, 1930 (Nematoda, Panagrolaimidae). Nematologica. 1988, 34: 144-155.
Nomination of Surtsey for the UNESCO World Heritage List. Edited by: Baldursson S, Ingadóttir Á. 2007, Reykjavík: Icelandic Institute of Natural History
[http://www.nematodes.org/nematodegenomes/index.php?title=About#tab=Welcome]
Blaxter ML, De Ley P, Garey JR, Liu LX, Scheldeman P, Vierstraete A, Vanfleteren JR, Mackey LY, Dorris M, Frisse LM, et al: A molecular evolutionary framework for the phylum Nematoda. Nature. 1998, 392: 71-75.
Erkut C, Penkov S, Khesbak H, Vorkel D, Verbavatz J-M, Fahmy K, Kurzchalia TV: Trehalose renders the dauer larva of Caenorhabditis elegans resistant to extreme desiccation. Curr Biol. 2011, 21: 1331-1336.
Parkinson J, Anthony A, Wasmuth J, Schmid R, Hedley A, Blaxter M: PartiGene--constructing partial genomes. Bioinformatics. 2004, 20: 1398-1404.
Klasson L, Walker T, Sebaihia M, Sanders MJ, Quail MA, Lord A, Sanders S, Earl J, O'Neill SL, Thomson N, et al: Genome evolution of Wolbachia strain wPip from the Culex pipien s group. Mol Biol Evol. 2008, 25: 1877-1887.
McLeod A, Smart CD, Fry WE: Characterization of 1,3-beta-glucanase and 1,3;1,4-beta-glucanase genes from Phytophthora infestans. Fungal Genet Biol. 2003, 38: 250-263.
Kikuchi T, Shibuya H, Jones JT: Molecular and biochemical characterization of an endo-beta-1,3-glucanase from the pinewood nematode Bursaphelenchus xylophilus acquired by horizontal gene transfer from bacteria. Biochem J. 2005, 389: 117-125.
Karim N, Jones JT, Okada H, Kikuchi T: Analysis of expressed sequence tags and identification of genes encoding cell-wall-degrading enzymes from the fungivorous nematode Aphelenchus avenae. BMC Genomics. 2009, 10: 525-
Kyndt T, Haegeman A, Gheysen G: Evolution of GHF5 endoglucanase gene structure in plant-parasitic nematodes: no evidence for an early domain shuffling event. BMC Evol Biol. 2008, 8: 305-
Jones JT, Furlanetto C, Kikuchi T: Horizontal gene transfer from bacteria and fungi as a driving force in the evolution of plant parasitism in nematodes. Nematology. 2005, 7: 641-646.
Mayer WE, Schuster LN, Bartelmes G, Dieterich C, Sommer RJ: Horizontal gene transfer of microbial cellulases into nematode genomes is associated with functional assimilation and gene turnover. BMC Evol Biol. 2011, 11: 13-
Wasmuth J, Schmid R, Hedley A, Blaxter M: On the extent and origins of genic novelty in the Phylum Nematoda. PLoS Negl Trop Dis. 2008, 2 (7): e258-
Yin Y, Martin J, Abubucker S, Wang ZY, Wyrwicz L, Rychlewski L, McCarter JP, Wilson RK, Mitreva M: Molecular determinants archetypical to the phylum Nematoda. BMC Genomics. 2009, 10: 114-
Elsworth B, Wasmuth J, Blaxter M: NEMBASE4: the nematode transcriptome resource. Int J Parasitol. 2011, 41: 881-894.
Gallin MY, Tan M, Kron MA, Rechnitzer D, Greene BM, Newland HS, White AT, Taylor HR, Unnasch TR: Onchocerca volvulus recombinant antigen--physical characterization and clinical correlates with serum reactivity. J Infect Dis. 1989, 160: 521-529.
Rao KVN, Eswaran M, Ravi V, Gnanasekhar B, Narayanan RB, Kaliraj P, Jayaraman K, Marson A, Raghavan N, Scott AL: The Wuchereria bancrofti orthologue of Brugia malayi SXP1 and the diagnosis of bancroftian filariasis. Mol Biochem Parasitol. 2000, 107: 71-80.
Wang SH, Zheng HJ, Dissanayake S, Cheng WF, Tao ZH, Lin SZ, Piessens WF: Evaluation of recombinant chitinase and SXP1 antigens as antimicrofilarial vaccines. Am J Trop Med Hyg. 1997, 56: 474-481.
Tsuji N, Suzuki K, Kasuga-Aoki H, Isobe T, Arakawa T, Matsumoto Y: Mice intranasally immunized with a recombinant 16-kilodalton antigen from roundworm Ascaris parasites are protected against larval migration of Ascaris suum. Infect Immun. 2003, 71: 5314-5323.
Fujiwara RT, Zhan B, Mendez S, Loukas A, Bueno LL, Wang Y, Plieskatt J, Oksov Y, Lustigman S, Bottazzi ME, et al: Reduction of worm fecundity and canine host blood loss mediates protection against hookworm infection elicited by vaccination with recombinant Ac-16. Clin Vaccine Immunol. 2007, 14: 281-287.
Jones JT, Smant G, Blok VC: SXP/RAL-2 proteins of the potato cyst nematode Globodera rostochiensis: secreted proteins of the hypodermis and amphids. Nematology. 2000, 2: 887-893.
Tytgat T, Vercauteren I, Vanholme B, De Meutter J, Vanhoutte I, Gheysen G, Borgonie G, Coomans A, Gheysen G: An SXP/RAL-2 protein produced by the subventral pharyngeal glands in the plant parasitic root-knot nematode Meloidogyne incognita. Parasitol Res. 2005, 95: 50-54.
Pujol N, Zugasti O, Wong D, Couillault C, Kurz CL, Schulenburg H, Ewbank JJ: Anti-fungal innate immunity in C. elegans is enhanced by evolutionary diversification of antimicrobial peptides. PLoS Pathog. 2008, 4 (7): e1000105-
Wasmuth JD, Blaxter ML: Prot4EST: translating expressed sequence tags from neglected genomes. BMC Bioinformatics. 2004, 5: 187-
Ward S, Burke DJ, Sulston JE, Coulson AR, Albertson DG, Ammons D, Klass M, Hogan E: Genomic organization of major sperm protein genes and pseudogenes in the nematode Caenorhabditis elegans. J Mol Biol. 1988, 199: 1-13.
Tarr DEK, Scott AL: MSP domain proteins. Trends Parasitol. 2005, 21: 224-231.
Blumenthal T, Squire M, Kirtland S, Cane J, Donegan M, Spieth J, Sharrock W: Cloning of a yolk protein gene family from Caenorhabditis elegans. J Mol Biol. 1984, 174: 1-18.
Spieth J, Blumenthal T: The Caenorhabditis elegans vitellogenin gene family includes a gene encoding a distantly related protein. Mol Cell Biol. 1985, 5: 2495-2501.
Kimble J, Sharrock WJ: Tissue-specific synthesis of yolk proteins in Caenorhabditis elegans. Dev Biol. 1983, 96: 189-196.
Jorcano JL, Ruizcarrillo A: H3.H4 tetramer directs DNA and core histone octamer assembly in the nucleosome core particle. Biochemistry. 1979, 18: 768-774.
Storch J, Xu Z: Niemann-Pick C2 (NPC2) and intracellular cholesterol trafficking. Biochim Biophys Acta Mol Cell Biol Lipids. 2009, 1791: 671-678.
Vanier MT, Millat G: Structure and function of the NPC2 protein. Biochim Biophys Acta Mol Cell Biol Lipids. 2004, 1685: 14-21.
Harcus YM, Parkinson J, Fernandez C, Daub J, Selkirk ME, Blaxter ML, Maizels RM: Signal sequence analysis of expressed sequence tags from the nematode Nippostrongylus brasiliensis and the evolution of secreted proteins in parasites. Genome Biol. 2004, 5: R39-
Schmid R, Blaxter ML: annot8r: GO, EC and KEGG annotation of EST datasets. BMC Bioinforma. 2008, 9: 180-
Kanehisa M, Araki M, Goto S, Hattori M, Hirakawa M, Itoh M, Katayama T, Kawashima S, Okuda S, Tokimatsu T, Yamanishi Y: KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008, 36: D480-D484.
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al: Gene ontology: tool for the unification of biology. Nat Genet. 2000, 25: 25-29.
Martinez A, Portero-Otin M, Pamplona R, Ferrer I: Protein targets of oxidative damage in human neurodegenerative diseases with abnormal protein aggregates. Brain Pathol. 2010, 20: 281-297.
Irvine GB, El-Agnaf OM, Shankar GM, Walsh DM: Protein aggregation in the brain: the molecular basis for Alzheimer's and Parkinson's diseases. Mol Med. 2008, 14: 451-464.
Hegde AN, Upadhya SC: The ubiquitin-proteasome pathway in health and disease of the nervous system. Trends Neurosci. 2007, 30: 587-595.
Bonifati V, Rizzu P, Squitieri F, Krieger E, Vanacore N, van Swieten JC, Brice A, van Duijn CM, Oostra B, Meco G, Heutink P: DJ-1 (PARK7), a novel gene for autosomal recessive, early onset parkinsonism. Neurol Sci. 2003, 24: 159-160.
Shendelman S, Jonason A, Martinat C, Leete T, Abeliovich A: DJ-1 is a redox-dependent molecular chaperone that inhibits alpha-synuclein aggregate formation. PLoS Biol. 2004, 2: 1764-1773.
Reardon W, Chakrabortee S, Pereira TC, Tyson T, Banton MC, Dolan KM, Culleton BA, Wise MJ, Burnell AM, Tunnacliffe A: Expression profiling and cross-species RNA interference (RNAi) of desiccation-induced transcripts in the anhydrobiotic nematode Aphelenchus avenae. BMC Mol Biol. 2010, 11: 6-
Browne J, Tunnacliffe A, Burnell A: Anhydrobiosis--plant desiccation gene found in a nematode. Nature. 2002, 416: 38-38.
Chakrabortee S, Boschetti C, Walton LJ, Sarkar S, Rubinsztein DC, Tunnacliffe A: Hydrophilic protein associated with desiccation tolerance exhibits broad protein stabilization function. Proc Natl Acad Sci USA. 2007, 104: 18073-18078.
de Nadal E, Alepuz PM, Posas F: Dealing with osmostress through MAP kinase activation. EMBO Rep. 2002, 3: 735-740.
Cuenda A, Rousseau S: p38 MAP-Kinases pathway regulation, function and role in human diseases. Biochim Biophys Acta, Mol Cell Res. 2007, 1773: 1358-1375.
Jonak C, Kiegerl S, Ligterink W, Barker PJ, Huskisson NS, Hirt H: Stress signaling in plants: a mitogen-activated protein kinase pathway is activated by cold and drought. Proc Natl Acad Sci USA. 1996, 93: 11274-11279.
Whitmarsh AJ: A central role for p38 MAPK in the early transcriptional response to stress. BMC Biol. 2010, 8: 47-
Brewster JL, Devaloir T, Dwyer ND, Winter E, Gustin MC: An osmosensing signal transduction pathway in yeast. Science. 1993, 259: 1760-1763.
Ferrigno P, Posas F, Koepp D, Saito H, Silver PA: Regulated nucleo/cytoplasmic exchange of HOG1 MAPK requires the importin beta homologs NMD5 and XPO1. EMBO J. 1998, 17: 5606-5614.
de Nadal E, Posas F: Multilayered control of gene expression by stress-activated protein kinases. EMBO J. 2010, 29: 4-13.
Stokoe D, Engel K, Campbell DG, Cohen P, Gaestel M: Identification of MAPKAP kinase-2 as a major enzyme responsible for the phosphorylation of the small mammalian heat-shock proteins. FEBS Lett. 1992, 313: 307-313.
Ito H, Okamoto K, Nakayama H, Isobe T, Kato K: Phosphorylation of alpha B-crystallin in response to various types of stress. J Biol Chem. 1997, 272: 29934-29941.
Dalle-Donne I, Rossi R, Milzani A, Di Simplicio P, Colombo R: The actin cytoskeleton response to oxidants: From small heat shock protein phosphorylation to changes in the redox state of actin itself. Free Radic Biol Med. 2001, 31: 1624-1632.
Strange K, Denton J, Nehrke K: Ste20-Type kinases: evolutionarily conserved regulators of ion transport and cell volume. Physiology. 2006, 21: 61-68.
Choe KP, Strange K: Evolutionarily conserved WNK and Ste20 kinases are essential for acute volume recovery and survival after hypertonic shrinkage in Caenorhabditis elegans. Am J Physiol Cell Physiol. 2007, 293: C915-C927.
Cheung WL, Turner FB, Krishnamoorthy T, Wolner B, Ahn SH, Foley M, Dorsey JA, Peterson CL, Berger SL, Allis CD: Phosphorylation of histone H4 serine 1 during DNA damage requires casein kinase II in S. cerevisiae. Curr Biol. 2005, 15: 656-660.
Knippschild U, Gocht A, Wolff S, Huber N, Lohler J, Stoter M: The casein kinase I family: participation in multiple cellular processes in eukaryotes. Cell Signal. 2005, 17: 675-689.
Gal TZ, Glazer I, Koltai H: Differential gene expression during desiccation stress in the insect-killing nematode Steinernema feltiae IS-6. J Parasitol. 2003, 89: 761-766.
Paradis S, Ruvkun G: Caenorhabditis elegans Akt/PKB transduces insulin receptor-like signals from AGE-1 PI3 kinase to the DAF-16 transcription factor. Genes Dev. 1998, 12: 2488-2498.
Padmanabhan S, Mukhopadhyay A, Narasimhan SD, Tesz G, Czech MP, Tissenbaum HA: A PP2A regulatory subunit regulates C. elegans Insulin/IGF-1 signaling by modulating AKT-1 phosphorylation. Cell. 2009, 136: 939-951.
Merida I, Avila-Flores A, Merino E: Diacylglycerol kinases: at the hub of cell signalling. Biochem J. 2008, 409: 1-18.
Arisz SA, Testerink C, Munnik T: Plant PA signaling via diacylglycerol kinase. Biochim Biophys Acta Mol Cell Biol Lipids. 2009, 1791: 869-875.
Sedding DG: FoxO transcription factors in oxidative stress response and ageing--a new fork on the way to longevity?. Biol Chem. 2008, 389: 279-283.
Smith GR, Shanley DP: Modelling the response of FOXO transcription factors to multiple post-translational modifications made by ageing-related signalling pathways. PLoS One. 2010, 5 (6): e11092-
Kirienko NV, Fay DS: SLR-2 and JMJC-1 regulate an evolutionarily conserved stress-response network. EMBO J. 2010, 29: 727-739.
Lildballe DL, Pedersen DS, Kalamajka R, Emmersen J, Houben A, Grasser KD: The expression level of the chromatin-associated HMGB1 protein influences growth, stress tolerance, and transcriptome in Arabidopsis. J Mol Biol. 2008, 384: 9-21.
Kranner I, Birtic S: A modulating role for antioxidants in desiccation tolerance. Integr Comp Biol. 2005, 45: 734-740.
Franca MB, Panek AD, Eleutherio ECA: Oxidative stress and its effects during dehydration. Comp Biochem Physiol A Mol Integr Physiol. 2007, 146: 621-631.
Wood ZA, Schroder E, Harris JR, Poole LB: Structure, mechanism and regulation of peroxiredoxins. Trends Biochem Sci. 2003, 28: 32-40.
Hayes JD, Flanagan JU, Jowsey IR: Glutathione transferases. Annu Rev Pharmacol Toxicol. 2005, 45: 51-88.
Corona M, Robinson GE: Genes of the antioxidant system of the honey bee: annotation and phylogeny. Insect Mol Biol. 2006, 15: 687-701.
Gallogly MM, Mieyal JJ: Mechanisms of reversible protein glutathionylation in redox signaling and oxidative stress. Curr Opin Pharmacol. 2007, 7: 381-391.
Meyer Y, Buchanan BB, Vignols F, Reichheld JP: Thioredoxins and gutaredoxins: unifying elements in redox biology. Annu Rev Genet. 2009, 43: 335-367.
Holmgren A: Thioredoxin and glutaredoxin systems. J Biol Chem. 1989, 264: 13963-13966.
Chang Q, Petrash JM: Disruption of aldo-keto reductase genes leads to elevated markers of oxidative stress and inositol auxotrophy in Saccharomyces cerevisiae. Biochim Biophys Acta Mol Cell Res. 2008, 1783: 237-245.
Malik AI, Storey KB: Activation of antioxidant defense during dehydration stress in the African clawed frog. Gene. 2009, 442: 99-107.
Lee SM, Koh HJ, Park DC, Song BJ, Huh TL, Park JW: Cytosolic NADP(+)-dependent isocitrate dehydrogenase status modulates oxidative damage to cells. Free Radic Biol Med. 2002, 32: 1185-1196.
Leterrier M, Del Rio LA, Corpas FJ: Cytosolic NADP-isocitrate dehydrogenase of pea plants: genomic clone characterization and functional analysis under abiotic stress conditions. Free Radic Res. 2007, 41: 191-199.
Adhikari BN, Wall DH, Adams BJ: Desiccation survival in an Antarctic nematode: molecular analysis using expressed sequenced tags. BMC Genomics. 2009, 10: 69-
Hundertmark M, Hincha DK: LEA (Late Embryogenesis Abundant) proteins and their encoding genes in Arabidopsis thaliana. BMC Genomics. 2008, 9: 22-
Bies-Ethève N, Gaubier-Comella P, Debures A, Lasserre E, Jobet E, Raynal M, Cooke R, Delseny M: Inventory, evolution and expression profiling diversity of the LEA (late embryogenesis abundant) protein gene family in Arabidopsis thaliana. Plant Mol Biol. 2008, 67: 107-124.
Tunnacliffe A, Wise MJ: The continuing conundrum of the LEA proteins. Naturwissenschaften. 2007, 94: 791-812.
Sharon MA, Kozarova A, Clegg JS, Vacratsis PO, Warner AH: Characterization of a group 1 late embryogenesis abundant protein in encysted embryos of the brine shrimp Artemia franciscana. Biochem Cell Biol. 2009, 87: 415-430.
Forster F, Liang CG, Shkumatov A, Beisser D, Engelmann JC, Schnolzer M, Frohme M, Muller T, Schill RO, Dandekar T: Tardigrade workbench: comparing stress-related proteins, sequence-similar and functional protein clusters as well as RNA elements in tardigrades. BMC Genomics. 2009, 10: 469-
Dure L: A repeating 11-mer amino-acid motif and plant desiccation. Plant J. 1993, 3: 363-369.
Browne JA, Dolan KM, Tyson T, Goyal K, Tunnacliffe A, Burnell AM: Dehydration-specific induction of hydrophilic protein genes in the anhydrobiotic nematode Aphelenchus avenae. Eukaryot Cell. 2004, 3: 966-975.
Dosztanyi Z, Csizmok V, Tompa P, Simon I: IUPred: web server for the prediction of intrinsically unstructured regions of proteins based on estimated energy content. Bioinformatics. 2005, 21: 3433-3434.
Kyte J, Doolittle RF: A simple method for displaying the hydropathic character of a protein. J Mol Biol. 1982, 157: 105-132.
Hunault G, Jaspard E: LEAPdb: a database for the late embryogenesis abundant proteins. BMC Genomics. 2010, 11: 9-
Hogg JS, Hu FZ, Janto B, Boissy R, Hayes J, Keefe R, Post JC, Ehrlich GD: Characterization and modeling of the Haemophilus influenzae core and supragenomes based on the complete genomic sequences of Rd and 12 clinical nontypeable strains. Genome Biol. 2007, 8: R103-
Morimoto RI: Proteotoxic stress and inducible chaperone networks in neurodegenerative disease and aging. Genes Dev. 2008, 22: 1427-1438.
Richter K, Haslbeck M, Buchner J: The heat shock response: life on the verge of death. Mol Cell. 2010, 40: 253-266.
Kabani M, Martineau CN: Multiple Hsp70 isoforms in the eukaryotic cytosol: Mere redundancy or functional specificity?. Curr Genomics. 2008, 9: 338-348.
Vos MJ, Hageman J, Carra S, Kampinga HH: Structural and functional diversities between members of the human HSPB, HSPH, HSPA, and DNAJ chaperone families. Biochemistry. 2008, 47: 7001-7011.
Sales K, Brandt W, Rumbak E, Lindsey G: The LEA-like protein HSP 12 in Saccharomyces cerevisiae has a plasma membrane location and protects membranes against desiccation and ethanol-induced stress. Biochim Biophys Acta Biomembr. 2000, 1463: 267-278.
Jonsson KI, Schill RO: Induction of Hsp70 by desiccation, ionising radiation and heat-shock in the eutardigrade Richtersius coronifer. Comp Biochem Physiol B Biochem Mol Biol. 2007, 146: 456-460.
Cho EK, Choi YJ: A nuclear-localized HSP70 confers thermoprotective activity and drought-stress tolerance on plants. Biotechnol Lett. 2009, 31: 597-606.
Hu XL, Liu RX, Li YH, Wang W, Tai FJ, Xue RL, Li CH: Heat shock protein 70 regulates the abscisic acid-induced antioxidant response of maize to combined drought and heat stress. Plant Growth Regulation. 2010, 60: 225-235.
Stengel F, Baldwin AJ, Painter AJ, Jaya N, Basha E, Kay LE, Vierling E, Robinson CV, Benesch JLP: Quaternary dynamics and plasticity underlie small heat shock protein chaperone function. Proc Natl Acad Sci USA. 2010, 107: 2007-2012.
Eyles SJ, Gierasch LM: Nature's molecular sponges: Small heat shock proteins grow into their chaperone roles. Proc Natl Acad Sci USA. 2010, 107: 2727-2728.
Liang P, Amons R, MacRae TH, Clegg JS: Purification, structure and in vitro molecular-chaperone activity of Artemia p26, a small heat-shock/alpha-crystallin protein. Eur J Biochem. 1997, 243: 225-232.
Sun Y, Bojikova-Fournier S, MacRae TH: Structural and functional roles for beta-strand 7 in the alpha-crystallin domain of p26, a polydisperse small heat shock protein from Artemia franciscana. FEBS J. 2006, 273: 1020-1034.
Schroder M: Endoplasmic reticulum stress responses. Cell Mol Life Sci. 2008, 65: 862-894.
Winter AD, McCormack G, Page AP: Protein disulfide isomerase activity is essential for viability and extracellular matrix formation in the nematode Caenorhabditis elegans. Dev Biol. 2007, 308: 449-461.
Karala AR, Psarrakos P, Ruddock LW, Klappa P: Protein disulfide isomerases from C. elegans are equally efficient at thiol-disulfide exchange in simple peptide-based systems but show differences in reactivity towards protein substrates. Antioxid Redox Signal. 2007, 9: 1815-1823.
Oda Y, Okada T, Yoshida H, Kaufman RJ, Nagata K, Mori K: Derlin-2 and Derlin-3 are regulated by the mammalian unfolded protein response and are required for ER-associated degradation. J Cell Biol. 2006, 172: 383-393.
Lu B, Gong ZH, Wang J, Zhang JH, Liang JS: Microtubule dynamics in relation to osmotic stress-induced ABA accumulation in Zea mays roots. J Exp Bot. 2007, 58: 2565-2572.
Bagniewska-Zadworna A: The root microtubule cytoskeleton and cell cycle analysis through desiccation of Brassica napus seedlings. Protoplasma. 2008, 233: 177-185.
Kubota H: Quality control against misfolded proteins in the cytosol: a network for cell survival. J Biochem. 2009, 146: 609-616.
Salomons FA, Menendez-Benito V, Bottcher C, McCray BA, Taylor JP, Dantuma NP: Selective accumulation of aggregation-prone proteasome substrates in response to proteotoxic stress. Mol Cell Biol. 2009, 29: 1774-1785.
Lamark T, Johansen T: Autophagy: links with the proteasome. Curr Opin Cell Biol. 2010, 22: 192-198.
Choe KP, Strange K: Genome-wide RNAi screen and in viv protein aggregation reporters identify degradation of damaged proteins as an essential hypertonic stress response. Am J Physiol Cell Physiol. 2008, 295: C1488-C1498.
Kovacs AL, Zhang H: Role of autophagy in Caenorhabditis elegans. FEBS Lett. 2010, 584: 1335-1341.
Barrett J, Butterworth PE: DNA stability in the anabiotic fourth-stage juveniles of Ditylenchus dipsaci (Nematoda). Ann Appl Biol. 1985, 106: 121-124.
Gusev O, Nakahara Y, Vanyagina V, Malutina L, Cornette R, Sakashita T, Hamada N, Kikawada T, Kobayashi Y, Okuda T: Anhydrobiosis-associated nuclear DNA damage and repair in the sleeping chironomid: linkage with radioresistance. PLoS One. 2010, 5 (11): e14008-
Neumann S, Reuner A, Brummer F, Schill RO: DNA damage in storage cells of anhydrobiotic tardigrades. Comp Biochem Physiol A Mol Integr Physiol. 2009, 153: 425-429.
Rebecchi L, Cesari M, Altiero T, Frigieri A, Guidetti R: Survival and DNA degradation in anhydrobiotic tardigrades. J Exp Biol. 2009, 212: 4033-4039.
Mattimore V, Battista JR: Radioresistance of Deinococcus radiodurans: functions necessary to survive ionizing radiation are also necessary to survive prolonged desiccation. J Bacteriol. 1996, 178: 633-637.
Ngo JK, Davies KJA: Mitochondrial Lon protease is a human stress protein. Free Radic Biol Med. 2009, 46: 1042-1048.
van West P, Kamoun S, van't Klooster JM, Govers F: Ric1, a Phytophthora infestans gene with homology to stress-induced genes. Curr Genet. 1999, 36: 310-315.
Navarre C, Goffeau A: Membrane hyperpolarization and salt sensitivity induced by deletion of PMP3, a highly conserved small protein of yeast plasma membrane. EMBO J. 2000, 19: 2515-2524.
Rasmussen M, Alexander RT, Darborg BV, Mobjerg N, Hoffmann EK, Kapus A, Pedersen SF: Osmotic cell shrinkage activates ezrin/radixin/moesin (ERM) proteins: activation mechanisms and physiological implications. Am J Physiol Cell Physiol. 2008, 294: C197-C212.
Sato N, Funayama N, Nagafuchi A, Yonemura S, Tsukita S: A gene family consisting of ezrin, radixin and moesin--its specific localization at actin filament plasma-membrane association sites. J Cell Sci. 1992, 103: 131-143.
Liu JJ, Sturrock R, Ekramoddoullah AKM: The superfamily of thaumatin-like proteins: its origin, evolution, and expression towards biological function. Plant Cell Rep. 2010, 29: 419-436.
Jin Y, Wang M, Fu JJ, Xuan N, Zhu Y, Lian Y, Jia ZW, Zheng J, Wang GY: Phylogenetic and expression analysis of ZnF-AN1 genes in plants. Genomics. 2007, 90: 265-275.
Haegeman A, Jacob J, Vanholme B, Kyndt T, Mitreva M, Gheysen G: Expressed sequence tags of the peanut pod nematode Ditylenchus africanus: The first transcriptome analysis of an Anguinid nematode. Mol Biochem Parasitol. 2009, 167: 32-40.
Enright AJ, Van Dongen S, Ouzounis CA: An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 2002, 30: 1575-1584.
Jacob J, Vanholme B, Haegeman A, Gheysen G: Four transthyretin-like genes of the migratory plant-parasitic nematode Radopholus similis: members of an extensive nematode-specific family. Gene. 2007, 402: 9-19.
Wang XC, Li WD, Zhao DF, Liu B, Shi Y, Chen BH, Yang HW, Guo PF, Geng X, Shang ZH, et al: Caenorhabditis elegans transthyretin-like protein TTR-52 mediates recognition of apoptotic cells by the CED-1 phagocyte receptor. Nat Cell Biol. 2010, 12: 655-669.
Dyson HJ, Wright PE: Intrinsically unstructured proteins and their functions. Nat Rev Mol Cell Biol. 2005, 6: 197-208.
Garay-Arroyo A, Colmenero-Flores JM, Garciarrubio A, Covarrubias AA: Highly hydrophilic proteins in prokaryotes and eukaryotes are common during conditions of water deficit. J Biol Chem. 2000, 275: 5668-5674.
Tompa P: The interplay between structure and function in intrinsically unstructured proteins. FEBS Lett. 2005, 579: 3346-3354.
Dunker AK, Brown CJ, Lawson JD, Iakoucheva LM, Obradovic Z: Intrinsic disorder and protein function. Biochemistry. 2002, 41: 6573-6582.
Goyal K, Tisi L, Basran A, Browne J, Burnell A, Zurdo J, Tunnacliffe A: Transition from natively unfolded to folded state induced by desiccation in an anhydrobiotic nematode protein. J Biol Chem. 2003, 278: 12977-12984.
Hand SC, Menze MA, Toner M, Boswell L, Moore D: LEA proteins during water stress: not just for plants anymore. Annu Rev Physiol. 2011, 73: 115-134.
Andres-Mateos E, Perier C, Zhang L, Blanchard-Fillion B, Greco TM, Thomas B, Ko HS, Sasaki M, Ischiropoulos H, Przedborski S, et al: DJ-1 gene deletion reveals that DJ-1 is an atypical peroxiredoxin-like peroxidase. Proc Natl Acad Sci USA. 2007, 104: 14807-14812.
Dubreuil G, Deleury E, Magliano M, Jaouannet M, Abad P, Rosso MN: Peroxiredoxins from the plant parasitic root-knot nematode, Meloidogyne incognita, are required for successful development within the host. Int J Parasitol. 2011, 41: 385-396.
Aalen RB: Peroxiredoxin antioxidants in seed physiology. Seed Sci Res. 1999, 9: 285-295.
Mowla SB, Thomson JA, Farrant JM, Mundree SG: A novel stress-inducible antioxidant enzyme identified from the resurrection plant Xerophyta viscosa Baker. Planta. 2002, 215: 716-726.
Oliver MJ: Desiccation tolerance in vegetative plant cells. Physiol Plant. 1996, 97: 779-787.
Adhikari BN, Wall DH, Adams BJ: Effect of slow desiccation and freezing on gene transcription and stress survival of an Antarctic nematode. J Exp Biol. 2010, 213: 1803-1812.
Denekamp NY, Reinhardt R, Albrecht MW, Drungowski M, Kube M, Lubzens E: The expression pattern of dormancy-associated genes in multiple life-history stages in the rotifer Brachionus plicatilis. Hydrobiologia. 2011, 662: 51-63.
Sambrook J, Russell DW: Molecular Cloning: A Laboratory Manual. 2001, New York: Cold Spring Harbor Laboratory, 3
Parkinson J, Guiliano DB, Blaxter M: Making sense of EST sequences by CLOBBing them. BMC Bioinforma. 2002, 3: 31-
Bairoch A: The ENZYME database in 2000. Nucleic Acids Res. 2000, 28: 304-305.
Jain E, Bairoch A, Duvaud S, Phan I, Redaschi N, Suzek BE, Martin MJ, McGarvey P, Gasteiger E: Infrastructure for the life sciences: design and implementation of the UniProt website. BMC Bioinforma. 2009, 10: 136-
Moriya Y, Itoh M, Okuda S, Yoshizawa AC, Kanehisa M: KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007, 35: W182-W185.
Lottaz C, Iseli C, Jongeneel CV, Bucher P: Modeling sequencing errors by combining Hidden Markov models. Bioinformatics. 2003, 19 (Suppl. 2): ii103-ii112.
Fukunishi Y, Hayashizaki Y: Amino acid translation program for full-length cDNA sequences with frameshift errors. Physiol Genomics. 2001, 5: 81-87.
Gasteiger E, Gattiker A, Hoogland C, Ivanyi I, Appel RD, Bairoch A: ExPASy: the proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 2003, 31: 3784-3788.
Winston PW, Bates DH: Saturated solutions for the control of humidity in biological research. Ecology. 1960, 41: 232-237.
Acknowledgements
This project was funded by Science Foundation Ireland (Projects 08/RFP/EOB166 and 09/RFP/EOB2506). The Irish Centre for High-End Computing (ICHEC) is supported by Science Foundation Ireland (08/HEC/I1450) and by the Higher Education Authority PRTLI4 programme (e-INIS project). The James Hutton Institute receives funding from the Scottish Government. The GenePool Genomics Facility is funded by the Scottish Universities' Life Science Alliance, The Natural Environment Research Council, The Darwin Trust of Edinburgh and the School of Biological Sciences, University of Edinburgh. BE is supported by a BBSRC PhD studentship. Computational analyses made use of the Edinburgh Compute and Data Facility (ECDF). This work benefited from interactions funded via COST 872.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
MS and JTJ prepared the P. superbus cDNA library and sequenced 4,224 cDNA clones. MB co-ordinated the sequencing of 4,992 cDNA clones. MB and TT processed the EST sequences through Partigene. SW, TT, GOM, AB and MP carried out BLAST analyses and sequence annotations. BE did the prot4EST translations and prepared the dataset for inclusion in NEMBASE4. MBD and TT did the ProtParam and IUPred analysis for the novel sequences. GOM carried out the KEGG analyses. SW did the TRIBE MCL analysis. SW, GOM and AB compared the P. superbus dataset to that of other anhydrobiotic nematodes. TT and AB analysed the LEA sequences. EDM carried out the qPCR experiments. TT and AB drafted the ms. MB and JTJ provided critical input into the first draft of the ms. AB co-ordinated the project. All authors read and approved the final manuscript.
Electronic supplementary material
13104_2011_1385_MOESM1_ESM.XLSX
Additional file 1:Most abundantly represented novel transcripts in a dataset of 7,606 ESTs prepared from a mixed stage unstressed culture of the anhydrobiotic nematode Panagrolaimus superbus. (XLSX 60 KB)
13104_2011_1385_MOESM2_ESM.DOC
Additional file 2:The representation of Gene Ontology (GO) terms recovered in BLAST searches of the P. superbus unigenes against the GO database. (DOC 406 KB)
13104_2011_1385_MOESM3_ESM.XLSX
Additional file 3:Putative anhydrobiotic and stress response genes constitutively expressed by an unstressed mixed stage population of Panagrolaimus superbus. Aa = Aphelenchus avenae; Da = Ditylenchus africanus; Pm = Plectus murrayi. (XLSX 54 KB)
13104_2011_1385_MOESM4_ESM.DOC
Additional file 4:An alignment of the Panagrolaimus superbus LEA sequence encoded by PSC00061 with a Caenorabditis briggsae LEA protein (Accession Number CAP25449). This C. briggsae sequence was most similar to PSC00061 in a BLASTx search of the NCBI nr database (Table 5). Putative 11-mer repeats are indicated in colour. (DOC 36 KB)
13104_2011_1385_MOESM5_ESM.XLSX
Additional file 5:The identities of the 67 TRIBE MCL unigene families which contain transcripts from each the anhydrobiotic nematodes Panagrolaimus superbus, Aphelenchus avenae, Ditylenchus africanus and Plectus murrayi, along with a list of the component P. superbus contigs contained in each family. (XLSX 49 KB)
13104_2011_1385_MOESM6_ESM.DOCX
Additional file 6:The primer sequences used for real time qPCR analysis of gene expression in Panagrolaimus superbus in response to desiccation stress. (DOCX 121 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Tyson, T., O'Mahony Zamora, G., Wong, S. et al. A molecular analysis of desiccation tolerance mechanisms in the anhydrobiotic nematode Panagrolaimus superbus using expressed sequenced tags. BMC Res Notes 5, 68 (2012). https://doi.org/10.1186/1756-0500-5-68
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1756-0500-5-68