
Abstract
Background
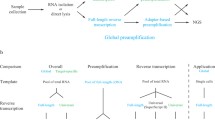
We assessed NanoString's nCounter™ Analysis System for its ability to quantify gene expression of forty-eight genes in a single reaction with 100 ng of total RNA or an equivalent amount of tissue lysate. In the nCounter™ System, multiplexed gene expression target levels are directly detected, without enzymatic reactions, via two sequence-specific probes. The individual mRNA is captured with one mRNA target sequence-specific capture probe that is used in a post-hybridization affinity purification procedure. The second mRNA target specific-sequence and fluorescent-labeled colored coded probe is then used in the detection with the 3-component complex separated on a surface via an applied electric field followed by imaging. We evaluated reproducibility, accuracy, concordance with quantitative RT-PCR, linearity, dynamic range, and the ability of the system to assay different inputs (matched samples of total RNA from Flash Frozen (FF) and Formalin Fixed Paraffin Embedded Tissues (FFPET), and crude tissue lysates (CTL)).
Findings
The nCounter™ Analysis System provided data equivalent to that produced by Taqman®-based assays for genes expressed within the ranges of the calibration curves (above ~0.5 mRNA copies per human cell based on an assumption of 10 pg of total RNA per cell). System response was linear over more than two orders of magnitude with typical CVs of ~6% for concentrations above 1 fM (105 molecules per mL). Profiling the industry-standard MAQC data set yielded correlation coefficients of >0.83 for intensity values and >0.99 for measured ratios. Ninety percent of nCounter™ ratio measurements were within 1.27–1.33 fold changes of the Taqman® data (0.34–0.41 in log2 scale) for FF total RNA samples.
Conclusion
The nCounter™ Analysis System generated robust data for multi-gene expression signatures across three different sample preparation conditions.
Similar content being viewed by others


Background
Analyses of gene expression from microarrays can be used to define a specific set of sequences (signatures) relevant to a particular biological phenomenon or response [1]. These signatures can comprise tens to hundreds of genes, a range that falls between the optimal economic and logistic space for two widely-used tools for measuring gene expression, RT-PCR and microarrays. A solution for follow-up would provide cost-effective, multiplexed measurements of gene expression for tens to hundreds of genes while producing data equivalent to that generated by microarrays and RT-PCR. This solution should also be able to analyze input materials of clinical relevance (e.g., total RNA from formalin-fixed, paraffin embedded tissues (FFPET) and crude tissue lysates (CTL)).
As part of an internally funded investigation, Merck scientists tested the ability of the nCounter™ Analysis System [2] to meet this need via an experimental design (see Table 1) that featured both synthetic spike-in and "natural" total RNA for a set of 48 probes. Microarray Quality Consortium (MAQC) [3] total RNAs, total RNA from matched flash frozen (FF) and formalin fixed, paraffin embedded tissues, and CTL prepared from the same FF tissue were used as source materials for the assay. The work was contracted to Nanostring and the data analyzed at Merck.
Methods
List of transcripts for the nCounter™ assay
The gene list for nCounter™ probes consisted of 14 human genes differentially regulated in our xenograft system, 25 human genes differentially regulated among the MAQC samples, and 9 sequences corresponding to synthetic transcripts used historically at Rosetta Inpharmatics as spike-ins for quality control (http://www.Agilent.com, Two-Color Microarray Spike-In Kit, part # 5188–5279, [4], Table 2). In addition, the nCounter™ Analysis System routinely includes spike-ins (cocktail #3 or #4,)[2]. These nCounter™ assay spike-in controls can be used for calibration and quality control purposes. For this work, the nCounter™ spike-in controls spanned 2.5 logs in concentration.
Samples
Samples (see Table 1) for the assay came from two sources: 1) EBC-1 lung cancer cell line xenograft tissues treated with vehicle or varying amounts of a compound; and 2) MAQC samples obtained from Ambion (Human Brain) and Stratagene (Universal Human Reference). Proportional mixes (25%:75% and 75%:25% UHR to Brain) were created. Crude tissue lysates (CTLs) were made by homogenizing 50–100 mg of FF xenograft tissues in 1 mL of Qiagen buffer RLT http://www.Qiagen.com and snap freezing a 100 μL aliquot (1/10th of total volume). Total RNA was purified from the remaining lysate using a Trizol-based protocol http://www.invitrogen.com. We isolated RNA from FFPE EBC1 xenografts using the Ambion RecoverAll protocol http://www.ambion.com. In all samples, one or the other set of Rosetta spike-ins were added to provide a measure of ratio accuracy. Samples were blinded before assaying at Nanostring Technologies, where the samples were processed to generate raw data (i.e. counts/gene). nCounter™ assay spike-ins control mixes #3 or #4 were added at random to each of the blinded RNA samples on the day the assay was performed.
The nCounter™ System assay
We performed the nCounter™ assay using 100 ng of total RNA or 2 μL of tissue lysate per replicate. Each assay was performed in triplicate to improve precision of the measurements. Details can be found in [2].
RT-PCR
Fourteen genes were assayed via a Taqman® quantitative RT-PCR protocol according to manufacturer's specifications using Applied Biosystem's High Capacity cDNA Reverse Transcription Kit (part # 4374967) and Taqman® Universal PCR Master Mix (part # 4364340). Taqman reporter probes were used (see Table 1 for a list of specific ABI assay identifiers). An aliquot of 400 ng of total RNA was reverse-transcribed and 1/80th of the reaction used for each replicate for each probe. The reaction volume for each replicate was 10 μL, with 0.5 μL of the Taqman 20× gene expression assay, 1 μL of sample, 5 μL of 2× Master Mix and 3.5 μL dH2O. All samples were assayed in quadruplicate for each probe according to Rosetta internal SOPs. Samples were run on an ABI 7900 HT system using the recommended ABI cycling protocol http://www3.appliedbiosystems.com/AB_Home/index.htm. See Table 2 for the specific Taqman® identifiers for the 14 probes used.
Data analysis
Eleven positive control nCounter™ spike-ins (spanning from 0.27 fM to 55 fM) were used to create the calibration curve for each nCounter™ array. Nine negative control spike-ins were used to assess the level of background (typically on the order of 10 counts). Mean of the negative controls was deducted from all other transcripts in the same assay prior to logarithmic transformation (log base 2). We used a standard linear regression model to find the least square fit of logarithm-transformed concentration on the logarithm-transformed number of molecules above background to generate the equation for the rest of the transcripts in the same assay. Each nCounter™ assay result was converted to an equivalent concentration using the assay standard curve. Use of the standard curve allows absolute measurements to be assigned to nCounter™ counts as needed.
To deduce the precision of the nCounter™ assay itself, we mean centered the data in log2 scale, resulting in a correction of approximately 1.08 fold. To achieve specified precision, NanoString recommends running each sample (by experiment) in triplicate. To mimic a typical experiment, therefore, we averaged triplicate assays for Rosetta spike-ins as well. Standard deviations of resulting mean values were used to calculate CVs.
To generate across-multiple-samples, gene-by-gene equivalency plots, both Taqman® and nCounter™ data were normalized to CUGBP1 as a reference gene for the xenograft samples. Originally four reference genes were identified from previous experiments as not varying significantly across our experimental conditions and were planned to be used as references in aggregate. However, three of the four did not reliably give signals above the lower limit of our standard curve and so were not used. This led us to deviate from generally accepted practice in which more than one reference gene is used to normalize data.
In the MAQC analysis, although the published Taqman® data were normalized to POLR2A, the nCounter™ normalization did not utilize any reference genes. This is because POLR2A was not one of the genes present in our genelist and so was unavailable as a common control. As a result of this approach, Taqman® data were normalized to mRNA amount, while nCounter™ normalization relied on the same amount of total RNA (100 ng) in each sample. This distinction is important because the MAQC study showed that UHR has 1.5-fold higher mRNA content than Brain (3% vs. 2%). To compensate for different mRNA content, 0.585 Ct, 0.46 Ct and 0.17 Ct were deducted from all genes of 0% Brain/100% UHR; 25% Brain/75% UHR; and 75% Brian/25% UHR samples, respectively.
For performance evaluations, a comparison was done for each possible pair of samples because we did not wish to artificially bias our data by arbitrarily assigning one sample as the "standard" to which other replicates would be compared. In these cases, we normalized to the mean of log intensity of the subset of genes in the corresponding sample for which measurements were above 0.27 fM in both samples of the pair. The same subset of genes was used to normalize Taqman®, using their mean Ct.
Assessment of the Spike-in performance and of the MAQC samples can be found in Additional File 1, Additional File 2, Additional File 3, and Additional File 4, and in Tables 3 and 4. Data from the analysis can be found in Additional File 5.
Results and discussion
Analysis of Xenograft-derived Samples
The sample set comprised four treatment conditions with four mouse xenografts per condition for a total of 16 samples. The 16 tissue samples were split and preserved by three methods (FF, FFPET, and CTL in Qiagen buffer RLT); total RNA was isolated from FF and FFPET for RT-PCR analysis. Fourteen genes were chosen for Taqman® comparison using samples that were either vehicle treated or treated with the highest level of compound. Ten genes were expected to change either up or down, and four reference genes were expected to remain constant. The genes chosen for Taqman® and Nanostring comparisons were picked based on internal Merck criteria. The differential expression in a previous microarray study of these samples showed relatively modest fold changes (~2 fold) at the highest compound treatment level used for this study (data for other, intermediate treatment levels is not shown).
A comparison of the compound and vehicle intensities between nCounter™ and Taqman® is shown in Figures 1, 2 and 3 for nine of the ten genes expected to change between treatments. Reference genes are not shown and one of the ten differentially expressed genes was not present at a sufficiently high level for accurate assessment. Figure 1 corresponds to FF tissue; Figure 2 to FFPET; and Figure 3 to CTL. In Figures 1, 2 and 3 only those points with expression levels within the calibration range are shown. The reference gene CUGBP1 was used to calculate the delta-delta CT for all genes.

Comparison of intensities derived from Taqman® and nCounter™ for FF-derived total RNA. The line in each case represents a slope of 1. Blue and black dots represent the two replicate measurements for each mouse sample. Only data points within the calibration curve are presented in these graphs. Units for the x-axis are delta CTs using CUGBP1 to normalize; units for the y-axis are log2 ratios of nCounter™ counts for a given gene and CUGPB1. Error bars represent standard deviations.

Comparison of intensities derived from Taqman® on FF total RNA and nCounter™ on FFPET-derived total RNA. The line in each case represents a slope of 1. Only data points within the calibration curve are presented in these graphs. Units for the x-axis are delta CTs using CUGBP1 to normalize; units for the y-axis are log2 ratios of nCounter™ counts for a given gene and CUGPB1. Error bars represent standard deviations.

Comparison of intensities derived from Taqman® on FF total RNA and nCounter™ on tissue lysates. The line in each case represents a slope of 1. Only data points within the calibration curve are presented in these graphs. Units for the x-axis are delta CTs using CUGBP1 to normalize; units for the y-axis are log2 ratios of nCounter™ counts for a given gene and CUGPB1. Error bars represent standard deviations.
As with the MAQC data, these sample sets show generally good agreement between the two platforms. The one outlier is NARG1 (top right graph in each Figure), which was consistently discordant in all Taqman® to nCounter™ comparisons. Since the region of NARG1 assayed by the Taqman® probe is at the junction between exons 1 and 2 and the region selected for the nCounter™ probe is close to the 3' end of the transcript, the two systems may be capturing valid but different transcript behavior of this gene. It should be noted that variability of measurements in the CTL samples (as represented by the error bars) was higher than for the other two sample types.
The FF and lysate data have a high degree of correlation, suggesting minor loss of data quality by using CTLs rather than purified total RNA. Taken together, the data in Figures 1, 2 and 3 suggest that the nCounter™ assay can be used to generate data from clinical samples with degraded RNA (FFPET, see Additional File 6 for representative quality) or from lysate preparations. It should be pointed out, however, that the FFPET model used (xenograft tissue) is not a perfect match for typical clinical samples and that not all degrees of degraded RNA will be amenable to this system
Performance metrics
To assess bias or compression in derived ratios, we plotted differences in ratios between nCounter™ and Taqman® versus average of ratios reported by both platforms (Figure 4). The scatter around the abscissa axis (y = 0) of differences suggests no bias in the measurement of ratios by nCounter™ relative to Taqman®, while the random scatter across degrees of differential expression (along the x-axis)implies no compression of ratios across ratio values.

Plot for ratio differences between nCounter™ and Taqman®. The differences in ratio between Taqman® and nCounter™ for the MAQC and xenograft data sets were plotted versus average ratio of two platforms. Markers in blue represent ratios derived for xenograft samples; markers in green represent ratios derived for MAQC samples.
Figure 5 displays a set of cumulative distribution function (CDF) curves for ratios reported by the nCounter™ assay for the MAQC and the three xenograft datasets (FF, FFPET and CTL) versus the Taqman®-derived ratios from the FF samples. The absence of ratio compression simplifies the subsequent analysis: since the magnitude of the ratio does not affect the concordance between two platforms by this method, we can combine all measurements together and determine how many measurements were off and by how much. Rather than depend solely on one references gene, we performed a comparison of all possible sample pairs; systematic shifts affecting all genes in each comparison were thereby attributed to the difference in total mass and subtracted out. As expected, ratios measured by nCounter™ and Taqman® were similar for MAQC and FF xenograft samples. The MAQC samples showed slightly less deviation between Taqman® and the nCounter™ System, which may reflect the controlled nature of the sample set and the smaller fraction of very-low intensity genes (see Table 3 for CV dependence on transcripts abundance). Overall the differences are small–for example, 90% of all nCounter™ measurements were within 1.27 and 1.33 fold changes of the Taqman® calculated ratio for the MAQC and cell line FF data, respectively. Interestingly, nCounter™ data from the CTL samples more closely mirrored the Taqman® data than did the FFPET total RNA data. However, even ratios reported by nCounter™ FFPET samples are close to Taqman® ratios obtained for FF samples: 90% of ratios were within 1.55 fold.

Cumulative distribution function plots for difference between Taqman® and nCounter™ derived ratios. Values on the x-axis are the absolute value of the difference between Taqman® and nCounter™ in log2 scale. The y-axis indicates the percentage of ratios which show a specific degree of difference between ratios or less.
Our impetus to assess the nCounter™ Analysis System was driven by its relative simplicity (that is, no need for amplification steps), its multiplexed format, and its potential to measure gene expression in samples from pre-clinical and clinical settings (e.g, fine-needle biopsies in lysate buffers, and FFPET materials). Our results confirmed the system has potential for pre-clinical and clinical measurements of multiple gene signatures in settings where the initial tissue collection would be conducive to FFPET or CTL preparations.
This platform could be used to fill an important and growing gap in drug development research. Microarray experiments routinely are used in basic research but often identify too many genes to allow higher-throughput downstream use of those signatures for screening or readouts. By allowing the cost effective and accurate measurement of expression of tens of genes from clinical samples, the nCounter™ system could facilitate translation of multi-gene expression based biomarkers into the clinic.
References
Van't Veer LJ, Dai HY, Vijver van de MJ, He YDD, Hart AAM, Mao M, Peterse HL, Kooy van der K, Marton MJ, Witteveen AT, Schreiber GJ, Kerkhoven RM, Roberts C, Linsley PS, Bernards R, Friend SH: Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002, 415: 530-536. 10.1038/415530a.
Geiss GK, Bumgarner RE, Birditt B, Dahl T, Dowidar N, Dunaway DL, Fell HP, Ferree S, George RD, Grogan T, James JJ, Maysuria M, Mitton JD, Oliveri P, Osborne JL, Peng T, Ratcliffe AL, Webster PJ, Davidson EH, Hood L, Dimitrov K: Direct multiplexed measurement of gene expression with color-coded probe pairs. Nature Biotechnology. 2007, 26: 317-325. 10.1038/nbt1385.
MAQC Consortium, Shi L, Reid LH, Jones WD, Shippy R, Warrington JA, Baker SC, Collins PJ, de Longueville F, Kawasaki ES, Lee KY, Luo Y, Sun YA, Willey JC, Setterquist RA, Fischer GM, Tong W, Dragan YP, Dix DJ, Frueh FW, Goodsaid FM, Herman D, Jensen RV, Johnson CD, Lobenhofer EK, Puri RK, Schrf U, Thierry-Mieg J, Wang C, Wilson M, Wolber PK, Zhang L, Amur S, Bao W, Barbacioru CC, Lucas AB, Bertholet V, Boysen C, Bromley B, Brown D, Brunner A, Canales R, Cao XM, Cebula TA, Chen JJ, Cheng J, Chu TM, Chudin E, Corson J, Corton JC, Croner LJ, Davies C, Davison TS, Delenstarr G, Deng X, Dorris D, Eklund AC, Fan XH, Fang H, Fulmer-Smentek S, Fuscoe JC, Gallagher K, Ge W, Guo L, Guo X, Hager J, Haje PK, Han J, Han T, Harbottle HC, Harris SC, Hatchwell E, Hauser CA, Hester S, Hong H, Hurban P, Jackson SA, Ji H, Knight CR, Kuo WP, LeClerc JE, Levy S, Li QZ, Liu C, Liu Y, Lombardi MJ, Ma Y, Magnuson SR, Maqsodi B, McDaniel T, Mei N, Myklebost O, Ning B, Novoradovskaya N, Orr MS, Osborn TW, Papallo A, Patterson TA, Perkins RG, Peters EH, Peterson R, Philips KL, Pine PS, Pusztai L, Qian F, Ren H, Rosen M, Rosenzweig BA, Samaha RR, Schena M, Schroth GP, Shchegrova S, Smith DD, Staedtler F, Su Z, Sun H, Szallasi Z, Tezak Z, Thierry-Mieg D, Thompson KL, Tikhonova I, Turpaz Y, Vallanat B, Van C, Walker SJ, Wang SJ, Wang Y, Wolfinger R, Wong A, Wu J, Xiao C, Xie Q, Xu J, Yang W, Zhang L, Zhong S, Zong Y, Slikker W: The MicroArray Quality Control (MAQC) project shows inter- and intraplatform reproducibility of gene expression measurements. Nat Biotechnol. 2006, 24: 1151-1161. 10.1038/nbt1239.
Hughes TR, Mao M, Jones AR, Burchard J, Marton MJ, Shannon KW, Lefkowitz SM, Ziman M, Schelter JM, Meyer MR, Kobayashi S, Davis C, Dai H, He YD, Stephaniants SB, Cavet G, Walker WL, West A, Coffey E, Shoemaker DD, Stoughton R, Blanchard AP, Friend SH, Linsley PS: Expression profiling using microarrays fabricated by an ink-jet oligonucleotide synthesizer. Nat Biotechnol. 2001, 19: 342-347. 10.1038/86730.
Acknowledgements
The authors would like to acknowledge the following individuals: Dr. Daniel Holder, Dr. Matthew Marton, Dr. Sergey Lezhnin, Dr. Jeffrey Sachs, Mr. Mark Parrish, Mr. Andrew Shoesmith, Ms. Sally Dow. These people helped in the preparation of materials, in providing useful advice and in reading the manuscript.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
GG and AM-H are employees of Nanostring.
Authors' contributions
VM performed the data transformations and analysis; KS designed and initiated the experiments and assembled the sample set, and also was the primary author of the manuscript; NB performed the Taqman assays; JW provided the xenograft model system and the tissue samples used, and also provided critical review of experimental design; GG and AM-H processed the samples at Nanostring and provided technical and critical review of the manuscript; TF provided overall project guidance, technical and critical review of the experiments and assisted in writing. All authors read and approved the final manuscript.
Electronic supplementary material
13104_2008_218_MOESM1_ESM.doc
Additional file 1: Performance of Nanostring spike-ins. Nanostring spike-ins span a reported range from 0.25 fM to 50 fM. All samples were assayed in triplicate; data are broken down by replicate. nCounter™ spike-ins consisted of two parallel sets of the same RNAs made into two different cocktails (cocktails #3 and #4). Some RNAs are set at different concentrations between these cocktails while others are at the same concentration (Table 3). (DOC 26 KB)
13104_2008_218_MOESM2_ESM.doc
Additional file 2: Performance of Rosetta spike-ins. The Rosetta spike-ins were originally constructed to measure dynamic range, ratio fidelity, and hybridization quality of an in-house, two-color microarray system [4]. Data from the nCounter™ spike-in controls were used to derive the precision profile and to provide a calibration curve for quantitation of other RNA transcripts. Spike-ins curves showed good linearity and reproducibility, and good conservation of relative measurements. Overall, the precision of the platform for the Rosetta Spike-ins had CVs of approximately 6%, except for concentrations below 1.5 fM (CVs were 10–30%). Error bars represent standard deviations. (DOC 24 KB)
13104_2008_218_MOESM3_ESM.doc
Additional file 3: Comparison of intensity values derived from Taqman® (x-axis) and Nanostring (y-axis) for each of the four MAQC samples (UHR, 25% Brain/75% UHR, 75% Brain/25% UHR, and Brain). Markers in red represent genes that were outside of the calibration curve. Pair-wise nCounter™ to Taqman® comparisons of measured expression levels of genes on a per sample basis showed correlation coefficients ranging from 0.835 to 0.886. Error bars represent standard deviations. (DOC 46 KB)
13104_2008_218_MOESM4_ESM.doc
Additional file 4: Comparison of calculated log 2 ratios for Brain vs UHR for Nanostring and Taqman®. Transcripts that were outside the calibration curve are shown in red and are labeled with their lowest measured concentration in either Brain or UHR (in fM). In the comparison of ratios, data for nCounter™ and Taqman® were first corrected for the known differences in mRNA content between Brain and UHR (2% to 3%). Additional File 4 shows the fit of the ratio comparisons to the line y = x for samples whose intensity falls within the standard curve (blue squares in Additional File 4), R = 0.995, demonstrating good linear agreement between Taqman® and nCounter™ ratio measurements. (DOC 32 KB)
13104_2008_218_MOESM5_ESM.xls
Additional file 5: Data from Nanostring nCounter experiment. This spreadsheet contains the raw nCounter™ data, the Taqman® data used for comparisons, and the normalized and averaged nCounter™ data. (XLS 536 KB)
13104_2008_218_MOESM6_ESM.doc
Additional file 6: Representative Bioanalyzer traces from FFPE-derived RNA. These traces demonstrate the degree of RNA degradation assayed by the nCounter™ system in our experiments. (DOC 34 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Malkov, V.A., Serikawa, K.A., Balantac, N. et al. Multiplexed measurements of gene signatures in different analytes using the Nanostring nCounter™ Assay System. BMC Res Notes 2, 80 (2009). https://doi.org/10.1186/1756-0500-2-80
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1756-0500-2-80