
Abstract
Background
Many methodologies have been used in research to identify the “intrinsic” subtypes of breast cancer commonly known as Luminal A, Luminal B, HER2-Enriched (HER2-E) and Basal-like. The PAM50 gene set is often used for gene expression-based subtyping; however, surrogate subtyping using panels of immunohistochemical (IHC) markers are still widely used clinically. Discrepancies between these methods may lead to different treatment decisions.
Methods
We used the PAM50 RT-qPCR assay to expression profile 814 tumors from the GEICAM/9906 phase III clinical trial that enrolled women with locally advanced primary invasive breast cancer. All samples were scored at a single site by IHC for estrogen receptor (ER), progesterone receptor (PR), and Her2/neu (HER2) protein expression. Equivocal HER2 cases were confirmed by chromogenic in situ hybridization (CISH). Single gene scores by IHC/CISH were compared with RT-qPCR continuous gene expression values and “intrinsic” subtype assignment by the PAM50. High, medium, and low expression for ESR1, PGR, ERBB2, and proliferation were selected using quartile cut-points from the continuous RT-qPCR data across the PAM50 subtype assignments.
Results
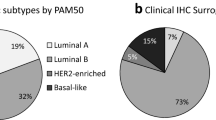
ESR1, PGR, and ERBB2 gene expression had high agreement with established binary IHC cut-points (area under the curve (AUC) ≥ 0.9). Estrogen receptor positivity by IHC was strongly associated with Luminal (A and B) subtypes (92%), but only 75% of ER negative tumors were classified into the HER2-E and Basal-like subtypes. Luminal A tumors more frequently expressed PR than Luminal B (94% vs 74%) and Luminal A tumors were less likely to have high proliferation (11% vs 77%). Seventy-seven percent (30/39) of ER-/HER2+ tumors by IHC were classified as the HER2-E subtype. Triple negative tumors were mainly comprised of Basal-like (57%) and HER2-E (30%) subtypes. Single gene scoring for ESR1, PGR, and ERBB2 was more prognostic than the corresponding IHC markers as shown in a multivariate analysis.
Conclusions
The standard immunohistochemical panel for breast cancer (ER, PR, and HER2) does not adequately identify the PAM50 gene expression subtypes. Although there is high agreement between biomarker scoring by protein immunohistochemistry and gene expression, the gene expression determinations for ESR1 and ERBB2 status was more prognostic.
Similar content being viewed by others


Background
For over a decade, research studies have used gene expression to classify invasive breast cancers into biologically and clinically distinct subtypes that have become known as Luminal A, Luminal B, HER2-Enriched (HER2-E) and Basal-like [1–3]. Subtype information has repeatedly shown to be an independent predictor of survival in breast cancer when used in multivariate analyses with standard clinical-pathological variables [3–6]. In 2009, Parker et al. derived a minimal gene set (PAM50) for classifying “intrinsic” subtypes of breast cancer [3, 7]. The PAM50 gene set has high agreement in classification with larger “intrinsic” gene sets previously used for subtyping [1, 3, 4, 8], and is now commonly employed [9–12].
There are several multi-gene expression tests clinically available for determining risk of relapse in early stage breast cancer, including the 21-gene recurrence score [13] (Oncotype Dx®, Genomic Health Inc, Redwood City, CA, http://www.oncotypedx.com), the 14-gene distant metastasis signature [14] (BreastOncPx™, US Labs, Irvine, CA, http://www.uslabs.net), the 97-gene histologic grade predictor [15] (MapQuant Dx™ Genomic Grade, Ipsogen, Marseilles, France and New Haven, CT, USA, http://www.ipsogen.com), and the 70-gene prognosis signature [16] (MammaPrint®, Agendia, Irvine, CA, http://www.agendia.com). The molecular signature of proliferation is perhaps the strongest variable in all these tests for determining outcome in ER + breast cancer.
In addition to gene expression profiling by microarray or RT-qPCR [2–4, 8, 17, 18], many studies have used immunohistochemical panels to identify subtypes [19–21]. For example, high grade ER+/HER2- tumors and ER+/HER2+ tumors are often considered Luminal B, while ER-/HER2+ are considered HER2-E subtype and triple negative tumors are considered Basal-like. In this study, we assess agreement between histopathology/IHC status and PAM50 classification for subtype, ESR1, PGR, ERBB2, and proliferation.
Methods
Samples and clinical data
There was ethical review and approval for all protocols used in this study from the respective centers involved and all subjects gave written informed consent to participate. A training set was developed using 171 breast samples, comprised of 16 “normal” breast tissue samples from reduction mammoplasties or grossly uninvolved breast tissue and 155 primary invasive breast cancers. These samples were collected from 2005–2009 under IRB approved protocols at the University of Utah and the University of North Carolina at Chapel Hill. Clinical-pathological information associated with the samples is based on the College of American Pathology (CAP) and American Joint Committee on Cancer (AJCC) standards at the time of collection (Additional file 1). Subtype classification and single and meta-gene (proliferation) scores were predicted on an independent test set of 814 samples from the GEICAM/9906 clinical trial, a randomized Phase 3 trial of fluorouracil, epirubicin, and cyclophosphamide alone or followed by paclitaxel [22]. Patients that were hormone receptor positive (ER and/or PR positive by IHC) were given adjuvant tamoxifen. The hormone receptor status for these samples was evaluated at a single site (Department of Pathology, Hospital General Universitario de Alicante) using immunohistochemistry (IHC) for progesterone receptor (PR) (clone PgR636, DAKO, Glostrup, Denmark) and estrogen receptor (ER) (clone 1D5, DAKO, Glostrup, Denmark) (Additional file 2). The scores for the proportion of dyed cells and intensity were summed to obtain a total Allred Score [23]. Measurement of HER2 expression was performed by Herceptest™ (DAKO, Glostrup, Denmark) and samples with scores of 2+ by IHC were confirmed by CISH, following the ASCO/CAP guidelines [24]. The clinical data for the training set and GEICAM/9906 test set are summarized in Table 1.
Measurement of PCR efficiency, limits of detection, and limits of quantification
Breast cancer cell lines (BT474, MCF7, MDA-MB-231, MDA-MB-436, MDA-MB-453, MDA-MB-468, SKBR3 and T47D) were cultured, pelleted and processed into FFPE tissue blocks. The RNA was extracted, pooled, reverse transcribed, and serially diluted at 2-fold increments from 2.56μg to 0.039ng per assay, which corresponds to a range of 7.11ng to 108.51fg of cDNA per reaction well. Each gene was measured in triplicate per RT-qPCR run on the Roche LC480 and 2 runs were performed for each of the 17 dilutions. A detailed description of methods used to calculate PCR efficiency, limits of detection and limits of quantification can be found in Additional file 3.
Selection of prototype samples for the RT-qPCR training set
Training set samples were run across 3 batches of PCR plates manufactured at ARUP Laboratories (ARUP Laboratories, Salt Lake City, UT, http://www.aruplab.com). The method to identify prototype samples representing the subtypes has been previously described [3]. Briefly, hierarchical clustering (median centered by gene, Pearson correlation, centroid-linkage) [25] was performed on the RT-qPCR data and SigClust was run at each node of the dendrogram beginning at the root and stopping when the test was no longer significant (p > 0.001). A “centroid” was generated for each subtype in the training set using the average expression for each gene across all prototype samples of a given subtype. Single sample subtype prediction was performed by calculating a Spearman rank correlation coefficient between the gene expression values of an individual sample compared to each of the centroid gene values for Luminal A, Luminal B, HER2-Enriched, Basal-like, and Normal. The subtype classification for the new sample is assigned to the centroid with the highest correlation.
10-Fold cross validation to determine stability of selected prototypes
The 154 prototype samples identified by SigClust were randomly split into 10 groups. Nine of the 10 groups were used to calculate new centroids for each of the 5 possible subtype assignments. Each sample from the remaining group was then assigned a subtype based on closest proximity to the newly calculated centroids using Spearman's Rho. The process of calculating centroids using 9 of the 10 groups and predicting on the remaining group was repeated leaving out a different group each time.
Measurements of assay reproducibility
Reproducibility of the PAM50 assay was determined using 3 cell lines (MCF7, ME16C and SKBR3) and a pool of Luminal A prototype samples that were each run 12 times (3 runs across 4 batches of PAM50 plates) over 30 days. Variation in each gene measurement was assessed using the difference between the mean calibrator crossing point (CP) and each sample replicate CP (ΔCP). The square root of the mean CV2 for ΔCP was used to estimate the variation for each gene within plate, within batch, and across batches. Higher gene CVs may be due to lower concentration of a single gene within a sample. We used the technical variability in measuring each gene to further assess the stability of the categorical subtype call in the GEICAM/9906 test set samples. Since the biology between subtypes is a continuum and some samples may have close proximity to more than 1 prototypic subtype, we used a Monte-Carlo simulation to introduce random error into the call to determine the frequency of switching subtype [26].
Scaling single and Meta-Gene scores
The PAM50 subtype assay can also provide quantitative and qualitative gene expression scores for the standard biomarkers usually measured semi-quantitatively by IHC: ESR1/ER, PGR/PR and ERBB2/HER2. In addition, the PAM50 contains many cell cycle regulated genes that can be combined into a meta-gene for proliferation (CENPF, ANLN, CDC20, CCNB1, CEP55, MYBL2, MKI67, UBE2C, RRM2, and KIF2C). The meta-gene for proliferation were selected because they had strong correlation within the associated dendrogram of the training set cluster. The quantitative scale of 1–10 for the single genes and proliferation was derived by rescaling the original log-expression ratios from the training set and included a 10% buffer on either side of the original values to allow for values that were higher or lower than what was encountered in the training set. Any new values that were less than 0 or greater than 10 were truncated at 0 and 10, respectively.
Fixed cut-points (low vs. intermediate/high) for the single genes (ESR1, PGR, and ERBB2) and proliferation were directly applied from the training set to the GEICAM/9906 test set. Receiver Operator Characteristic (ROC) curves were generated by dichotomizing IHC data and treating RT-qPCR data as a continuous variable.
Results
Training set, subtype stability, and classification accuracy
We identified 154 prototypic samples from the RT-qPCR data by hierarchical clustering of the PAM50 classifier genes, and statistical selection from the dendrogram by SigClust [27]. The training set was comprised of 53 Luminal A, 27 Luminal B, 32 HER2-enriched, 38 Basal-like and 4 Normal-like (Figure 1). The 10-fold cross validation had 91.6% concordance (multi-rater kappa score of 0.885) with the initial SigClust subtype assignments (Additional file 4).

Clinical PAM50 RT-qPCR breast cancer training set. Hierarchical clustering of RT-qPCR data for the PAM50 classifier genes normalized to the 5 control genes using 171 FFPE procured breast samples. Statistical selection using SigClust identified the 5 significant groups previously identified and designated as Luminal A (dark blue), Luminal B (light blue), HER2-E (pink), Basal-like (red), and Normal (green). The 16 non-neoplastic samples (grey), from reduction mammoplasty and grossly uninvolved breast tissues, all Clustered together and away from the invasive cancers. SigClust identified 4 reduction mammoplasty samples (green) that were used to train the Normal subtype.
Interference from normal breast tissue contamination
A major source of subtype misclassification comes from having normal tissue within the tumor sample [28]. We assessed the effect of having contaminating normal tissue within the tumor sample by diluting out RNA from tumor subtypes with pooled RNA from “normal” reduction mammoplasties (0%, 25%, 50% and 75%). Primary tumors were pooled to represent Luminal A and HER2-E samples while cell lines were used to represent Luminal B (MCF7) and Basal-like (ME16C). The changes in subtype classification occurred in a systematic fashion with all subtypes switching directly to a classification of Normal, with the exception of Luminal B, which switched to Luminal A. The switch from Luminal B to Luminal A required 50% contribution from the normal breast tissue signature. Interference data from the introduction of normal breast tissue RNA into each of the subtypes is provided in Additional file 5. During the dilution series for HER2-E with “normal” there was switching in the ESR1 score between intermediate and low suggesting that both samples had similar ESR1 expression near the cut-off for those scores.
Subtype, immunohistochemistry, and RT-qPCR gene scores
The RT-qPCR values for ESR1, PGR, ERBB2, and proliferation were evaluated across prototypic samples in the training set. High, intermediate, and low cut points were made based on the continuous distribution of expression across the tumor subtypes. The cut-points for each of the scores and how they were determined is presented in Table 2. Figure 2 shows the expression and cut-points for ESR1 in the training set and how these compare within the GEICAM/9906 test set. Additional single and meta-gene cut-points for the training and test sets can be found in Additional file 6. Comparisons between the gene expression and IHC data for GEICAM/9906 gave good overall agreement with a high area under the curve (AUC) for ESR1/ER (AUC = 0.90), PGR/PR (AUC = 0.90), and ERBB2/HER2 (AUC = 0.95) (Figure 3). Rather than re-optimize the cut-points on the test set, the fixed cut-points based on the training set were used and it showed high sensitivity/specificity, although a slightly higher false positive rate for ERBB2 than would have been selected by eye.

ESR1 score cut-offs using training set and the GEICAM/9906 testing set. The ESR1 score is provided as a qualitative call of high, intermediate, or low. The cut-offs were based on the continuous expression of ESR1 across prototype samples in the training set. Each circle on the box plot represents an individual sample that is color coded according to IHC status. The cut-points between high, intermediate, and low classes were individually derived from the training set samples (A). Data from ESR1 gene expression over the GEICAM 9906 samples (B) are plotted on the same scale as the training set. Samples are colored according to ER IHC positivity (red) or negativity (blue) determined at a central facility.

Receiver Operator Characteristic (ROC) curves for ESR1 , PGR , and ERBB2 for the GEICAM/9906 test set. ROC curves for the GEICAM/9906 test set were generated using the clinical IHC status (positive vs. negative) for ER, PR, and HER2/neu as compared to the continuous RT-qPCR data for ESR1, PGR, and ERBB2. The cut-points for sensitivity/specificity are based on the training set.
Ninety-two percent (497/538) of Luminal (A/B) tumors were ER + by IHC and 99% (530/538) had an intermediate-high ESR1 score (Tables 3 and 4). Luminal A tumors more frequently expressed PR/PGR than Luminal B tumors using either IHC (94% vs 74%) or qPCR (95% vs 61%).
Although the HER2-E subtype is often thought of as being ER-, only 36% (63/174) were ER- by IHC and 44% (76/174) were low ESR1 score. Seventeen percent of HER2-E samples were called triple-negative. Of the clinically HER2+ group by IHC/CISH, approximately two-thirds (69/113 = 61%) were HER2-E and one-third were Luminal B (37/113 = 33%) subtype (Figure 4). Using the qPCR cut-off for ERBB2 expression, we found that 98% (609/624) of samples that were low ERBB2 were also HER2- by IHC/CISH, while 53% (109/190) of tumors with intermediate-high ERBB2 expression were HER2+. However, analyses just within the HER2-E subtype showed 71% (66/93) of tumors with high ERBB2 gene expression were HER2+ by IHC/CISH.

Association between HER2 status and “intrinsic” subtype. Figure (A) shows the subtype distribution within HER2+ samples by IHC/CISH. Figure (B) shows the ER/HER2 status for samples only within the HER2-E subtype.
Ninety percent (63/70) of Basal-like tumors were ER- by IHC and 96% (67/70) were low ESR1 score. Furthermore, 81% (57/70) of Basal-like tumors were triple negative (ER-/PR-/HER2-) by IHC/CISH and 86% (60/70) were low in all 3 genes by qPCR (Table 5). Conversely, only 56% (57/101) and 67% (60/90) of triple negatives defined by IHC/CISH or qPCR were Basal-like, respectively. There was no difference (p > 0.05) in ESR1, PGR or ERBB2 expression by qPCR in Basal-like tumors, regardless of being triple-negative or non-triple negative by IHC/CISH (Figure 5).

Relative transcript abundance for ESR1 , PGR , and ERBB2 in the Basal-like subtype. There was no difference (p > 0.05) in (A) ESR1, (B) PGR, or (C) ERBB2 expression by qPCR in Basal-like tumors, regardless of being called triple-negative or non-triple negative by IHC/CISH.
Additional file 7 shows a comparison of unsupervised hierarchical clustering with supervised subtype assignment and single marker scores for GEICAM/9906. In general, the supervised classification agreed with the sample associated dendrogram clusters. The side branches of the dendrogram clusters are less correlated to other samples and reflect the continuum in the biology, especially between Luminal A, Luminal B and HER2-E subtypes. The HER2-E and Basal-like subtypes cluster away from the Luminal tumors and have similar gene expression profiles overall; however, standard IHC/CISH biomarkers poorly define these subtypes.
Prognostic significance of gene expression versus standard methods for ER and HER2 status
Although there was high agreement between IHC/CISH and RT-qPCR measurements for ER/ESR1 and HER2/ERBB2, we wanted to assess whether the two different methods provided equivalent prognostic information. When tested in a multivariate Cox model for overall survival, only the RT-qPCR assignments were selected in the final Cox model in the GEICAM/9906 test set (Table 6). When all patients with locally advanced breast cancer were stratified, regardless of chemotherapy regimen (FEC vs FEC-T), both classifications for assessing ER/ESR1 and HER2/ERBB2 status were significantly associated with outcome (Figure 6). Since endocrine therapy was based on ER status determined by IHC, those ER + samples that were ESR1- (29/154 = 19%) would have received adjuvant tamoxifen and conversely those patients with ER- tumors that were ESR1+ (45/660 =7%) would not have received therapy. When separating outcome based on agreement and disagreement between the methods, we find that women with ER+/ESR1+ tumors have similar outcomes to women with ER-/ESR1+ tumors, and women with ER-/ESR1- tumors have similar outcomes to women with ER+/ESR1- tumors. This shows that the RT-qPCR assignment is more prognostic and accurate than IHC for ER.

Kaplan-Meier plots of overall survival in GEICAM 9906 data set. When stratifying all patients with locally advanced breast cancer, regardless of chemotherapy regimen (FEC vs FEC-T), both RT-qPCR (A) and IHC/CISH (B) molecular classifications for assessing ESR1/ER and ERBB2/Her2 status were significant. However, the separation of the survival curves suggests that ER-status as assessed by qPCR has prognostic superiority to IHC (C).
Discussion
Many studies have tried to identify the gene expression-based “intrinsic” subtypes using a variety of methods for the sake of simplicity, cost, and available technologies. Methods that can be used from formalin-fixed, paraffin-embedded tissues are optimal since this is how samples are procured and archived in most pathology departments. The two preferred technologies for gene expression profiling from FFPE tissues are RT-qPCR [17, 18] and Nanostring nCounter [29]. The nCounter system uses color-coded probes that bind directly to the RNA transcript without reverse transcription and PCR amplification. While these methods have high agreement for gene quantification, other methodologies may lead to different conclusions and treatment decisions. For instance, in the NCIC.CTG MA.12 clinical trial that randomized pre-menopausal women with primary breast cancer to tamoxifen versus placebo it was found that a panel 6 IHC antibodies for subtyping was not prognostic but the PAM50 RT-qPCR subtypes were prognostic [30]. In another randomized study (NCIC.CTG MA.5) that assessed PAM50 subtype sensitivity to anthracycline-based chemotherapy, it was shown that the HER2-E subtype received the most benefit, while women with Basal-like tumors had no benefit from this aggressive treatment [31]. This study and the MA.5 trial found that only about two-thirds of clinically Her2+ tumors are classified as HER2-E and about the same percent of triple negatives are classified as Basal-like. Thus, only a subset of the IHC defined groups overlap with PAM50 subtype classification, which may have ramifications for clinical trial findings and predicting therapy benefit.
Receiver Operator Characteristic (ROC) curves are commonly used in medicine to optimize the sensitivity/specificity of an assay depending on the purpose of the test (i.e. screening, monitoring, prognosis, etc.) [32]. In clinical pathology, ROC curves are often used to validate a new methodology against an existing “gold” standard. A major limitation to this approach is that cut-offs are then determined by comparison to an often less than perfect reference. We used an approach for selecting single (ESR1, PGR, ERBB2) and meta-gene (proliferation) cut-offs that was based on the distribution of expression of these markers across the different subtypes. This method showed to be reproducible in an independent test set.
The ROC curves showed high agreement between RT-qPCR and the standard IHC biomarkers. ESR1 had high sensitivity although the cut-off for ER + status was 10% positive staining nuclei, whereas the new recommendation for determining ER status is 1% [33]. These borderline cases for ER positivity may be better characterized by the overall subtype biology. For ERBB2, there was high specificity, which is optimal since confirmatory CISH or FISH would only be performed when it was uncertain if the gene was truly amplified [34]. It has been suggested that the use of single gene RT-qPCR measurement for ERBB2 is insufficient for determining HER2 positive samples that may benefit from trastuzumab/Herceptin® therapy [35]. Dabbs et al. found that the negative predictive value for determining HER2/ERBB2 status was high between the HercepTest and the GHI Oncotype Dx qPCR assay (99%); but the concordance for positive HER2/ERBB2 samples was only 28%. In contrast, we showed that the concordance between HER2 (IHC/CISH) and ERBB2 (RT-qPCR) is greater than 90% when restricted to the HER2-E subtype.
In order to determine if there was a prognostic difference between the RT-qPCR and IHC we included both methods in a Cox proportional hazards model and showed that gene expression remained significant in the multivariate analysis and replaced IHC. Furthermore, the outcome plots for women with tumors scored positive for ER by IHC but negative for ESR1 had outcomes similar to women that were ER-/ESR1-. Conversely, women with ER- tumors by IHC but positive for ESR1 had similar outcomes to women with ER+/ESR1+ disease. Thus, despite the fact that patients were treated in favor of the IHC diagnosis (i.e. ER + disease was treated with adjuvant tamoxifen) the course of disease was in agreement with the gene expression determination. The better prognosis seen in the ESR1+ but ER- subtype is curious since these patients would not have been given adjuvant endocrine blockade therapy. However, gene expression for ESR1 may be identifying the “true” luminal origin of these tumors which have a better prognosis, regardless of therapy [30]. In addition, the patients included in the test set were locally advanced and received chemotherapy that can cause chemotherapy induced amenorrhea and a reduction in ovarian function [36], which again may benefit the luminal subtype most.
The Normal subtype was developed from reduction mammoplasty “normal” breast tissue and serves as a quality control measure since these cases would be considered to have an insufficient amount of tumor tissue to make a tumor subtype call. Interference studies showed that the introduction of “normal” breast tissue RNA caused a systematic shift in subtype assignment with subtypes switching to Normal, except Luminal B which changed to Luminal A.
None of the assignment switches occurred until the introduction of 50% “normal” breast tissue RNA. The greatest risk of misclassification would come from Luminal B subtypes masquerading as Luminal A tumors because of “normal” tissue contamination [28]; however, these tumors maintain a high proliferation score suggesting they are still a high risk Luminal tumor.
A fifth tumor type that has often been referred to as “Normal-like” has been suggested to be an artifact of having too few tumor cells and a large background of normal breast cells in the sample. Our mixing experiments here support this hypothesis and show that when increasing amounts of “normal” tissue RNA is added to a tumor it switches into the Normal-like group. It is, however, suspected that some tumors now called Normal-like may be put into the recently described Claudin-low classification [37]. The Claudin-low subtype is mostly triple-negative, shares biomarkers in common with normal breast epithelial cells and Basal-like tumors, and may be caused by deficiency in either BRCA1or p53, or both; however there is no clinical indication for Claudin-low, and most are typically classified as Basal-like. There are now many more groups of tumors being identified with transcriptome and copy number variance analyses [38, 39]. The overlap between these new groups, existing subtypes, and standard biomarkers already in practice should allow for more personalized treatments and better outcomes in the future.
Conclusions
Compiling small biomarker panels for the purpose of “intrinsic” subtyping is of limited value in identifying PAM50 based subtypes. Gene expression scoring for ESR1 and ERBB2 has good agreement with the corresponding protein biomarkers (ER and HER2) and may have more prognostic power.
References
Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA, et al: Molecular portraits of human breast tumours. Nature. 2000, 406 (6797): 747-752. 10.1038/35021093.
Sorlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, Hastie T, Eisen MB, van de Rijn M, Jeffrey SS, et al: Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A. 2001, 98 (19): 10869-10874. 10.1073/pnas.191367098.
Parker JS, Mullins M, Cheang MC, Leung S, Voduc D, Vickery T, Davies S, Fauron C, He X, Hu Z, et al: Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol. 2009, 27 (8): 1160-1167. 10.1200/JCO.2008.18.1370.
Hu Z, Fan C, Oh DS, Marron JS, He X, Qaqish BF, Livasy C, Carey LA, Reynolds E, Dressler L, et al: The molecular portraits of breast tumors are conserved across microarray platforms. BMC Genomics. 2006, 7: 96-10.1186/1471-2164-7-96.
Nielsen TO, Parker JS, Leung S, Voduc D, Ebbert M, Vickery T, Davies SR, Snider J, Stijleman IJ, Reed J, et al: A comparison of PAM50 intrinsic subtyping with immunohistochemistry and clinical prognostic factors in tamoxifen-treated estrogen receptor-positive breast cancer. Clin Cancer Res. 2010, 16 (21): 5222-5232. 10.1158/1078-0432.CCR-10-1282.
Cheang MCU, Voduc KD, Dongsheng T, Jiang S, Leung S, Chia SK, Shepherd L, Levine MN, Pritchard KI, Davies SR, et al: Responsiveness of intrinsic subtypes to adjuvant anthracycline substitution in the NCIC.CTG MA.5 randomized trial. Clin Cancer Res. in press, .
Tibshirani R, Hastie T, Narasimhan B, Chu G: Diagnosis of multiple cancer types by shrunken centroids of gene expression. Proc Natl Acad Sci U S A. 2002, 99 (10): 6567-6572. 10.1073/pnas.082099299.
Sorlie T, Tibshirani R, Parker J, Hastie T, Marron JS, Nobel A, Deng S, Johnsen H, Pesich R, Geisler S, et al: Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc Natl Acad Sci U S A. 2003, 100 (14): 8418-8423. 10.1073/pnas.0932692100.
Ellis MJ, Suman VJ, Hoog J, Lin L, Snider J, Prat A, Parker JS, Luo J, DeSchryver K, Allred DC, et al: Randomized phase II neoadjuvant comparison between letrozole, anastrozole, and exemestane for postmenopausal women with estrogen receptor-rich stage 2 to 3 breast cancer: clinical and biomarker outcomes and predictive value of the baseline PAM50-based intrinsic subtype--ACOSOG Z103. J Clin Oncol. 2011, 29 (17): 2342-2349. 10.1200/JCO.2010.31.6950.
Esserman LJ, Berry DA, Cheang MC, Yau C, Perou CM, Carey L, DeMichele A, Gray JW, Conway-Dorsey K, Lenburg ME, et al: Chemotherapy response and recurrence-free survival in neoadjuvant breast cancer depends on biomarker profiles: results from the I-SPY 1 TRIAL (CALGB 150007/150012; ACRIN 6657). Breast Cancer Res Treat. 2012, 132 (3): 1049-1062. 10.1007/s10549-011-1895-2.
Gonzalez-Angulo AM, Iwamoto T, Liu S, Chen H, Do KA, Hortobagyi GN, Mills GB, Meric-Bernstam F, Symmans WF, Pusztai L: Gene expression, molecular class changes, and pathway analysis after neoadjuvant systemic therapy for breast cancer. Clin Cancer Res. 2012, 18 (4): 1109-1119. 10.1158/1078-0432.CCR-11-2762.
Iwamoto T, Booser D, Valero V, Murray JL, Koenig K, Esteva FJ, Ueno NT, Zhang J, Shi W, Qi Y, et al: Estrogen receptor (ER) mRNA and ER-related gene expression in breast cancers that are 1% to 10% ER-positive by immunohistochemistry. J Clin Oncol. 2012, 30 (7): 729-734. 10.1200/JCO.2011.36.2574.
Paik S, Shak S, Tang G, Kim C, Baker J, Cronin M, Baehner FL, Walker MG, Watson D, Park T, et al: A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N Engl J Med. 2004, 351 (27): 2817-2826. 10.1056/NEJMoa041588.
Tutt A, Wang A, Rowland C, Gillett C, Lau K, Chew K, Dai H, Kwok S, Ryder K, Shu H, et al: Risk estimation of distant metastasis in node-negative, estrogen receptor-positive breast cancer patients using an RT-PCR based prognostic expression signature. BMC Cancer. 2008, 8: 339-10.1186/1471-2407-8-339.
Sotiriou C, Wirapati P, Loi S, Harris A, Fox S, Smeds J, Nordgren H, Farmer P, Praz V, Haibe-Kains B, et al: Gene expression profiling in breast cancer: understanding the molecular basis of histologic grade to improve prognosis. J Natl Cancer Inst. 2006, 98 (4): 262-272. 10.1093/jnci/djj052.
van de Vijver MJ, He YD, van't Veer LJ, Dai H, Hart AA, Voskuil DW, Schreiber GJ, Peterse JL, Roberts C, Marton MJ, et al: A gene-expression signature as a predictor of survival in breast cancer. N Engl J Med. 2002, 347 (25): 1999-2009. 10.1056/NEJMoa021967.
Perreard L, Fan C, Quackenbush JF, Mullins M, Gauthier NP, Nelson E, Mone M, Hansen H, Buys SS, Rasmussen K, et al: Classification and risk stratification of invasive breast carcinomas using a real-time quantitative RT-PCR assay. Breast Cancer Res. 2006, 8 (2): R23-10.1186/bcr1399.
Mullins M, Perreard L, Quackenbush JF, Gauthier N, Bayer S, Ellis M, Parker J, Perou CM, Szabo A, Bernard PS: Agreement in breast cancer classification between microarray and quantitative reverse transcription PCR from fresh-frozen and formalin-fixed, paraffin-embedded tissues. Clin Chem. 2007, 53 (7): 1273-1279. 10.1373/clinchem.2006.083725.
Nielsen TO, Hsu FD, Jensen K, Cheang M, Karaca G, Hu Z, Hernandez-Boussard T, Livasy C, Cowan D, Dressler L, et al: Immunohistochemical and clinical characterization of the basal-like subtype of invasive breast carcinoma. Clin Cancer Res. 2004, 10 (16): 5367-5374. 10.1158/1078-0432.CCR-04-0220.
Morrison DH, Rahardja D, King E, Peng Y, Sarode VR: Tumour biomarker expression relative to age and molecular subtypes of invasive breast cancer. Br J Cancer. 2012, .
Caudle AS, Yu TK, Tucker SL, Bedrosian I, Litton JK, Gonzalez-Angulo AM, Hoffman K, Meric-Bernstam F, Hunt KK, Buchholz TA, et al: Local-regional control according to surrogate markers of breast cancer subtypes and response to neoadjuvant chemotherapy in breast cancer patients undergoing breast conserving therapy. Breast Cancer Res. 2012, 14 (3): R83-10.1186/bcr3198.
Martin M, Rodriguez-Lescure A, Ruiz A, Alba E, Calvo L, Ruiz-Borrego M, Munarriz B, Rodriguez CA, Crespo C, de Alava E, et al: Randomized phase 3 trial of fluorouracil, epirubicin, and cyclophosphamide alone or followed by Paclitaxel for early breast cancer. J Natl Cancer Inst. 2008, 100 (11): 805-814. 10.1093/jnci/djn151.
Allred DC, Harvey JM, Berardo M, Clark GM: Prognostic and predictive factors in breast cancer by immunohistochemical analysis. Modern Pathol. 1998, 11 (2): 155-168.
Wolff AC, Hammond ME, Schwartz JN, Hagerty KL, Allred DC, Cote RJ, Dowsett M, Fitzgibbons PL, Hanna WM, Langer A, et al: American Society of Clinical Oncology/College of American Pathologists guideline recommendations for human epidermal growth factor receptor 2 testing in breast cancer. J Clin Oncol. 2007, 25 (1): 118-145.
Eisen MB, Spellman PT, Brown PO, Botstein D: Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci U S A. 1998, 95 (25): 14863-14868. 10.1073/pnas.95.25.14863.
Ebbert MT, Bastien RR, Boucher KM, Martin M, Carrasco E, Caballero R, Stijleman IJ, Bernard PS, Facelli JC: Characterization of uncertainty in the classification of multivariate assays: application to PAM50 centroid-based genomic predictors for breast cancer treatment plans. J Clin Bioinforma. 2011, 1: 37-10.1186/2043-9113-1-37.
Liu Y, Hayes DN, Nobel A, Marron JS: Statistical Significance of Clustering for High-Dimension, Low-Sample Size Data. J Am Stat Assoc. 2008, 103 (483): 1281-1293. 10.1198/016214508000000454.
Elloumi F, Hu Z, Li Y, Parker JS, Gulley ML, Amos KD, Troester MA: Systematic Bias in Genomic Classification Due to Contaminating Non-neoplastic Tissue in Breast Tumor Samples. BMC Med Genomics. 2011, 4: 54-10.1186/1755-8794-4-54.
Geiss GK, Bumgarner RE, Birditt B, Dahl T, Dowidar N, Dunaway DL, Fell HP, Ferree S, George RD, Grogan T, et al: Direct multiplexed measurement of gene expression with color-coded probe pairs. Nat Biotechnol. 2008, 26 (3): 317-325. 10.1038/nbt1385.
Chia SK, Bramwell VH, Tu D, Shepherd LE, Jiang S, Vickery T, Mardis E, Leung S, Ung K, Pritchard KI, et al: A 50-Gene Intrinsic Subtype Classifier for Prognosis and Prediction of Benefit from Adjuvant Tamoxifen. Clin Cancer Res. 2012, 18 (16): 4465-4472. 10.1158/1078-0432.CCR-12-0286.
Cheang MC, Voduc KD, Tu D, Jiang S, Leung S, Chia SK, Shepherd LE, Levine MN, Pritchard KI, Davies S, et al: Responsiveness of intrinsic subtypes to adjuvant anthracycline substitution in the NCIC.CTG MA.5 randomized trial. Clin Cancer Res. 2012, 18 (8): 2402-2412. 10.1158/1078-0432.CCR-11-2956.
Soreide K: Receiver-operating characteristic curve analysis in diagnostic, prognostic and predictive biomarker research. J Clin Pathol. 2009, 62 (1): 1-5. 10.1136/jcp.2008.061010.
Hammond ME, Hayes DF, Dowsett M, Allred DC, Hagerty KL, Badve S, Fitzgibbons PL, Francis G, Goldstein NS, Hayes M, et al: American Society of Clinical Oncology/College Of American Pathologists guideline recommendations for immunohistochemical testing of estrogen and progesterone receptors in breast cancer. J Clin Oncol. 2010, 28 (16): 2784-2795. 10.1200/JCO.2009.25.6529.
Garcia-Caballero T, Grabau D, Green AR, Gregory J, Schad A, Kohlwes E, Ellis IO, Watts S, Mollerup J: Determination of HER2 amplification in primary breast cancer using dual-colour chromogenic in situ hybridization is comparable to fluorescence in situ hybridization: a European multicentre study involving 168 specimens. Histopathology. 2010, 56 (4): 472-480. 10.1111/j.1365-2559.2010.03503.x.
Dabbs DJ, Klein ME, Mohsin SK, Tubbs RR, Shuai Y, Bhargava R: High False-Negative Rate of HER2 Quantitative Reverse Transcription Polymerase Chain Reaction of the Oncotype DX Test: An Independent Quality Assurance Study. J Clin Oncol. 2011, 29 (32): 4279-4285. 10.1200/JCO.2011.34.7963.
Swain SM, Jeong JH, Geyer CE, Costantino JP, Pajon ER, Fehrenbacher L, Atkins JN, Polikoff J, Vogel VG, Erban JK, et al: Longer therapy, iatrogenic amenorrhea, and survival in early breast cancer. N Engl J Med. 2010, 362 (22): 2053-2065. 10.1056/NEJMoa0909638.
Prat A, Parker JS, Karginova O, Fan C, Livasy C, Herschkowitz JI, He X, Perou CM: Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res. 2010, 12 (5): R68-10.1186/bcr2635.
Ha G, Roth A, Lai D, Bashashati A, Ding J, Goya R, Giuliany R, Rosner J, Oloumi A, Shumansky K, et al: Integrative analysis of genome-wide loss of heterozygosity and mono-allelic expression at nucleotide resolution reveals disrupted pathways in triple negative breast cancer. Genome Res. 2012, -Epub
Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ, Speed D, Lynch AG, Samarajiwa S, Yuan Y, et al: The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012, 486 (7403): 346-352.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1755-8794/5/44/prepub
Acknowledgements
This work was supported by the Huntsman Cancer Institute (HCI)/Foundation, the ARUP Institute for Clinical and Experimental Pathology, and NCI grants U01 CA114722-01 and P30 CA42014-19. A. Prat is supported by the Translational Oncology fellowship of the Sociedad Española de Oncologia Médica (SEOM). We thank the TRAC facility and Research Informatics at HCI, and the UNC Tissue Procurement Facility for contributing samples. We also appreciate the guidance of Dr. Joel S. Parker.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
ARUP Laboratories Inc. has a financial interest in the commercial offering of the subject matter. PSB receives research funding from the ARUP Institute for Clinical and Experimental Pathology, although he is not an employee of ARUP. PSB, CMP, and MJE have equity interest in Bioclassifier LLC, which has sublicensed the PAM50 signature from the University of Utah.
Authors’ contributions
RRLB participated in design of the study, generating data, and drafting the manuscript. ARL participated in recruiting patients, collecting samples and clinical data, and reviewing the manuscript. MTWE participated in design of the study, bioinformatics and statistical analysis, and drafting the manuscript. AP participated in bioinformatics and statistical analysis. BM participated in recruiting patients, collecting samples and clinical data, and reviewing the manuscript. LR participated in design of the study. PM participated in design of the study. MRB participated in recruiting patients, collecting samples and clinical data, and reviewing the manuscript. DA participated in design of the study. BL participated in manufacturing of the PCR plates. IA performed IHQ/CISH on GEICAM/9906 samples and participated in reviewing the manuscript. TD participated in manufacturing of the PCR plates. DW participated in manufacturing of the PCR plates. MAS participated in recruiting patients, collecting samples and clinical data, and reviewing the manuscript. LB participated in manufacturing of the PCR plates. KMB participated in statistical analysis. EA participated in recruiting patients, collecting samples and clinical data, and reviewing the manuscript. LP participated in statistical analysis. CAD participated in sample preparation and organization. IA participated in recruiting patients, collecting samples and clinical data, and reviewing the manuscript. CF participated in robotics design. IJS participated in data generation and reviewing the manuscript. JP participated in recruiting patients, collecting samples and clinical data, and reviewing the manuscript. AA participated in recruiting patients, collecting samples and clinical data, and reviewing the manuscript. EC participated in design of the study and reviewing the manuscript. RC participated in design of the study, managing the collection of samples and central laboratory activity, and reviewing the manuscript. MJE participated in reviewing the manuscript. TON participated in reviewing the manuscript. CMP participated in collecting samples and clinical data, and reviewing the manuscript. MA participated in the conceiving and design of the study. PSB participated in conceiving and design of the study, and drafting the manuscript. MM participated in design of the study, recruiting patients, collecting samples and clinical data, and reviewing the manuscript. All authors read and approved the final manuscript.
Philip S Bernard and Miguel Martín contributed equally to this work.
Electronic supplementary material
12920_2012_315_MOESM1_ESM.xlsx
Additional file 1: Clinical-pathological information associated with training set subtypes. Clinical-pathological information associated with the 171 samples included in the training set. (XLSX 23 kb) (XLSX 23 KB)
12920_2012_315_MOESM2_ESM.xls
Additional file 2: Clinical-pathological information and PAM50 data associated with GEICAM/9906 test set. Clinical-pathological information and PAM50 RT-qPCR results associated with the 814 samples included in the GEICAM9906 test set. (XLS 1106 kb) (XLS 1 MB)
12920_2012_315_MOESM4_ESM.xlsx
Additional file 4: 10-fold cross validation of training set. Each gene was measured in triplicate per RT-qPCR run on the Roche LC480 and 2 runs were performed for each of the 17 dilutions. The prototype samples identified by SigClust were split into 10 groups and nine of the 10 groups were used to calculate new centroids for each of the 5 possible subtype assignments. (XLSX 12 kb) (XLSX 12 KB)
12920_2012_315_MOESM5_ESM.xlsx
Additional file 5: Interference in subtype call and single/meta-gene scores from normal contamination. Interference by normal cell contamination of subtype call and single and meta-gene classes is shown. The changes in subtype classification occurred in a systematic fashion with all subtypes switching to a classification of Normal/Insufficient, with the exception of Luminal B, which switched to Luminal A. (XLSX 10 kb) (XLSX 10 KB)
12920_2012_315_MOESM6_ESM.pdf
Additional file 6: Single and meta-gene cutoffs. Data from single and meta-gene expression score over the GEICAM 9906 samples are plotted on the 1–10 scale. The cut-points between high, intermediate, and low classes were individually derived from the training set. Samples are color-coded according to immunohistochemistry positivity (red) or negativity (blue), except in the case of the training set proliferation score where samples are colored by high, intermediate or low proliferation class. Luminal score samples are colored as being ER+/PR+, ER + or PR + (positive, red), and ER-/PR- (negative, blue). (PDF 1524 kb) (PDF 1 MB)
12920_2012_315_MOESM7_ESM.pdf
Additional file 7: Hierarchical clustering for GEICAM 9906. A comparison of unsupervised hierarchical clustering with supervised subtype assignment and single marker scores for GEICAM 9906. (PDF 1616 kb) (PDF 2 MB)
12920_2012_315_MOESM8_ESM.xlsx
Additional file 8: PCR Efficiency, limits of detection, and limits of quantification. Supplemental table listing the efficiency of PCR, limits of detection, and limits of quantification for the 50 classifier and 5 housekeeper genes of the PAM50. Data are from 34 runs across 17 dilutions from a mixture of 8 breast cancer cell lines. (XLSX 11 KB)
12920_2012_315_MOESM9_ESM.xlsx
Additional file 9: Reproducibility of PAM50 gene measurements.Within plate, within plate batch and across plate batch coefficient of variation for the 50 classifier and 5 housekeeper genes of the PAM50 were calculated using cell lines and a tumor samples. (XLSX 11 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Bastien, R.R., Rodríguez-Lescure, Á., Ebbert, M.T. et al. PAM50 Breast Cancer Subtyping by RT-qPCR and Concordance with Standard Clinical Molecular Markers. BMC Med Genomics 5, 44 (2012). https://doi.org/10.1186/1755-8794-5-44
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1755-8794-5-44