
Abstract
Background
In cancer research, the association between a gene and clinical outcome suggests the underlying etiology of the disease and consequently can motivate further studies. The recent availability of published cancer microarray datasets with clinical annotation provides the opportunity for linking gene expression to prognosis. However, the data are not easy to access and analyze without an effective analysis platform.
Description
To take advantage of public resources in full, a database named "PrognoScan" has been developed. This is 1) a large collection of publicly available cancer microarray datasets with clinical annotation, as well as 2) a tool for assessing the biological relationship between gene expression and prognosis. PrognoScan employs the minimum P-value approach for grouping patients for survival analysis that finds the optimal cutpoint in continuous gene expression measurement without prior biological knowledge or assumption and, as a result, enables systematic meta-analysis of multiple datasets.
Conclusion
PrognoScan provides a powerful platform for evaluating potential tumor markers and therapeutic targets and would accelerate cancer research. The database is publicly accessible at http://gibk21.bse.kyutech.ac.jp/PrognoScan/index.html.
Similar content being viewed by others


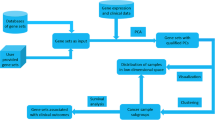
Background
A number of genes are recognized as being potentially relevant to cancers. One way to evaluate such genes is to assess their relationship to prognosis. At present, many cancer microarray datasets with clinical annotation have become available in the public domain and provide vast opportunities to link gene expression to prognosis. However, the data are not easy to access and analyze without an effective analysis platform.
Standard survival analysis consists of two steps: 1) grouping patients and 2) comparing the risk difference of the groups. When conducting survival analysis based on continuous measurement such as gene expression, determination of the appropriate cutpoints for groupings remains a critical and difficult task. Thus, although two pioneer databases, ITTACA [1] and REMBRANDT http://rembrandt.nci.nih.gov, have provided survival analysis functionality with user defined cutpoints for several focused cancer microarray datasets, researchers without prior biological knowledge or assumptions for the gene may end up using an arbitrary threshold (e.g. median, tertile, quartile) that does not necessarily reflect the biology of the gene or may laboriously test a number of possible cutpoints.
The minimum P-value approach is a comprehensive method to find the optimal risk separation cutpoint in continuous measurements and have shown the utility in the analyses of tumor size [2], cell cycle phase estimation measurement [3], and gene copy number [4]. In addition, it is intuitive for oncologists, and thus, a systematic application of this approach to gene expression from microarray seems logical. Recent studies have reported expression thresholds at which the gene becomes a contributor to the development of the cancer such as Bub1 for tumorigenesis [5], HOXB4 for cellular transformation [6], and MYC for tumor maintenance [7], and provided a rationale for the application to gene expression. Thus, we developed "PrognoScan", a database featuring a large collection of publicly available cancer microarray datasets with clinical annotation and a tool for assessing the relationship between gene expression and prognosis using the minimum P-value approach. This database enables systematic meta-analysis of the prognostic value of a gene in multiple datasets and consequently will accelerate cancer research.
Construction and content
Data collection
Cancer microarray datasets with clinical annotation were intensively collected from the public domain including Gene Expression Omnibus (GEO) [8], ArrayExpress [9] and individual laboratory web sites, under the following criteria: 1) includes patient information on survival event and time, 2) contains large enough sample sizes to enable survival analysis, 3) is derived from a 'whole genome' platform and has no values missing so quantile normalization will function properly and 4) is derived from a platform for which probe annotation for a public identifier (e.g. gene symbol, GenBank accession number, UniGene ID) is available. As of February 2009, the collection included more than 40 datasets of various cancer types spanning a wide range of cancers including bladder, blood, breast, brain, esophagus, head and neck, kidney, lung, and ovarian (Table 1) [10–35], far more comprehensive than both ITTACA, which focuses on bladder cancer, breast cancer and uveal melanoma, and REMBRANDT, which specializes in brain cancers. Because some samples were used more than once by more than one study, the origin of the samples was checked. Sample duplications within a dataset were dealt with by leaving one representative arbitrary. Sample overlaps among datasets were accepted, because the study design designated by each contributor may be of value. The collected microarray datasets were standardized by using quantile normalization. Probe annotations were retrieved from GEO and ArrayExpress. Each probe was mapped to an Entrez Gene ID by querying the accompanied public identifier in UniGene database. The information in the dataset was manually curated and includes 1) study design-cohort, cancer type, subtype, endpoint, therapy history and pathological parameters-and 2) experimental procedure-sample preparation, storage, array type and signal processing method. To assess prognostic value of genes in various contexts, available endpoints such as overall survival (OS), recurrence free survival (RFS), event free survival (EFS), and distant-metastasis free survival (DMFS) were adopted as much as possible. All tables were relationally linked and stored in the MySQL server.
Data analysis
Survival analysis in PrognoScan employs the minimum P-value approach [2] to find the cutpoint in continuous gene expression measurement for grouping patients. First, patients are ordered by expression value of a given gene. Next, patients are divided into two (high and low) expression groups at all potential cutpoint, and the risk differences of the two groups are estimated by log-rank test. Then, optimal cutpoint that gives the most pronounced P-value (P min) is selected.
This exploratory approach, however, is known to cause inflation of a type I error because it conducts multiple correlated testing [36–38]. Thus, P-value correction is conducted to control the error rate using the following formula [39].

where z is the (1 - P min /2)-quantile of the standard normal distribution, φ denotes the standard normal density function, and [ε, 1 - ε] denote the range of the quantile considered to be cutpoints. PrognoScan uses ε = 0.1 to avoid small groupings from cutpoints of < 0.1 or > 0.9 quantile. For any given gene, this cutpoint determination and prognostic value assessment can be applied to all possible combinations of dataset, endpoint and probe. For convenience, we term each combination as "test". Note that, because probe design for each gene differs, the number of possible tests varies according to the gene. For statistical analysis and visualization, R packages http://www.r-project.org are used.
Utility
The top page of PrognoScan is quite simple and the user need only input gene identifier(s) (Fig. 1A). To show the features of the database and its utility, we give three meta-analysis examples. The first example is MKI67, a well known tumor proliferation marker. The prognostic value of MKI67 protein expression has been reported for many types of malignant tumor including brain, breast, and lung cancer and a few exceptions for certain tumors such as non-Hodgkin's lymphoma [40]. When given the gene, PrognoScan displays a summary in table format of tests for the gene with columns for dataset, cancer type, subtype, endpoint, cohort, contributor, array type, probe ID, number of patient, optimal cutpoint, P min and P cor as Fig. 1B for MKI67 (shown in full in Additional file 1). In the table, 52 out of 152 tests showed an association between microarray expression and cancer prognosis (bladder 3/5, blood 6/28, breast 39/83, brain 3/8, esophagus 0/1, head and neck 0/4, kidney 0/1, lung 1/16, ovarian 0/6) with 5% significance level. Clicking the probe ID in the list reveals a detailed report, which includes further annotations for the dataset (Fig. 2A) and four intuitive visualization panels (Fig. 2B–E). The example of the Rotterdam cohort for DMFS depicts that patients can be dichotomized at the 34 percentile to give the minimum P-value and the group with high MKI67 expression has poorer survival (P cor = 0.0078). We found all tests but one for B-cell lymphoma OS showed a positive correlation to poorer survival, consistent with previous study results [40]. We further confirmed that the expressions of other well known proliferation markers TOP2A, PCNA and Aurora A also showed association with poorer survival in various tests (Additional file 2).

PrognoScan screenshot and sample search results (part 1). (A) The top page is quite simple and only requires entering the gene identifier(s). (B) Summary table for MKI67, shown here in part (See Additional file 1 for the full table.). Column headings include dataset, cancer type, subtype, endpoint, cohort, contributor, array type, probe ID, number of patients, optimal cutpoint, P min and P cor. A statistically significant value of P cor is given in red font. Each dataset has a link to the public domain where the raw data is archived. By clicking a probe ID in the summary table, a detailed report for the test is displayed. The table can be downloaded in a tab delimited file from the button at bottom.

PrognoScan screenshot and sample search results (part 2). (A) Annotation table. Row headings are color-coded. For example, headings of details such as therapy history, sample type and pathological parameters are highlighted in yellow and basic attributes in blue. (B) Expression plot. Patients are ordered by the expression values of the given gene. The X-axis represents the accumulative number of patients and the Y-axis represents the expression value. Straight lines (cyan) show the optimal cutpoints that dichotomize patients into high (red) and low (blue) expression groups. (C) Expression histogram. The distribution of the expression value is presented where the X-axis represents the number of patients and the Y-axis represents the expression value on the same scale as the expression plot. The line of the optimal cutpoint is also shown (cyan). (D) P-value plot. For each potential cutpoint of expression measurement, patients are dichotomized and survival difference between high and low expression groups is calculated by log-rank test. The X-axis represents the accumulative number of patients on the same scale as the expression plot and the Y-axis represents raw P-values on a log scale. The cutpoint to minimize the P-value is determined and indicated by the cyan line. The gray line indicates the 5% significance level. (E) Kaplan-Meier plot. Survival curves for high (red) and low (blue) expression groups dichotomized at the optimal cutpoint are plotted. The X-axis represents time and the Y-axis represents survival rate. 95% confidence intervals for each group are also indicated by dotted lines.
The second example is SIX1, emerging as a tumor-susceptible gene. This homeobox gene has been shown to promote tumor progression through direct activation of Cyclin A1 [41, 42] and to associate with prognosis of late-stage ovarian cancer [43] and hepatocellular carcinoma [44]. It has also been reported that SIX1 can be amplified and/or overexpressed in breast cancers [45, 46]. Nonetheless, to our knowledge, association with breast cancer prognosis has not yet been demonstrated. And so we tested SIX1. For ovarian cancer, a clear association was not observed in three tests available in PrognoScan. For this cancer type, further subgrouping based on stage may be needed, as reported [43]. On the other hand, SIX1 expression was positively associated with 5 out of 28 breast cancer tests (Fig. 3; Uppsala cohort; P cor = 0.0002, 0.0006 and 0.0449, Uppsala+Oxford cohort; P cor = 0.0346, Stockholm cohort; P cor = 0.0354) with statistical significance, indicative of its contribution to breast cancer malignancy. In addition, SIX1 expression showed nonsignificant trend toward worse prognosis in the GUYT2 and MGH cohorts (P cor = 0.0601, 0.0729, respectively). Using PrognoScan, SIX1 expression was correlated to breast cancer prognosis in multiple tests for the first time.

Kaplan-Meier plots for high and low SIX1-expressing groups in breast cancers.
The third example is MCTS1, a candidate oncogene amplified in T cell lymphoma. MCTS1 in a xenograft model causes transformation of NIH 3T3 mouse fibroblasts [47] and increases tumorgenicity by promoting angiogenesis and inhibiting apoptosis [48]. Similar to SIX1, prognostic analysis of this gene has not been reported for any cancers. PrognoScan depicted statistical significance in several tests: blood 2/7, breast 4/21, brain 1/2, lung 2/5 (Fig. 4). In all these 9 tests, a higher expression of MCTS1 associated with poorer survival, suggesting proactive involvement of this gene in the malignancy in the cancers. Again, this prognostic analysis was the first to show these relationships.

Kaplan-Meier plots for high and low MCTS1-expressing groups in breast, lung, blood and brain cancers.
Discussion and conclusion
PrognoScan is a database that focuses on the prognostic value of individual genes and differs conceptually from gene signatures. van't Veer et al. showed that the '70 gene signature' can predict risk of breast cancer recurrence, and that pattern analysis of multifactorial gene signature has greater potential for improving cancer subtype classification and risk prediction [49]. On the other hand, the prognostic value of an individual gene, for which pattern analysis is not applicable, suggests underlying relevance of the gene to cancer etiology and in turn stimulates research. With the number of public cancer microarray datasets with clinical annotation currently available, it is reasonable to utilize those assets to link gene expression to prognosis. Actually, Mehra et al., Paulson et al., and Kim et al. interrogated published cancer microarray datasets to evaluate targeted genes, GATA3, HBP1 and CUL7, respectively [50–52]. In this study, candidate oncogene SIX1 was correlated to breast cancer prognosis and MCTS1 to brain, blood, breast and lung cancer prognosis for the first time. PrognoScan aims to fulfill such substantial practical requirements.
Regarding survival analysis using publicly available microarray datasets, several considerations exist:
-
1)
Cohorts. Datasets come from a number of different institutions around the world, and patient backgrounds differ. In addition, several datasets are based on specific subpopulations, for example, dataset GSE2034 is from lymph node-negative breast cancers, and GSE5287 is from cisplatin-containing chemotherapy-treated bladder cancers. Hence, it is possible that the specific association between gene expression and prognosis is found in a certain cohort. To give an example, Dai et al. reported that cell cycle genes are highly prognostic in groups with high ER expression for their age but less or nonprognostic in other groups [53].
-
2)
Quality of care. It has been reported that the hospital itself could be a factor in clinical outcome [54–56]. This means, even if cohorts were equivalent at the time of profiling, subsequent care may affect the clinical course of a patient.
-
3)
Experimental factors. Expression measurement of microarray is subject to various factors at the experiment level. Microdissection (e.g. GSE1378) would reduce contamination of mRNAs from non-cancer cells [57]. Formalin fixation of a sample (e.g. GSE2873) influences the quality of mRNAs [58]. Array type (e.g. Affymetrix, cDNA microarrays) and data processing method (e.g. MAS, RMA) can also influence gene expression measurements [59]. In addition, it is known that a substantial number of incorrect probes are used in microarrays [60].
-
4)
Random error. Even though there may be no relation between a gene expression and prognosis, false positives may be detected by chance.
Thus, one needs to regard the results from PrognoScan in the context of complex conditions. Currently, PrognoScan provides curated information such as cohort, therapy history, pathological parameters and array type to aid in the interpretation of the results. As a next step, developing an "interpreter" for complex meta-analysis result is tempting and we are now contemplating the challenge. In the meantime, we will continue collecting published datasets and will update PrognoScan every 6 months. Increased data content will help the judgment of the robustness of the prognostic value of a gene.
Further plans for PrognoScan also include development of the algorithm for finding multiple cutpoints. From the limited computational resources, cutpoint selection is currently done for two-way (high and low) expression grouping. For clinical practice, three-way (high, intermediate, and low) expression grouping can also be used. Thus, we are trying to develop a grid search algorithm, demonstrated as the "X-Tile" tool [61]. In summary, this new database provides a powerful platform for evaluating potential tumor markers and therapeutic targets, and as a result, will accelerate cancer research.
Availability and requirements
PrognoScan requires nothing other than a web browser and is available from the server at Kyushu Institute of Technology (KIT): http://gibk21.bse.kyutech.ac.jp/PrognoScan/index.html.
References
Elfilali A, Lair S, Verbeke C, La Rosa P, Radvanyi F, Barillot E: ITTACA: a new database for integrated tumor transcriptome array and clinical data analysis. Nucleic Acids Res. 2006, 34: D613-6. 10.1093/nar/gkj022.
Abel U, Berger J, Wiebelt H: CRITLEVEL: an exploratory procedure for the evaluation of quantitative prognostic factors. Methods Inf Med. 1984, 23: 154-6.
Kronqvist P, Kuopio T, Collan Y: Quantitative thresholds for mitotic counts in histologic grading: confirmation in nonfrozen samples of invasive ductal breast cancer. Ann Diagn Pathol. 2000, 4: 65-70. 10.1016/S1092-9134(00)90013-3.
Jensen KC, Turbin DA, Leung S, Miller MA, Johnson K, Norris B, Hastie T, McKinney S, Nielsen TO, Huntsman DG, Gilks CB, West RB: New cutpoints to identify increased HER2 copy number: analysis of a large, population-based cohort with long-term follow-up. Breast Cancer Res Treat. 2008, 112: 453-9. 10.1007/s10549-007-9887-y.
Jeganathan K, Malureanu L, Baker DJ, Abraham SC, van Deursen JM: Bub1 mediates cell death in response to chromosome missegregation and acts to suppress spontaneous tumorigenesis. J Cell Biol. 2007, 179: 255-67. 10.1083/jcb.200706015.
Will E, Speidel D, Wang Z, Ghiaur G, Rimek A, Schiedlmeier B, Williams DA, Baum C, Ostertag W, Klump H: HOXB4 inhibits cell growth in a dose-dependent manner and sensitizes cells towards extrinsic cues. Cell Cycle. 2006, 5: 14-22.
Shachaf CM, Gentles AJ, Elchuri S, Sahoo D, Soen Y, Sharpe O, Perez OD, Chang M, Mitchel D, Robinson WH, Dill D, Nolan GP, Plevritis SK, Felsher DW: Genomic and proteomic analysis reveals a threshold level of MYC required for tumor maintenance. Cancer Res. 2008, 68: 5132-42. 10.1158/0008-5472.CAN-07-6192.
Barrett T, Troup DB, Wilhite SE, Ledoux P, Rudnev D, Evangelista C, Kim IF, Soboleva A, Tomashevsky M, Edgar R: NCBI GEO: mining tens of millions of expression profiles – database and tools update. Nucleic Acids Res. 2007, 35: D760-5. 10.1093/nar/gkl887.
Parkinson H, Kapushesky M, Shojatalab M, Abeygunawardena N, Coulson R, Farne A, Holloway E, Kolesnykov N, Lilja P, Lukk M, Mani R, Rayner T, Sharma A, William E, Sarkans U, Brazma A: ArrayExpress – a public database of microarray experiments and gene expression profiles. Nucleic Acids Res. 2007, 35: D747-50. 10.1093/nar/gkl995.
Als AB, Dyrskjøt L, Maase von der H, Koed K, Mansilla F, Toldbod HE, Jensen JL, Ulhøi BP, Sengeløv L, Jensen KM, Orntoft TF: Emmprin and survivin predict response and survival following cisplatin-containing chemotherapy in patients with advanced bladder cancer. Clin Cancer Res. 2007, 13: 4407-14. 10.1158/1078-0432.CCR-07-0109.
Metzeler KH, Hummel M, Bloomfield CD, Spiekermann K, Braess J, Sauerland MC, Heinecke A, Radmacher M, Marcucci G, Whitman SP, Maharry K, Paschka P, Larson RA, Berdel WE, Büchner T, Wörmann B, Mansmann U, Hiddemann W, Bohlander SK, Buske C, Cancer and Leukemia Group B; German AML Cooperative Group: An 86-probe-set gene-expression signature predicts survival in cytogenetically normal acute myeloid leukemia. Blood. 2008, 112: 4193-201. 10.1182/blood-2008-02-134411.
Raponi M, Lancet JE, Fan H, Dossey L, Lee G, Gojo I, Feldman EJ, Gotlib J, Morris LE, Greenberg PL, Wright JJ, Harousseau JL, Löwenberg B, Stone RM, De Porre P, Wang Y, Karp JE: A 2-gene classifier for predicting response to the farnesyltransferase inhibitor tipifarnib in acute myeloid leukemia. Blood. 2008, 111: 2589-96. 10.1182/blood-2007-09-112730.
Hummel M, Bentink S, Berger H, Klapper W, Wessendorf S, Barth TF, Bernd HW, Cogliatti SB, Dierlamm J, Feller AC, Hansmann ML, Haralambieva E, Harder L, Hasenclever D, Kühn M, Lenze D, Lichter P, Martin-Subero JI, Möller P, Müller-Hermelink HK, Ott G, Parwaresch RM, Pott C, Rosenwald A, Rosolowski M, Schwaenen C, Stürzenhofecker B, Szczepanowski M, Trautmann H, Wacker HH, Spang R, Loeffler M, Trümper L, Stein H, Siebert R, Molecular Mechanisms in Malignant Lymphomas Network Project of the Deutsche Krebshilfe: A biologic definition of Burkitt's lymphoma from transcriptional and genomic profiling. N Engl J Med. 2006, 354: 2419-30. 10.1056/NEJMoa055351.
Jais JP, Haioun C, Molina TJ, Rickman DS, de Reynies A, Berger F, Gisselbrecht C, Brière J, Reyes F, Gaulard P, Feugier P, Labouyrie E, Tilly H, Bastard C, Coiffier B, Salles G, Leroy K, Groupe d'Etude des Lymphomes de l'Adulte: The expression of 16 genes related to the cell of origin and immune response predicts survival in elderly patients with diffuse large B-cell lymphoma treated with CHOP and rituximab. Leukemia. 2008, 22: 1917-24. 10.1038/leu.2008.188.
Zhan F, Huang Y, Colla S, Stewart JP, Hanamura I, Gupta S, Epstein J, Yaccoby S, Sawyer J, Burington B, Anaissie E, Hollmig K, Pineda-Roman M, Tricot G, van Rhee F, Walker R, Zangari M, Crowley J, Barlogie B, Shaughnessy JD: The molecular classification of multiple myeloma. Blood. 2006, 108: 2020-8. 10.1182/blood-2005-11-013458.
Chin K, DeVries S, Fridlyand J, Spellman PT, Roydasgupta R, Kuo WL, Lapuk A, Neve RM, Qian Z, Ryder T, Chen F, Feiler H, Tokuyasu T, Kingsley C, Dairkee S, Meng Z, Chew K, Pinkel D, Jain A, Ljung BM, Esserman L, Albertson DG, Waldman FM, Gray JW: Genomic and transcriptional aberrations linked to breast cancer pathophysiologies. Cancer Cell. 2006, 10: 529-41. 10.1016/j.ccr.2006.10.009.
Schmidt M, Böhm D, von Törne C, Steiner E, Puhl A, Pilch H, Lehr HA, Hengstler JG, Kölbl H, Gehrmann M: The humoral immune system has a key prognostic impact in node-negative breast cancer. Cancer Res. 2008, 68: 5405-13. 10.1158/0008-5472.CAN-07-5206.
Ma XJ, Wang Z, Ryan PD, Isakoff SJ, Barmettler A, Fuller A, Muir B, Mohapatra G, Salunga R, Tuggle JT, Tran Y, Tran D, Tassin A, Amon P, Wang W, Wang W, Enright E, Stecker K, Estepa-Sabal E, Smith B, Younger J, Balis U, Michaelson J, Bhan A, Habin K, Baer TM, Brugge J, Haber DA, Erlander MG, Sgroi DC: A two-gene expression ratio predicts clinical outcome in breast cancer patients treated with tamoxifen. Cancer Cell. 2004, 5: 607-16. 10.1016/j.ccr.2004.05.015.
Pawitan Y, Bjöhle J, Amler L, Borg AL, Egyhazi S, Hall P, Han X, Holmberg L, Huang F, Klaar S, Liu ET, Miller L, Nordgren H, Ploner A, Sandelin K, Shaw PM, Smeds J, Skoog L, Wedrén S, Bergh J: Gene expression profiling spares early breast cancer patients from adjuvant therapy: derived and validated in two population-based cohorts. Breast Cancer Res. 2005, 7: R953-64. 10.1186/bcr1325.
Wang Y, Klijn JG, Zhang Y, Sieuwerts AM, Look MP, Yang F, Talantov D, Timmermans M, Meijer-van Gelder ME, Yu J, Jatkoe T, Berns EM, Atkins D, Foekens JA: Gene-expression profiles to predict distant metastasis of lymph-node-negative primary breast cancer. Lancet. 2005, 365: 671-9.
Sotiriou C, Wirapati P, Loi S, Harris A, Fox S, Smeds J, Nordgren H, Farmer P, Praz V, Haibe-Kains B, Desmedt C, Larsimont D, Cardoso F, Peterse H, Nuyten D, Buyse M, Vijver Van de MJ, Bergh J, Piccart M, Delorenzi M: Gene expression profiling in breast cancer: understanding the molecular basis of histologic grade to improve prognosis. J Natl Cancer Inst. 2006, 98: 262-72.
Bild AH, Yao G, Chang JT, Wang Q, Potti A, Chasse D, Joshi MB, Harpole D, Lancaster JM, Berchuck A, Olson JA, Marks JR, Dressman HK, West M, Nevins JR: Oncogenic pathway signatures in human cancers as a guide to targeted therapies. Nature. 2006, 439: 353-7. 10.1038/nature04296.
Miller LD, Smeds J, George J, Vega VB, Vergara L, Ploner A, Pawitan Y, Hall P, Klaar S, Liu ET, Bergh J: An expression signature for p53 status in human breast cancer predicts mutation status, transcriptional effects, and patient survival. Proc Natl Acad Sci USA. 2005, 102: 13550-5. 10.1073/pnas.0506230102.
Ivshina AV, George J, Senko O, Mow B, Putti TC, Smeds J, Lindahl T, Pawitan Y, Hall P, Nordgren H, Wong JE, Liu ET, Bergh J, Kuznetsov VA, Miller LD: Genetic reclassification of histologic grade delineates new clinical subtypes of breast cancer. Cancer Res. 2006, 66: 10292-301. 10.1158/0008-5472.CAN-05-4414.
Loi S, Haibe-Kains B, Desmedt C, Wirapati P, Lallemand F, Tutt AM, Gillet C, Ellis P, Ryder K, Reid JF, Daidone MG, Pierotti MA, Berns EM, Jansen MP, Foekens JA, Delorenzi M, Bontempi G, Piccart MJ, Sotiriou C: Predicting prognosis using molecular profiling in estrogen receptor-positive breast cancer treated with tamoxifen. BMC Genomics. 2008, 9: 239-10.1186/1471-2164-9-239.
Zhou Y, Yau C, Gray JW, Chew K, Dairkee SH, Moore DH, Eppenberger U, Eppenberger-Castori S, Benz CC: Enhanced NF kappa B and AP-1 transcriptional activity associated with antiestrogen resistant breast cancer. BMC Cancer. 2007, 7: 59-10.1186/1471-2407-7-59.
Desmedt C, Piette F, Loi S, Wang Y, Lallemand F, Haibe-Kains B, Viale G, Delorenzi M, Zhang Y, d'Assignies MS, Bergh J, Lidereau R, Ellis P, Harris AL, Klijn JG, Foekens JA, Cardoso F, Piccart MJ, Buyse M, Sotiriou C, TRANSBIG Consortium: Strong time dependence of the 76-gene prognostic signature for node-negative breast cancer patients in the TRANSBIG multicenter independent validation series. Clin Cancer Res. 2007, 13: 3207-14. 10.1158/1078-0432.CCR-06-2765.
Anders CK, Acharya CR, Hsu DS, Broadwater G, Garman K, Foekens JA, Zhang Y, Wang Y, Marcom K, Marks JR, Mukherjee S, Nevins JR, Blackwell KL, Potti A: Age-specific differences in oncogenic pathway deregulation seen in human breast tumors. PLoS ONE. 2008, 3: e1373-10.1371/journal.pone.0001373.
Chanrion M, Negre V, Fontaine H, Salvetat N, Bibeau F, Mac Grogan G, Mauriac L, Katsaros D, Molina F, Theillet C, Darbon JM: A gene expression signature that can predict the recurrence of tamoxifen-treated primary breast cancer. Clin Cancer Res. 2008, 14: 1744-52. 10.1158/1078-0432.CCR-07-1833.
Murat A, Migliavacca E, Gorlia T, Lambiv WL, Shay T, Hamou MF, de Tribolet N, Regli L, Wick W, Kouwenhoven MC, Hainfellner JA, Heppner FL, Dietrich PY, Zimmer Y, Cairncross JG, Janzer RC, Domany E, Delorenzi M, Stupp R, Hegi ME: Stem cell-related "self-renewal" signature and high epidermal growth factor receptor expression associated with resistance to concomitant chemoradiotherapy in glioblastoma. J Clin Oncol. 2008, 26: 3015-24. 10.1200/JCO.2007.15.7164.
Phillips HS, Kharbanda S, Chen R, Forrest WF, Soriano RH, Wu TD, Misra A, Nigro JM, Colman H, Soroceanu L, Williams PM, Modrusan Z, Feuerstein BG, Aldape K: Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell. 2006, 9: 157-73. 10.1016/j.ccr.2006.02.019.
Chung CH, Parker JS, Ely K, Carter J, Yi Y, Murphy BA, Ang KK, El-Naggar AK, Zanation AM, Cmelak AJ, Levy S, Slebos RJ, Yarbrough WG: Gene expression profiles identify epithelial-to-mesenchymal transition and activation of nuclear factor-kappaB signaling as characteristics of a high-risk head and neck squamous cell carcinoma. Cancer Res. 2006, 66: 8210-8. 10.1158/0008-5472.CAN-06-1213.
Beer DG, Kardia SL, Huang CC, Giordano TJ, Levin AM, Misek DE, Lin L, Chen G, Gharib TG, Thomas DG, Lizyness ML, Kuick R, Hayasaka S, Taylor JM, Iannettoni MD, Orringer MB, Hanash S: Gene-expression profiles predict survival of patients with lung adenocarcinoma. Nat Med. 2002, 8: 816-24.
Tomida S, Koshikawa K, Yatabe Y, Harano T, Ogura N, Mitsudomi T, Some M, Yanagisawa K, Takahashi T, Osada H, Takahashi T: Gene expression-based, individualized outcome prediction for surgically treated lung cancer patients. Oncogene. 2004, 23: 5360-70. 10.1038/sj.onc.1207697.
Raponi M, Zhang Y, Yu J, Chen G, Lee G, Taylor JM, Macdonald J, Thomas D, Moskaluk C, Wang Y, Beer DG: Gene expression signatures for predicting prognosis of squamous cell and adenocarcinomas of the lung. Cancer Res. 2006, 66: 7466-72. 10.1158/0008-5472.CAN-06-1191.
Altman DG, Lausen B, Sauerbrei W, Schumacher M: Dangers of using "optimal" cutpoints in the evaluation of prognostic factors. J Natl Cancer Inst. 1994, 86: 829-35. 10.1093/jnci/86.11.829.
Mazumdar M, Glassman JR: Categorizing a prognostic variable: review of methods, code for easy implementation and applications to decision-making about cancer treatments. Stat Med. 2000, 19: 113-32. 10.1002/(SICI)1097-0258(20000115)19:1<113::AID-SIM245>3.0.CO;2-O.
Holländer N, Schumacher M: On the problem of using 'optimal' cutpoints in the assessment of quantitative prognostic factors. Onkologie. 2001, 24: 194-9. 10.1159/000050315.
Miller R, Siegmund D: Maximally selected chi square statistics. Biometrics. 1982, 38: 1011-16. 10.2307/2529881.
Brown DC, Gatter KC: Ki67 protein: the immaculate deception?. Histopathology. 2002, 40: 2-11. 10.1046/j.1365-2559.2002.01343.x.
Coletta RD, Christensen K, Reichenberger KJ, Lamb J, Micomonaco D, Huang L, Wolf DM, Muller-Tidow C, Golub TR, Kawakami K, Ford HL: The Six1 homeoprotein stimulates tumorigenesis by reactivation of cyclin A1. Proc Natl Acad Sci USA. 2004, 101: 6478-83. 10.1073/pnas.0401139101.
Coletta RD, Christensen KL, Micalizzi DS, Jedlicka P, Varella-Garcia M, Ford HL: Six1 overexpression in mammary cells induces genomic instability and is sufficient for malignant transformation. Cancer Res. 2008, 68: 2204-13. 10.1158/0008-5472.CAN-07-3141.
Behbakht K, Qamar L, Aldridge CS, Coletta RD, Davidson SA, Thorburn A, Ford HL: Six1 overexpression in ovarian carcinoma causes resistance to TRAIL-mediated apoptosis and is associated with poor survival. Cancer Res. 2007, 67: 3036-42. 10.1158/0008-5472.CAN-06-3755.
Ng KT, Man K, Sun CK, Lee TK, Poon RT, Lo CM, Fan ST: Clinicopathological significance of homeoprotein Six1 in hepatocellular carcinoma. Br J Cancer. 2006, 95: 1050-5. 10.1038/sj.bjc.6603399.
Ford HL, Kabingu EN, Bump EA, Mutter GL, Pardee AB: Abrogation of the G2 cell cycle checkpoint associated with overexpression of HSIX1: a possible mechanism of breast carcinogenesis. Proc Natl Acad Sci USA. 1998, 95: 12608-13. 10.1073/pnas.95.21.12608.
Reichenberger KJ, Coletta RD, Schulte AP, Varella-Garcia M, Ford HL: Gene amplification is a mechanism of Six1 overexpression in breast cancer. Cancer Res. 2005, 65: 2668-75. 10.1158/0008-5472.CAN-04-4286.
Prosniak M, Dierov J, Okami K, Tilton B, Jameson B, Sawaya BE, Gartenhaus RB: A novel candidate oncogene, MCT-1, is involved in cell cycle progression. Cancer Res. 1998, 58: 4233-7.
Levenson AS, Thurn KE, Simons LA, Veliceasa D, Jarrett J, Osipo C, Jordan VC, Volpert OV, Satcher RL, Gartenhaus RB: MCT-1 oncogene contributes to increased in vivo tumorigenicity of MCF7 cells by promotion of angiogenesis and inhibition of apoptosis. Cancer Res. 2005, 65: 10651-6. 10.1158/0008-5472.CAN-05-0845.
van't Veer LJ, Dai H, Vijver van de MJ, He YD, Hart AA, Mao M, Peterse HL, Kooy van der K, Marton MJ, Witteveen AT, Schreiber GJ, Kerkhoven RM, Roberts C, Linsley PS, Bernards R, Friend SH: Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002, 415: 530-6. 10.1038/415530a.
Mehra R, Varambally S, Ding L, Shen R, Sabel MS, Ghosh D, Chinnaiyan AM, Kleer CG: Identification of GATA3 as a breast cancer prognostic marker by global gene expression meta-analysis. Cancer Res. 2005, 65: 11259-64. 10.1158/0008-5472.CAN-05-2495.
Paulson KE, Rieger-Christ K, McDevitt MA, Kuperwasser C, Kim J, Unanue VE, Zhang X, Hu M, Ruthazer R, Berasi SP, Huang CY, Giri D, Kaufman S, Dugan JM, Blum J, Netto G, Wazer DE, Summerhayes IC, Yee AS: Alterations of the HBP1 transcriptional repressor are associated with invasive breast cancer. Cancer Res. 2007, 67: 6136-45. 10.1158/0008-5472.CAN-07-0567.
Kim SS, Shago M, Kaustov L, Boutros PC, Clendening JW, Sheng Y, Trentin GA, Barsyte-Lovejoy D, Mao DY, Kay R, Jurisica I, Arrowsmith CH, Penn LZ: CUL7 is a novel antiapoptotic oncogene. Cancer Res. 2007, 67: 9616-22. 10.1158/0008-5472.CAN-07-0644.
Dai H, van't Veer L, Lamb J, He YD, Mao M, Fine BM, Bernards R, Vijver van de M, Deutsch P, Sachs A, Stoughton R, Friend S: A cell proliferation signature is a marker of extremely poor outcome in a subpopulation of breast cancer patients. Cancer Res. 2005, 65: 4059-66. 10.1158/0008-5472.CAN-04-3953.
Schrag D, Cramer LD, Bach PB, Cohen AM, Warren JL, Begg CB: Influence of hospital procedure volume on outcomes following surgery for colon cancer. JAMA. 2000, 284: 3028-35. 10.1001/jama.284.23.3028.
Bilimoria KY, Bentrem DJ, Ko CY, Tomlinson JS, Stewart AK, Winchester DP, Talamonti MS: Multimodality therapy for pancreatic cancer in the U.S. Cancer. 2007, 110: 1227-34. 10.1002/cncr.22916.
Vernooij F, Heintz AP, Witteveen PO, Heiden-van der Loo van der M, Coebergh JW, Graaf van der Y: Specialized care and survival of ovarian cancer patients in The Netherlands: nationwide cohort study. J Natl Cancer Inst. 2008, 100: 399-406. 10.1093/jnci/djn033.
Mizuarai S, Takahashi K, Kobayashi T, Kotani H: Advances in isolation and characterization of homogeneous cell populations using laser microdissection. Histol Histopathol. 2005, 20: 139-46.
Mazumder A, Wang Y: Gene-expression signatures in oncology diagnostics. Pharmacogenomics. 2006, 7: 1167-73. 10.2217/14622416.7.8.1167.
Irizarry RA, Bolstad BM, Collin F, Cope LM, Hobbs B, Speed TP: Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 2003, 31: e15-10.1093/nar/gng015.
Draghici S, Khatri P, Eklund AC, Szallasi Z: Reliability and reproducibility issues in DNA microarray measurements. Trends Genet. 2006, 22: 101-9. 10.1016/j.tig.2005.12.005.
Camp RL, Dolled-Filhart M, Rimm DL: X-tile: a new bio-informatics tool for biomarker assessment and outcome-based cut-point optimization. Clin Cancer Res. 2004, 10: 7252-9. 10.1158/1078-0432.CCR-04-0713.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1755-8794/2/18/prepub
Acknowledgements
We thank K. Kitajima of KIT for his technical support for database development and D. Schmitt and F. Ford of Chugai Pharmaceuticals for their helpful discussions and checking of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
HM and AS designed the database. KK and KN aided in the conception and design of the database. HM and KK participated in writing the manuscript. All authors read and approved the final manuscript.
Electronic supplementary material
12920_2008_82_MOESM1_ESM.xls
Additional file 1: Full summary table for MKI67. A well known tumor proliferation marker MKI67 was assessed with PrognoScan and the summary table was indicated. (XLS 182 KB)
12920_2008_82_MOESM2_ESM.xls
Additional file 2: Number of statistically significant tests for four proliferation markers among nine cancer types. Tumor proliferation markers, TOP2A, PCNA and Aurora A were assessed with PrognoScan. Together with the result for MKI67, associations with nine cancer types were indicated. (XLS 18 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Mizuno, H., Kitada, K., Nakai, K. et al. PrognoScan: a new database for meta-analysis of the prognostic value of genes. BMC Med Genomics 2, 18 (2009). https://doi.org/10.1186/1755-8794-2-18
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1755-8794-2-18