
Abstract
Background
Mycobacterium tuberculosis, the causative agent of tuberculosis (TB), infects ~8 million annually culminating in ~2 million deaths. Moreover, about one third of the population is latently infected, 10% of which develop disease during lifetime. Current approved prophylactic TB vaccines (BCG and derivatives thereof) are of variable efficiency in adult protection against pulmonary TB (0%–80%), and directed essentially against early phase infection.
Methods
A genome-scale dataset was constructed by analyzing published data of: (1) global gene expression studies under conditions which simulate intra-macrophage stress, dormancy, persistence and/or reactivation; (2) cellular and humoral immunity, and vaccine potential. This information was compiled along with revised annotation/bioinformatic characterization of selected gene products and in silico mapping of T-cell epitopes. Protocols for scoring, ranking and prioritization of the antigens were developed and applied.
Results
Cross-matching of literature and in silico-derived data, in conjunction with the prioritization scheme and biological rationale, allowed for selection of 189 putative vaccine candidates from the entire genome. Within the 189 set, the relative distribution of antigens in 3 functional categories differs significantly from their distribution in the whole genome, with reduction in the Conserved hypothetical category (due to improved annotation) and enrichment in Lipid and in Virulence categories. Other prominent representatives in the 189 set are the PE/PPE proteins; iron sequestration, nitroreductases and proteases, all within the Intermediary metabolism and respiration category; ESX secretion systems, resuscitation promoting factors and lipoproteins, all within the Cell wall category. Application of a ranking scheme based on qualitative and quantitative scores, resulted in a list of 45 best-scoring antigens, of which: 74% belong to the dormancy/reactivation/resuscitation classes; 30% belong to the Cell wall category; 13% are classical vaccine candidates; 9% are categorized Conserved hypotheticals, all potentially very potent T-cell antigens.
Conclusion
The comprehensive literature and in silico-based analyses allowed for the selection of a repertoire of 189 vaccine candidates, out of the whole-genome 3989 ORF products. This repertoire, which was ranked to generate a list of 45 top-hits antigens, is a platform for selection of genes covering all stages of M. tuberculosis infection, to be incorporated in rBCG or subunit-based vaccines.
Similar content being viewed by others


Background
Mycobacterium tuberculosis (Mtb), the causative agent of tuberculosis (TB), remains a major health threat. Each year, 8 million new TB cases occur and 2 million individuals die of TB [1]. Moreover, it is estimated that one third of the population is latently infected with Mtb, of which ~10% will develop active disease during lifetime. The development of active TB occurs when the balance between natural immunity and the pathogen changes (e.g. upon waning of protective immune response during adolescence and in HIV patients, [2]). In addition, at present ~50 million individuals are probably infected with multi drug-resistance (MDR) strains of Mtb (WHO, 2006), rendering antibiotic treatment difficult.
The current vaccine, introduced over 80 years ago, is the live attenuated bacterium Mycobacterium bovis Bacillus Calmette-Geurin (BCG), designed as a prophylactic vaccine for pre-infection administration. BCG is known to protect young children against severe forms of TB however it does not efficiently and consistently protect adults against the most prevalent form of the disease, namely, pulmonary TB (variable protective efficacy ranges from 0% to 80%), nor does BCG offer protection from establishment of latent TB and subsequent reactivation [3–9].
In principle, current putative vaccination strategies against TB can be divided into two groups: (a) prophylactic vaccines (aimed at disease prevention), based on BCG with or without antigens secreted by replicating bacteria recognized during the early stage of infection (e.g. ESAT-6 or Ag85A/B), or protective mutants (live attenuated BCG substitutes) and (b) as yet undefined post-exposure vaccines (boosting BCG) aimed at elimination/containment of latent TB and prevention of reactivation. Ideally, a prime-boost approach comprising of a prophylactic vaccine with subunit, viral vectored or DNA-based vaccines, based on late-stage antigens induced in the dormant stage (transition from replicating to non-replicating stage, latency antigens) or resuscitation/reactivation stage, should have maximum impact on all stages of Mtb infection [2, 6, 10–15].
In the past few years there have been important breakthroughs in the development of improved prophylactic TB vaccines. Novel vaccine candidates, mostly selected as single gene products, include: rBCG vaccines (e.g. rBCG30 – expressing Ag85B or ΔureCHly+rBCG – an urease deficient strain expressing listeriolysin), virus-based recombinant vaccines (e.g. MVA85A – Rv3804c expressed in replication-deficient vaccinia virus), live attenuated Mtb strains and subunit vaccines comprising of dominant secreted antigens which could boost the immune response after priming with BCG (e.g. Mtb72F, Ag85B-ESAT-6 fusion), (reviewed by [1, 4–7, 10, 16]).
Post-exposure vaccine development requires identification of gene products participating in adaptation of Mtb to the intracellular habitat as Mtb changes from replication to dormancy or from dormancy to resuscitation. Moreover, in the absence of effective prediction models and animal models assessing protective immunity, evaluation of a large number of individual antigens remains laborious [12]. In vitro gene expression studies under conditions which mimic dormancy and/or reactivation constitute the major source of information. Availability of whole genome sequences of diverse mycobacterial strains in conjunction with data from global analyses such as whole-genome DNA microarray and proteomic technologies have been the subject of intensive research in the recent years, both at site of pulmonary TB and in ex vivo macrophages [11, 17]. To identify genes expressed specifically during latency, different in vitro conditions have been suggested to model the harsh environment within macrophages or granulomas. These include oxygen deprivation, nutrient starvation and iron limitation. Together with expression studies in lungs/granulomas, in phagocytized bacteria inside activated macrophages or in murine/guinea pig infection models, these datasets provide better understanding of the transition of bacteria through stages of active multiplication, dormancy and resuscitation. Based on bioinformatic studies and analysis of multiple dormancy-related datasets, novel drug targets against the dormant phase of Mtb infection have been recently identified [18].
Several attempts have been made to modify the immunogenicity or antigenicity of BCG by generating recombinant strains expressing cytokines, pore-forming listeriolysin/perfringolysin, immunodominant antigens or additional antigens missing from the genome of the avirulent M. bovis BCG genome. Over 200 genes located in 14 gene segments assigned to defined regions of difference (RDs) are missing in the vaccine strain. In principle, potency of BCG vaccines could be improved by supplementing with missing immunodominant RD genes; however critical evaluation is required since RD regions are associated with virulence. Indeed, when BCG was supplemented by the classical RD-1 antigen ESAT-6 (with or without Ag85B [19]), enhanced protection was observed in mice however the recombinant strain was more virulent as compared to the wild-type. In a more recent study, Kalra & Grover [20] demonstrated the enhanced prophylactic potential of selected combinations of RD antigens supplementing BCG, improving protection in aerosol exposed mice.
Mtb is an intracellular pathogen and as such cell mediated immunity rather than antibody-mediated immunity is essential for the control of bacterial replication and subsequent protection against TB [7]. It is thought that a coordinated response of the cellular immune response is fundamental to the protective immunity [21–23], including both CD4 and CD8 T-cells and several cytokines such as IFNγ and TNFα. It has been suggested that CD4 T-cells are mainly crucial during the early phases of infection, while the CD8 T-cells play an important role in the chronic phase of the disease [21–23], however little is known about antigen-specific human T-cell responses against persisting mycobacteria, particularly in the context of latent infection and protection against disease, with the exception of some products of the DosR regulon [24, 25]. Therefore, rational selection of sequences that may function as T-cell epitopes in vaccine formulations is crucial. Relatively few human CD8 T-cell epitopes have been found by conventional methods. The availability of the genomic sequence [26] provides new horizons in analyzing the potential immunome of the bacilli, using in silico identification of CTL binders. Several prediction screens of Mtb T-cell epitopes were reported to date [25, 27–32], nevertheless these analyses were mostly restricted either to few MHC alleles or to a limited number of preselected subset of genes.
In this study, we have combined multiple published datasets from: (a) gene expression and DNA microarray experiments mimicking conditions leading to dormancy or at the dormant state; (b) genome-wide insertional mutagenesis examining gene essentiality under different conditions; (c) genes expressed in macrophages under conditions mimicking persistence; (d) genes expressed in lungs/granulomas; (e) expression under iron limiting conditions; (f) identification of genes products reported to elicit humoral and cellular response or potential vaccine candidates; (g) in silico identification of T-cell epitopes by dedicated epitope-mapping algorithms and databases compiling relevant experimental data. We designed a whole-genome scoring, ranking and prioritization algorithm and used it to analyze the combined dataset. Accordingly, we selected a set of 189 putative vaccine candidate proteins covering all disease phases, from which we further derived a shorter list of 45 top-ranking antigens for vaccine studies.
Methods
Genome sequences and annotations of Mycobacterium tuberculosis H37Rv [GenBank: AL123456] were downloaded from the NCBI [33]. Sequence similarity searches were conducted using the Blast algorithm, vs. the non-redundant (nr, NCBI) database. Comparisons against secondary databases of domains and motifs were performed as follows: CDD [34], SMART [35], Pfam [36], Interpro [37], Prosite [38]. Secretion signals were analyzed by SignalP [39, 40] for the secretion via the SecA pathway, and TATfind for searching of the Twin-Arginine Translocation (Tat) motif of the TAT pathway [41]. Transmembrane helical segments were predicted by Tmpred [42]. The database of bacterial lipoproteins, Dolop [43] was searched for putative lipoproteins. Predictions of CTL epitopes were carried out using the NetCTL integrative approach [44], which combines predictions of MHC class I binding, TAP transport efficiency, and proteasomal cleavage. The analysis was conducted for each of 12 HLA supertypes. A threshold of 1.25 was set on the combined prediction score. The results were parsed by in-house perl scripts. Immune epitope information was complemented by querying the IEDB database [45] for documented B- and T-cell epitopes. The following Mtb-related servers and databases were screened for additive relevant data: TB-sgc – The TB Structural Genomics Consortium [46], Tuberculist-the database on Mycobacterium tuberculosis genetics [47], TBDB – an integrated platform for TB drug discovery [48], MTBreg – The Database of Conditionally Regulated Proteins in Mycobacterium tuberculosis [49] and BioHealthBase – the Biodefense and Public Health Database [50]. Functional categories provided for the selected ORF products were as implemented in [26, 51], and their assignment to each of the antigen was derived from the Tuberculist database (see above). Classes/phases of Mtb infection were assigned as well to the selected antigens: the well established classical antigens, DosR-regulated antigens (as reported in [52, 53]), reactivation antigens (antigens listed by Talaat and his coworkers [54], as well as documented putative implications of individual antigens), and the 5 known resuscitation antigens [55]; antigens not belonging to any of the above classes, were denoted as "Others". p-values were calculated using the binomical distribution of frequencies.
Results and Discussion
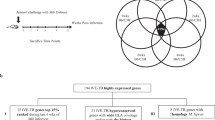
The strategy for whole genome-based selection of candidate genes to be included in a vaccine platform, consisted of: (1) compilation of documented data originating from global analyses pertaining to criteria relevant to vaccine and to M. tuberculosis (Mtb) pathogenesis (2) selection of a subset of genes for further evaluation, based on the accumulated data, by cross matching the data in the different criteria; (3) bioinformatic analyses aimed at both further characterization of the candidate genes, in terms of annotation, gene context and cellular localization and; immunoinformatic analysis conducted for the prediction of T-cell epitopes; (4) development and application of a ranking scheme, based on qualitative and quantitative measures, as a tool for prioritizing the selected candidates. A schematic presentation of the various steps, as well as the reductive scheme generating a list of best-hit antigens, is given in Figure 1.

Flowchart of the selection procedure – data mining and bioinformatic analyses. A schematic presentation of the reductive approach applied for the whole-genome selection of Mtb vaccine candidates.
Generation of a comprehensive, literature-based dataset
The first step towards the selection of vaccine candidates consisted of accumulating published experimental data, by an extensive literature scan of documented analyses, with emphasize on global analyses. Target genes to be involved in a future vaccine are either ORF products which have the potential to elicit an immune response (humoral and/or cellular), and/or involved in various manifestations and phases of the infection and/or virulence (as a basis for an attenuated strain). Accordingly, we assembled all evidences into the following general 11 categories (Table 1):
Macrophages
Numerous comprehensive studies related to the infection of the macrophages by the bacilli were conducted, aiming at identifying the genes involved in processes as adhesion to the host cell, phagocytosis-mediated entry and survival in the macrophage, adaptation to the environment and resistance to both phagocytosis and lysis, and to free radicals. Different approaches were applied to characterize both the profile of expression at various relevant conditions and the growth/survival of wild-type Mtb and selected mutants. These include subtractive hybridization, transcriptomics, proteomics and mutagenesis. In our screen, we included representative studies covering the abovementioned aspects, as detailed in Table 1[56–64].
MprAB
This two component system is one of two major families of transcriptional regulators necessary for stress adaption processes. The system is induced in response to various stress conditions and was shown to be required for growth in vivo during the persistent stage of infection. Comparative DNA microarray analysis of Mtb H37Rv and an mprA mutant strain enabled to profile the gene expression in each of the strains and to identify 221 genes which were presumably regulated by MprA under stress conditions [65].
Hypoxia
One of the well established models of the tuberculi non-replicating dormant state, is the Wayne model [66], which is based on culturing Mycobacteria in decreasing concentrations of oxygen, mimicking the environment in which the bacteria transform to a non-replicating form, residing in granulomas, which are of hypoxic nature. In this respect, growth under low O2 and NO concentration or under depletion of NO synthase are the major conditions implemented to identify the expression profile related to hypoxic conditions. Sherman and his coworkers [67] used a whole genome microarray to identify 101 genes whose expression is significantly altered by hypoxic conditions, of which 47 are induced. A similar approach was followed by Voskuil and coworkers [52], resulting in a set of 48 genes affected by low O2 concentration or by the presence of nontoxic concentration of NO (known to inhibit bacterial respiration). Likewise, mutants deficient in NO synthase were used to profile the genes expressed during hypoxia-like conditions [62].
Reactivation
Very little is known about the genetic basis and the signals which are involved in the reactivation from the dormant state of the mycobacterium. In an attempt to decipher the factors which govern the phase of reactivation, some researchers adopted an immunossupressive model, in which reactivation of latent infection occurs following treatment with the immunossupressive reagent dexamethazone [68, 69]. Using this model, Talaat and coworkers [54] recently identified a total of 174 genes that were up-regulated during the reactivation phase of tuberculosis. A family of 5 proteins, denoted resuscitation promoting factors (Rpfs), has been shown to be involved in regulating mycobacterial growth [55, 70]. Single deletions of the Rpf members were used to determine the effect of these proteins on the kinetics of reactivation [71].
Dormancy
The shifting to the non-replicating persistence state ("dormancy") of the bacteria is accompanied by modulation of expression of genes related to the adaptation to the non-replicating conditions. The adaptive processes include: starvation of essential nutrients, cessation of growth in stationary phase, and depletion of oxygen. Of these, the later is the most investigated and constitutes the basis for the in vitro model used in dormancy studies. The set of genes found to be induced by hypoxia, nitric oxide and adaptation to the non-replicating conditions, under the regulation of DosR (Dormancy survival regulator, Rv3133c), were denoted as the dormancy/DosR regulon [52, 53]. Transcription profiles were determined by a whole-genome comparative DNA microarray analysis of the exponential growth at time points in the stationary phase, as well as under specific non-replicating persistent conditions of low oxygen and nitric oxide [52, 53, 62]. In a separate analysis, the differential expression of genes under aerobic and anaerobic conditions was deciphered by a proteome (2D-PAGE) analysis, revealing proteins which were unique or more abundant in the anaerobic conditions [72].
Lungs
As the first point of entry of the M. tuberculosis upon infection, following implantation the bacteria reside and replicate in granulomas in the lungs. Various strategies were employed to depict the pattern of expression both in human granulomas and in guinea pig and mouse lungs, and these revealed an extensive list of genes necessary for growth and survival, as well as identification of genes expressed during infection in the lungs (detailed in Table 1, [17, 71, 73–78]).
Acr-coregulated
The 16 kDa α-crystallin (Acr) protein was shown to be induced and required inside macrophages and highly expressed in the presence of NO, low O2 concentrations and in the stationary phase ([79–81] and references therein). The Acg gene product was later identified as a novel macrophage-induced gene, whose expression is co-regulated with that of acr [79]. Aiming at identifying other genes which are under the same regulation, an acr-coregulated gene (ACG) family was denoted, following the identification of a conserved sequence motif found in the promoter region of the 15 family members [82].
Secreted
Proteins which are exported from the cytoplasm and either secreted to the milieu or anchored to the cell wall, are of a major importance as targets of the immune system, in view of their exposure in the host. The secretion pathways in M. tuberculosis are less established than in other bacilli, in particular those implicated in virulence factor secretion. In addition to the classical pathways (the SecA and TAT systems), the pathogen harbors a unique virulence-related secretion system – ESX system and probably additional systems which remain to be identified. Identification of secreted proteins by a whole genome global analysis was conducted by Gomez and coworkers [83]. A predictive approach of all proteins harboring a secretion signal of the classical SecA pathway, but lacking membrane spanning segments, was carried out, resulting in the identification of 52 proteins, the location of which was further confirmed by fusion to a marker of subcellular localization.
Immunogenicity
Data on immune response was compiled from global analyses covering both elicitation of humoral and cellular responses following vaccination of model animals, as well as seroreactivity profiles of selected antigens with sera from healthy and TB patients (see Table 1 and data pertaining to individual ORF products, in the text, [30, 84–89]).
Iron regulated
As for many other pathogens, Mycobacterium tuberculosis infection is dependent on the availability of iron in the depleted macrophage, and therefore iron sequestration from the host is necessary for virulence [90, 91]. Iron is also known to influence both the innate and adaptive immune responses to Mtb [92]. Failure to assemble the iron acquisition machinery or to repress iron uptake has deleterious effects for Mtb [90, 91]. In addition to expressing iron-uptake systems during iron deficiency, Mtb displays several changes in gene expression in response to iron availability. All changes taking place during the response to iron deficiency are controlled by iron-dependent regulatory networks. The IdeR gene product is a dual functional regulator controlling transcription of genes involved in iron acquisition, iron storage and macrophage survival [93, 94]. ideR is an essential gene in M. tuberculosis and its deletion results in deregulated siderophore biosynthesis and sensitivity to oxidative stress [94]. The transcription profile of genes whose expression is modulated by iron levels was mapped by a DNA microarray analysis, and revealed 155 genes which were iron-regulated; one third of these were regulated by IdeR [94].
Vaccine
Data pertaining to vaccine potential of the antigens was derived from publications describing global analyses of preselected antigens for their potential to induce an immunoprotective response [25, 95–97]. These include mostly evaluation of DNA vaccine candidates, as listed in Table 1. For this specific category, a large number of evidence was also derived from information pertaining to particular antigens studied both as DNA or protein vaccine candidates [14, 20, 24, 25, 71, 84, 98–141], among which are obviously the well established classical antigens.
Selection of a dataset of ~200 vaccine candidates
Of the total 3989 ORF products, 1209 had documented evidence in at least one of the sources listed in Table 1. Cross-matching of the compiled data for all 1209 ORF products, allowed for a preliminary selection of 344 antigens, for which either more than two values above the threshold exist, or, alternatively, one value above the threshold together with at least two other values with lower values (below the threshold) were present. The threshold for each literature source was set as the median of the experimental values extracted from the particular literature source.
The literature sources which were used to generate the dataset (Table 1) were grouped within each category (e.g. all 9 studies in the Macrophage category were treated as a single piece of evidence). Following the threshold selection described above (step I in Figure 1), each ORF product was assigned a "+" sign in a given category if experimental data has been reported at least in one study. Summation of the "+" signs across all categories, for each ORF product, produced a total, un-weighted score, with a maximal possible total score of 11 (according to the number of categories). Among the 344 antigens, 143 had a relatively high total score of 3–8. Of the remaining 201 low scoring antigens, 46 had documented information related to vaccine potential and/or a critical role in the pathogenesis traits of the bacilli. As such, these 46 antigens were added to the 143 high scoring antigens (having a total score ≥ 3), generating the final list of 189 candidates for further evaluation (Additional file 1: List of 189 selected antigens).
Bioinformatics analysis of the 189 vaccine candidates
Inspection of the list of 189 candidates reveals that, according to the annotation deposited at the NCBI, 69 antigens have no assigned function. In an attempt to further characterize the candidate genes, we followed an in-depth bioinformatic analysis which was aimed at revising the existing annotation, as well as determining the cellular localization and the presence of secretion signals, and protein repeats patterns. The revised annotation was thus based on sequence similarity searches against updated databases, and domain/motif assignment following querying secondary databases (see: Methods). Also, Mtb-related servers were screened to extract function-related information (see: Methods). Additive information was derived from the accumulating data in the literature, and specifically on functional characterization of particular genes or their mycobacterial orthologs. These in silico analyses and data mining resulted in the assignment of a putative function to 60% of the 69 candidates with unknown function, and the addition of further functional details to 15 candidates (Additional file 1: List of 189 selected antigens).
Immunoinformatic analysis of the 189 vaccine candidates
As mentioned above, the significance of the cellular immune response in protection against intracellular pathogens in general and Mtb in particular, is well documented. We therefore conducted a comprehensive mapping of the T-cell immunity potential of the selected antigens, by compiling both the results of an in-silico search for putative T-cell epitopes and experimental published data.
Using NetCTL as an integrative approach for prediction of 9-mer CTL epitopes [44], we analyzed the 189 proteins for the presence of MHC binding peptides. The analysis was conducted on 12 HLA supertypes, representing ~120 human MHC alleles and providing a population coverage of 99.8% worldwide. For our large scale analysis, we have chosen a relatively high combined score threshold of 1.25, which gives preferentiality to true binders. At this threshold, the specificity is 0.993 and the sensitivity is 0.54 (as reported by the NetCTL server). Most potent candidates were considered as those having binders for as many supertypes (this argument was further used to rank the antigens at a later stage, see below). Compared to in silico studies of the Mtb antigens immunomic potential [25, 27–29, 31, 32, 125], the analysis described herein is inclusive both with respect to its scope and to the number of HLA alleles covered.
In addition to the theoretical analysis, experimental evidences for T-cell epitopes were retrieved both from the IEDB database [45], by querying each of the antigens as well as data for cellular immunoreactivity of individual gene products from the extensive study of Leyten et al. [24] and other literature sources [24, 25, 84–86, 88, 99, 102, 106, 115, 119, 124, 127, 142–144]. The compiled T-cell immunity data was added as yet an additional criterion for further evaluation and ranking of the 189 antigens, leading to a total of 14 criteria.
Assignment of a qualitative score to the 189 vaccine candidates
The compilation of the experimental data with the bioinformatic and immunoinformatic analyses, together with biological reasoning resulted in a comprehensive knowledge-based dataset of the 189 vaccine candidates (Additional file 2: "Raw data of the 189 selected antigens"), according to a total of 14 criteria (Table 1). In order to rank the list, and further prioritize the antigens, we developed a scheme which is based on both a qualitative and a quantitative score calculated for each antigen. The scores are calculated according to 14 criteria, encompassing the 11 literature-based categories as well as T-cell immunome data, cell wall and repeats-derived information. The first scoring iteration assigns an equal weight to each criterion. A "+" sign is indicated for each indication in each of the criteria, and an arithmetic score is calculated, by summing the "+" signs (See Additional file 3: 'Qualitative scores for 189 selected antigens'). The scores obtained ranged between 9-1, exhibiting a normal distribution of number of antigens per scores, as follows: scores 9–8: 11.6%; scores 7–6: 32.8%; scores 5–4: 45.5% and; scores 3-1: 12%. This rather naïve scoring is of value in rendering a preliminary ranking, which allows to limit the bias that may arise in favor of antigens with an extensive coverage in the literature. Nevertheless, it suffers from some inherent limitations: (1) as described above, an equal weight was assigned to each criterion, yet their differential relevance to vaccine development is not reflected; (2) any data in a specific criterion contributed equally to the total qualitative score, regardless of the relative potency of the actual result reported in the publication from which the data was extracted, or for data obtained from our analyses. (3) in spite of a broad range of qualitative scores, their distributions reveal large clusters of genes. To note, groups of 17 and 19 genes harbor a qualitative score of 8 and 7, respectively, while a group of 44 genes have a qualitative score of 6. This implies that in order to prioritize the equally-scored antigens within the groups, we had to further refine our scoring system and apply additional measures.
Employing a numerical scheme for the assignment of quantitative scores
To address the above-described limitations of the qualitative measure, we introduced weighted internal scores to each of the 14 criteria, as presented in Table 2. The actual data for determination of the internal scores was extracted from the relevant experimental studies. The internal score scaling was based on the number of sources and/or the intensity of the results which contributed to the particular criterion. Accordingly, the maximal internal score was 2 for most criteria, except for Macrophage and Vaccine, which were up-weighted due to their significance/relevance to vaccine development, by giving a maximal internal score of 3. These scoring rules were also useful for the final ranking among high scoring genes. The final quantitative score calculated for each ORF product, is the total of the internal scores assigned in each of the 14 criteria for a particular ORF product. As expected, the quantitative scores allowed dissecting the large groups of equally qualitatively scored antigens, and ranking further the candidates. These scores can be used as a tool for further trimming down the number of vaccine candidates, and selecting for the top-hit targets. This process generated a list of 55 antigens with a qualitative score value of 6 and above. Of this list, 10 antigens were predicted to harbor more than 5 membrane-spanning domains (Rv0286, Rv0290, Rv0450c, Rv0754, Rv1196, Rv1348, Rv1736c, Rv1737c, Rv1997, Rv2123). Although potentially valuable vaccine targets, these antigens were removed from the list due to the potential technical problems which may occur during cloning and production of such recombinant proteins. Consequently, a list of 45 top ranking antigens is provided (Table 3). In this list, the antigens are sorted by their qualitative score and consecutively – by their quantitative score. According to this type of ranking, the antigens are clustered into groups, by limiting both the qualitative and the quantitative lower value scores in a certain group. For instance, Group I of antigens includes candidates having a qualitative score of 8 and above, provided that the quantitative score is not lower than 12. According to such an approach for ranking, the antigens were delineated into three groups: the first includes the 12 top best-hit antigens both in terms of qualitative and quantitative scores and the following second and third groups comprise of 20 and 13 antigens, respectively (Table 3). There are other modes of sorting which could be considered as well, giving more weight to the quantitative score vs. the qualitative score and vice versa. It should be mentioned though, that using these different ranking approaches had marginal consequences on the grouping, affecting mainly those antigens at the edges of each of the groups.
Functional categories of the selected genes
According to the revised gene annotation and the accumulating data from the literature, the 189 selected antigens were assigned a functional category (listed in Additional file 1: List of 189 selected antigens) following the classification implemented previously for the Mtb complete genome sequence by Cole and coworkers (and re-annotated by Camus and co-workers, [26, 51]). Figure 2 presents a graphic view of the gene distribution according to the functional categories, in the repertoire of the 189 selected genes. When compared to their fraction in the whole genome [51], it appears that the Conserved hypothetical proteins category is under-represented in the list of 189 selected antigens, while the Lipid metabolism and Virulence, detoxification and adaptation categories are over-represented in the list of 189 selected antigens. To note, this higher fraction is a bona fide observation, and not a result of antigens which were re-assigned to a new functional category following the updated annotation.

Distribution of Mtb genes according to functional categories. The categories are adapted from Cole and his coworkers [26]. For practical reasons, the categories of "Conserved hypothetical proteins" and "Proteins of unknown function" were joined (category (III) in the Figure). Category (VI) encompasses the functional classes which were either less abundant or non existent in the list of 189 antigens selected herein. The percentage of genes in each of the functional categories is provided, as retrieved from Camus and his coworkers for the whole genome (A) [51], and calculated for this study 189 selected antigens (B) and 45 top-ranking antigens (C).
(1) Conserved hypothetical proteins
In the complete genome, 33% of the genes were assigned a functional category of either Conserved hypothetical proteins or Proteins of unknown function [51]. The proportion of this class of antigens is reduced to 14% in the list of 189 candidates presented in this study, resulting in 27 proteins for which the function remains unknown. This observation is evidently the consequence of the revision of annotation carried out in this study for the 189 selected genes, a process which, as detailed above, was based on cross-matching the in silico analysis against updated databases and accumulating data from the literature. The revised annotation contributed mostly to antigens which were consequently assigned as Intermediary metabolism and respiration (~75% of the re-annotated antigens). For example, of the 69 antigens which were re-annotated in this study, proteins Rv1461 and Rv1462, originally marked as hypothetical proteins, were found (both by domain analysis and literature search) to be part of the SUF machinery (mycobacterial system of [Fe-S] cluster assembly) identified by Huet and coworkers [145, 146]. Likewise, most Usp proteins selected in this study were annotated as hypothetical proteins, and the functional assignment permitted to associate them with the stress protein family (see below). The potential contribution of ORFs with unassigned function to immunogenicity and vaccine is clearly demonstrated by the fact that of the 27 proteins in this category, 22% (6 antigens) are included in the list of 45 top-hits antigens (Table 3). All the six antigens are DosR regulon antigens and are also among the top-best T-cell antigens determined in this study (see below).
(2) Lipid metabolism
Accumulating information suggests that fatty acids, rather than carbohydrates, constitute the dominant carbon substrate utilized by Mtb during infection [147]. Bacteria isolated from lungs of infected mice were shown to oxidize preferentially fatty acids [148]. Moreover, the genes encoding fatty acid catabolic β-oxidation cycle enzymes are extensively duplicated in the genome [26, 51]. In view of the nature of the dataset and the sources of information mostly related to aspects of the intracellular lifestyle of the bacilli and its survival, it is not surprising that the fraction of antigens related to lipid metabolism is as high as 16% (30 out of the 189 selected), compared to 6% (~240 antigens out of 3989) in the whole genome (p-value = 1.02 × 10-4) (Figure 2). Among the proteins involved in these processes, are many proposed vaccine candidates and drug targets. Several antigens have also shown promising cellular and humoral immune responses. Four out of the 30 proteins selected are among the 45 top-hit antigens. Basically, lipid metabolism can be divided into two main divisions:
(I) Lipid biosynthesis, modification and degradation
In this division are included 22 out of the 30 proteins in the category. Of the two discrete types of enzymes involved in fatty acid biosynthesis in bacteria, the type I and type II fatty acid synthases (FAS-I and FAS-II, respectively), condensing enzymes that catalyze the formation of carbon-carbon bonds, have received considerable interest, two of which are included in the 189 selected genes: kasA and kasB (Rv2245 and Rv2246, respectively) [26, 149]. The kasA-kasB operon (β-ketoacyl-ACP-synthases) is necessary for synthesis of fatty acids, mycolic acid and polyketides, by initiating the subsequent rounds of acyl extension [150, 151]. Few antigens selected in this study are related to biosynthesis of polyketides: a pks-associated protein (papA3, Rv1182) and a putative regulator of pks expression (annotated originally as a hypothetical protein, Rv1186c), the mycobactin biosynthesis operon (Rv2377c–Rv2386c, discussed below, in the section related to iron acquisition) as well as the polyketide non-peptide synthase (Rv0101c). Disruption of Rv0101 interrupts the virulence-related, cell wall constituent picothocerol dimycocereosate (DIM) biosynthesis, producing an avirulent strain with vaccine properties comparable to BCG [152]. The protein fadD26 (Rv2930) included in our selection, is a necessary part of the operon for biosynthesis of DIM [153], and its mutant is attenuated in mice and induces better protection than BCG when administered sc. [154]. The mutant is also associated with early attenuation of growth in macrophages [134]. Proteins involved in lipid modification include the Acyl-acyl carrier protein (ACP) desaturases, which convert saturated to unsaturated fatty acids. Three desaturases, desA1-A3, part of a multi-protein iron-enzyme complex (Rv0824c, Rv1094 and Rv3229c) were selected in this study. As for lipid degradation: fatty acid degradation genes are extensively duplicated in the genome (e.g. 36 paralogs capable of performing the first step, [26]), of which 6 were selected in this study (Rv0243, Rv3546, Rv2590, Rv3515c, Rv3139, Rv3140). Five proteins that are functionally related to lipid degradation are listed under either Intermediary metabolism and respiration (Rv1130 (prpD), Rv1131 (gltA1, citrate synthase), Rv0467 (icl) and Rv0211 (pckA)) or in PE and PPE proteins (Rv1169c). Rv1130, annotated originally as conserved hypothetical protein, is a homolog of S. typhimurium prpD and mmge – genes involved in β-oxidation of fatty acids, upregulated in macrophages [62], and essential for ex vivo intracellular growth. Rv0467 a member of the glyoxalate pathway, was one of the first genes reported to contribute to persistence [155–157]. Rv0211 encodes a gluconeogenetic rate-limiting enzyme – suggesting that fatty acids are in part converted into sugars via gluconeogenesis. In addition, Rv1169c (PE11), classified as a PPE and PPE protein, is one of numerous lipases which may be involved in host cell wall degradation, shows homology to triacylglycerol lipase, a paralog of which has been recently shown to be immunogenic [158, 159].
(II) Lipid cell wall composition
Mycolic acids are high molecular weight α-alkyl, β-hydroxy fatty acids crucial for the architecture and permeability of the mycobacterial cell envelope. Free-standing lipids, lipoglycans, and proteins also intercalate within this complex. This layer is covered by a capsule consisting of polysaccharides, proteins and lipids. The consequences of these structural unique features are an extremely sturdy and impermeable cell envelope [151, 160–162]. Mycolic acids containing glycolipids, in particular the prominent trehalose-6,6'-dimycolate (TDM), stimulate cellular and humoral immune response and granuloma formation [163]. Several gene products involved in mycolic acid biosynthesis and modification were selected in this study. Mycolyl transferases (Ag85 complex) are a family of proteins responsible for synthesis of cell-wall components – such as cord factor biosynthesis in Mtb (a dominant immunogenic and immunomodulatory structure necessary for maintenance of cell-wall integrity/virulence and an inhibitor of phagosome-lysozome fusion, [163, 164]). Mycolyl transferases are also termed: fibronectin binding proteins, and have been shown to be involved in host entry. Two representatives are included in our selection, Ag85A (Rv3804) and Ag85B (Rv1886), both classical, well documented B-cell and T-cell immunogens and vaccine candidates. Another protein selected herein is cmA2 (Rv0503c), which plays a role in the modulation of the host immune response, by modifying the cord factor, hence forming a lipid molecule which suppress inflammation [165, 166]. Finally, antigen Rv3130c, originally annotated as a hypothetical protein is actually tgs – member of a novel class of diacylglycerol acetyltransferases responsible for accumulation of triacylglycerol in Mtb as it enters a dormancy-like state in culture [167]. This antigen is constitutively over expressed in the epidemic Mtb of the W-Beijing lineage strain in comparison to non W-Beijing strains [168].
(3). Virulence, detoxification, adaptation
Inspection of Figure 2 reveals that antigens included in this functional category are enriched in the list of 189 antigens: 12% (23 antigens) as compared to only 2% (~80 antigens out of 3989) in the whole genome (p-value = 1.2 × 10-5). This is a consequence of the emphasis given to selection of antigens related to the pathogenesis traits. Of the 23 antigens, 6 are included in the list of 45 top-hit antigens. A total of 11 candidates in this category are stress proteins, belonging to the Universal stress protein (Usp) and the Heat shock protein (Hsp) superfamilies. The Usps encompass a conserved group of proteins involved in stress resistance, adaptation to energy deficiency, cell motility and adhesion [169]. The paradigm usp gene, uspA of Escherichia coli is transcriptionally activated by a large variety of stresses and is one of the most abundant proteins in growth arrested cells [170]. In our analysis, 6 out of the eight usp genes in the Mtb genome [171] were selected as vaccine candidates: Rv1636 (single Usp domain), Rv2028c, Rv3134c (a Usp domain followed by a typical C-terminal domain) and Rv1996, Rv2005c, and Rv2623 (harboring two Usp domains, and possessing a conserved ATP-binding motif and no signal sequence). Rv2623, Rv2005c, Rv2028c, and Rv3134c have been shown to be up-regulated under hypoxia [67], suggesting that they play a role in adaptation and survival of mycobacteria during growth arrest caused by oxygen limitation. The Hsp subfamily includes the extensively studied M. tuberculosis α-crystallin (acr) family-related proteins. The Mtb genome harbors 2 paralogs belonging to the acr family (hsp20/acr, Rv2031c and acr2, Rv0251). Rv2031c is regulated by the two-component regulatory system (DosR regulon), as detailed above, and is a latency stage disease marker [111]. While acr is induced by hypoxia or nitric oxide, acr2 is reported to be the protein most up-regulated under heat-shock as well as expressed under nitric oxide or during uptake by macrophages. Heat shock proteins assist in Mtb survival but also provide a signal to the immune response. acr2 has been shown to elicit a strong cellular response in cattle during early primary infection and thus has been suggested as a candidate for stage specific TB vaccine [172]. This protein is also tightly regulated by MprAB [173]. While analyzing immunogenicity of eight dormancy-regulon encoded proteins as DNA vaccines in a mouse model, Roupie and coworkers [25] have shown that Rv2031c induces the strongest Th1 response, and that mice persistently infected with Mtb developed an immune response against this gene. Both acr and acr2 have been documented as contributing to persistence [174]. Other Hsp proteins selected by this study are the endopeptidase Rv0384c (ClpB), Rv0350 – dnaK-Hsp70, and Rv0351-grpE (Hsp-70 cofactor), which are up-regulated in anaerobic stationary phase in M. smegmatis. PBMCs from TB patients and vaccinated individuals have been recently checked for cytokine profiles in response to DnaK [98]. To note, 4 out of the 6 candidates in the Virulence, detoxification and adaptation category included in the list of 45 top-ranking, are stress proteins (Rv1996, Rv2005c, Rv2031c and Rv2623).
Antigens Rv2006 and Rv3372 are part of the OstAB pathway, which was found to be essential for Mtb growth and virulence in mice [175, 176]. This pathway involves the generation of Trehalose, a major cell-wall constituent of glycolipids. Rv3372 was recently reported to be an immunodominant antigen, inducing both humoral and cellular immune response. The enzyme was also recognized by patient's sera and BCG vaccinated donors; however no protection data was mentioned [177]. Rv1908c (KatG, a catalase/peroxidase/peroxinitritase) has been mostly studied in the context of resistance to isoniazid as well as in development and detection of MDR strains [178, 179]. Intra-macrophage expression of katG is associated with growth and persistence of Mtb in mice and guinea pigs [180]; immunization with a multivalent combination DNA vaccine (containing the ESAT-6, MPT-64, MPT-63, and KatG) generated antigen-specific cell-mediated and humoral responses and elicited a strong protective response relative to BCG [122].
Another virulence-related family of proteins is Nlpc_p60, which belongs to the Firmicutes CHAP-related superfamily, consisting mainly of peptidoglycan hydrolases involved in cell separation. Recent evidences attribute a γ-glutamate-meso-diaminopimelate muropeptidase activity to Nlpc_p60 proteins [181]. Two members of this family, located in a single operon, are present in the Mtb genome, both selected herein: Rv1477 and Rv1478. It has been shown the M. marinum orthologs (iipA and iipB) are essential for virulence in-vivo, invasion and intra-cellular persistence in macrophages [182]. Moreover, it has been specified that the N-terminal sequence of Rv1477 is required for full virulence in vivo and in macrophages. In a recent study, Rv1477 was identified as an RpfB interacting protein (resuscitation promoting factor B, an important cell-wall hydrolase, see below), suggesting a role in cell division during reactivation/resuscitation [183].
Unlike the three functional categories described above, for which a major difference exists between their relative distribution in the list of 189 selected antigens as compared to that in the whole genome, a similar fractional distribution is observed in both lists with respect to the functional categories: Intermediary metabolism and respiration and the Cell wall and cell processes. The former includes 46 antigens, and obviously a thorough discussion of each of these antigens is beyond the scope of this paper, yet we would like to address three major clusters:
Iron acquisition
Iron acquisition-related gene products identified as candidates in this study, include a total of 16 antigens, 7 of which are classified as Intermediary metabolism and respiration proteins and are part of the SUF (mobilization of sulfur) machinery or iron storage proteins, while the remainder are siderophores-related proteins, classified either under the categories of Lipid metabolism or in the Cell wall and cell processes (ABC transporters, see below): (1) Siderophores: Mtb synthesize siderophores of the salicylate group, named mycobactins [90, 184, 185]. These are produced exclusively under iron limitation both as a membrane bound as well as soluble forms. The mbt-1 locus contains the siderophore core biosynthetic apparatus [90, 91], members of which were selected by this study the non-ribosomal peptide synthetases MbtB and MbtE (Rv2383c, Rv2380c) two polyketide synthetases MbtD and MbtC (Rv2381c and Rv2382c), an isochorismate synthase (MbtI, Rv2386c), a hydroxylase (MbtG, Rv2378c) and the conserved protein MbtH (Rv2377c). MbtB mutants impaired in siderophore biosynthesis are unable to replicate in infected macrophages [186]. The mbt-1 locus genes were included in the Lipid metabolism category, given that these are polyketide biosynthesis components. Also selected as part of the iron acquisition apparatus are the ABC transporters IrtA and IrtB (Rv1348 and Rv1349, respectively) which actively transport the iron-siderophore complex. Inactivation of these genes drastically affects the ability of Mtb to replicate. irtAB mutant is attenuated for survival in infected macrophages and lungs of infected mice [187]; (2) The SUF (mobilization of sulfur) machinery: A locus of seven proteins was functionally identified by Huet and coworkers [145, 146] as harboring the Mtb suf machinery – an exclusive mycobacterial system of [Fe-S] cluster assembly, probably essential for survival via its implication in bacterial resistance to low iron limitation and oxidative stress. The system is required for maturation of physiologically important metalloproteins. Sassetti and coworkers [61] defined this operon as necessary for in-vitro mycobacterial growth. Of this locus, our selection disclosed 5 components (Rv1461 (SufB), Rv1462 (SufD), Rv1464 (SufS), Rv1465 (NifU) and Rv1466. Rv1466 presents no homology with any documented suf gene but probably acts as a predicted metal-sulfur biosynthesis enzyme. Consistently, Rv1461 to Rv1466 are over-expressed in Mtb grown on low iron [94]. Moreover Rv1460, Rv1463 and Rv1464 have been described as stress response genes transcriptionally up-regulated in anaerobic stationary phase in M. smegmatis; (3) Iron storage proteins – bacterioferritin: Ferritins constitute the major non-heme iron storage proteins in animals, plants and microorganisms, playing an important role in ferric iron homeostasis. The Mtb genome harbors 2 paralogs: bfrA (Rv1876) and bfrB (Rv3841) both selected in this study. The bfrB antigen has been shown to be abundant under hypoxic conditions [188], a dominant T-cell antigen in infected mice [189] and antigenic in human disease [190].
Of the 16 candidate genes related to iron acquisition, only the two ABC transporters classified as Cell wall and cell processes category, Rv1348 and Rv1349, received relatively high score (qualitative scores of 7), yet the former was not included in the list of 45 top-hit candidates, because of a high number of transmembrane segments.
Nitroreductases
One of the activities induced in Mtb by hypoxia is the dissimilatory reduction of nitrate to nitrite which serves as an energy source as bacteria adapt to anaerobiosis [66, 191]. Five nitroreductase-related proteins are included in the list of the selected 189 antigens, 4 of which are classified under Intermediary metabolism and respiration. The Mtb genome contains 2 loci homologous to prokaryotic respiratory NAR (nitrate reductase) genes [26]. The first-narGHJI is not represented in the list of 189 genes, while the second locus contains two antigens, both selected in this study: nitrate reductase narX (Rv1736), exhibiting strong similarity to the narGHJI cluster (yet probably inactive as a classical reductase [192]) and narK2 – a nitrite extrusion protein (Rv1737c), a member of the devR regulon (acting as a redox sensor [191] and an hypoxia induced antigen [67]). Three other selected ORF products, listed as putative nitroreductases, are part of a family of 8 unusual nitroreductases identified by Purkayashta and coworkers [79]: The hypoxia-induced acg (Rv2032, necessary for survival) and two paralogs (Rv3127 and Rv3131c), all harboring parts of the 5 nitroreductase order-conserved motifs identified [79]. Interestingly, of the five nitroreductase-related antigens selected, three (Rv2032, Rv3127 and Rv3131c) are included in the list of 45 top-hits.
Proteases
Although many proteases are documented as virulence factors in other pathogenic bacteria, little information is available regarding their role in mycobacteria pathogenesis. The Mtb genome harbors ~60 putative proteases [48], of which only 7 are included in the 189 list (all except one are classified under Intermediary metabolism and respiration). Among these are proteases being evaluated in vaccine studies: the classical Rv0125, a DegQ-like serine protease (HtrA-family), a construct of which with Rv1196c demonstrated protective efficacy equal to BCG in diverse animal models [135, 193] and; the zinc-metalloprotease Rv0198c (pepO, an M13 peptidase resembling eukaryotic neprilysin [194]), which was evaluated as a vaccine candidate by Vipond and coworkers [96, 97]. Rv2625c, originally annotated as a conserved transmembrane Ala and Leu-rich protein, is probably a zinc protease and part of the DosR regulon. Rv2869c is a homolog of the eukaryotic site two protease (S2P) controlling membrane compositions. In Mtb it has been recently found that the enzyme controls cell envelope composition, in-vivo growth and persistence [195]. Knockout mutants of Rv2869c exhibited reduced expression of genes related to lipid biosynthetic and degrading genes, including the resuscitation factor rpfC. Rv2224c was identified as a putative virulence gene by high throughput techniques and attenuation studies [61, 76], and shown to be preferentially expressed in human macrophages and up-regulated after the onset of starvation. Similar to Rv2869c, Rv2224c may modify envelope composition or, alternatively, hydrolyze fatty acyl esters with mycobactericidal activity [196]. Another selected protease gene is the HslV protease Rv2110c (prcB, proteosome β-subunit), a core component of the proteosome, known to play a critical role in bacterial stress defense. None of the proteases selected in this study are represented in the list of 45 top-hits.
It is well established, that the characteristic mycobacterial cell envelope is a dominant feature of the biology of M. tuberculosis and other mycobacterial pathogens (as described above under lipid synthesis). Indeed, the second largest category which has a similar representation in the list of 189 selected antigens as in the whole genome, is the Cell wall and cell processes (a total of 38 antigens selected in this study). Most strikingly, 37% of the antigens under this functional category are included in the list of 45 top-hits candidates. This group contains three clusters of key antigens: the ESX secretion system, the resuscitation antigens and lipoproteins
The Esat-6 system (ESX)
The ESX is one of the 4 secretion systems which have been identified so far in Mycobacterium tuberculosis, and constitutes a major determinant in mycobacterial pathogenesis and immunogenicity. ESX-derived antigens have been described as immunogens in infected cattle [124, 197], dominant early-phase T-cell antigens [102, 106, 119, 127, 136, 142, 198], putative subunit vaccine candidates [85, 107, 120], as well as potentiators of BCG vaccines [20]. These antigens have been also extensively studied as promising immunodiagnostic markers [84, 85], and in mechanistic/regulatory studies ([199–201] and references therein).
The complete genome of Mtb H37Rv harbors five ESX copies (not necessarily identical, [202]). The ESX1 locus, the most well studied, is located in the extended region of difference 1 (extRD1) of the genome, and therefore absent from all vaccine strains of M. bovis BCG. The products of this system locus are suggested to modulate early events during infection (e.g. [102, 199–201] and references therein) and involved in phenotypes as growth in macrophages, suppression of the macrophage inflammatory and immune response, phagosome maturation arrest [203] and virulence factor delivery. Sixteen ESX proteins are represented in the 189 selected antigens (9 of these are listed under the Cell wall and cell processes functional category). These include the classical well documented antigens: ESAT-6 (esxA, Rv3875) and 2 of its paralogs (the well studied vaccine candidate Rv0288 (esxH) and Rv1793 (esxN)), and CFP10 (esxB, Rv3874) and its paralog (esxG, Rv0287). In addition, the following ATPases were shown to be core components of the ESX system: Rv3871, Rv0282, Rv0283, and Rv0284, Rv3876 as well as the Ala-rich protein Rv3878 and Rv3879c, the transporters Rv0289 and Rv0290 and Rv0292, a transmembrane protein. Rv3616c (EspB) a recently identfied effector of this secretion system, was selected herein as well. In total, three ESX components (Rv0288, Rv1793 and Rv3875) are part of the 45 top-hit antigens.
Resuscitation promoting factors (Rpfs)
The Rpfs, secreted or membrane-anchored peptidoglycan/glycosyl hydrolases [204, 205] were originally identified in Micrococcus luteus, promoting the recovery of bacteria from latency to a replicating phase [206]. In a later study the presence of similar autocrine growth factors in Mtb was reported [123, 207]. Five Mtb gene products encoding Rpf homologs were identified (Rv0867c-rpfA, Rv1009-rpfB, Rv1884c-rpfC, Rv2389c-rpfD and Rv2450c-rpfE, [55]) and documented to have resuscitation activity [70]. Two strains of Mtb in which three rpfs were deleted were both attenuated in mice and did not reactivate in an in vitro reactivation assay [208]. Also, s strain lacking rpfB exhibited a delayed reactivation in a mouse dormancy model and reduced mouse lung colonization [71]. The Rpf proteins are secreted or membrane anchored, which renders them candidates for recognition by the host immune system. Affinity purified antibodies were shown to inhibit bacterial growth in vitro and it has been suggested that sequestration of Rpfs might also provide a means to limit/prevent bacterial multiplication in vivo [123, 207]. When administered as subunit vaccines to mice, 4 out of 5 Mtb Rpfs were found to be highly immunogenic, eliciting both IgG1 and IgG2a responses (highest for Rv2389), T-cell proliferation and cytokine production. Vaccination of mice with RpfE (Rv2450c) results in significant protection against subsequent high-dose challenge with a virulent strain (in survival times and bacterial multiplication in lungs and spleen) to levels comparable to Ag85B and Esat-6 [88]. As mentioned, all the five known resuscitation promoting factors are included in the 45 top-ranking hits; three of which (RpfE, RpfB and RpfA) belong to the highest scoring group (GroupI, Table 3).
Lipoproteins
Lipoproteins are reported to affect both innate and adaptive immunity as well as bacterial in vivo growth and virulence. Of 48 predicted lipoproteins in the Mtb genome [209], 6 were selected in this study, and at least four are directly relevant for vaccine design. The classical 19 kDA antigen (Rv3763), is a well studied glycosylated lipoprotein acting as a phagocytosis stimulating adhesion [210] factor through TLR2 signaling in the macrophage, thus facilitating the bacilli persistence [209]. The lipoprotein LprG (Rv1411c) required for growth in mice [211], is a B and T-cell antigen [212], inducing protective T-cell response within the lungs of infected mice sufficient for early control of Mtb [140]. It is probably acting as a TLR-2 ligand, inhibiting MHC class II antigen processing [213, 214]. In a more recent study, a fusion protein of Rv1411c with ESAT-6 (where Rv1411c acts as a natural adjuvant) was constructed, resulting in protection of mice exposed to low dose aerosol challenge [214]. RpfB (Rv1009), the only lipoprotein in the resuscitation factor family, is another prominent representative described above. In addition to these clusters of antigens, a promising cell wall antigen is the secreted T-cell antigen TB8.4 (Rv1174c). This protein, detected in human macrophages and sputum of TB patients [215], affects growth of M. bovis BCG in human macrophages and mice [216], probably via its role in both resistance to intracellular stress and adaptation to hypoxia [217]. The Rv1174c knockout mutants are attenuated in vivo and Rv1174c-derived peptides were found to resuscitate one year old cultures [218]. Moreover, immunization with either plasmid DNA or the Rv1174c recombinant protein induces a strong and protective T-cell response in mice [105].
Another functional cluster of proteins is the unique surface-associated PE & PPE family, which encompasses ~4.5% of the Mtb genome [219–222] and a similar percentage in the 189 selected list (Figure. 2). The PE family harbors a Pro-Glu motive at their N-terminus (positions 8 and 9 in a 110 residue long conserved domain). A subgroup of the PE family is the PE-PGRS, where the PE domain is followed by multiple tandem repeats of GGA or GGN [223, 224]. In the PPE family (ProProGlu N-terminal motif, positions 7–9 in a 180 residue conserved domain) the repeat domain is followed by a variable C-terminus [221]. Originally members of this family were used as strain differentiation markers [225]. PE/PPE genes are widely present in pathogenic bacteria yet missing in non-pathogenic species, and therefore are assumed to fulfill important functions related to survival within different environmental niches. Indeed, the first evidence came from the observation that inactivation orthologs of the PE gene product wag22 in M. marinum results in a replication defect in macrophages and decreased survival in granulomas [226]. Certain PE/PPE proteins play a role in immune evasion and antigenic variation [26, 219, 223, 227], others are virulence related [224, 226, 228, 229]. Evidence that at least some PE_PGRS genes are antigens expressed during infection in the host comes from serological studies. Many of the PE/PPE proteins elicit strong immune response [104, 159, 227, 230–232]. Of the antigens selected in this study, 10 belongs to the PE/PPE family (equivalent to its fraction in the genome), of which three (Rv1169, Rv3347c and Rv3873) are included in the top-ranking 45 antigens (see Additional file 1: List of 189 selected antigens, and Table 3), most of them having particular indications as for their involvement in virulence and/or immune response, as follows: PPE55 (Rv3347c) is recognized by antibodies elicited during sub-clinical infections of guinea-pigs and could be applied to the differentiation between latent and incipient TB [233]. PPE protective subunit (Rv3812) was recently identified as T-cell antigens with vaccine potential [103]. Rv1169c elicits a strong but differential B-cell response among different categories of TB patients [234]. PPE68 (Rv3873) which is a member of the RD1 region, was shown to act as both a T-cell and B cell antigen in mice [106] and is also a constituent of the ESX secretion system, as are the selected PE5 (Rv0285) and PPE4 (Rv0286).
Distribution of immunopotent T-cell antigens
Inspection of the combined weighed scores of experimental data and predictions in the T-cell immunogenicity category revealed that 29 antigens harbor the highest scores in this category (a total quantitative score 3 or 4). Of these, 70% (20 antigens) are included in the list of 45 top-ranking candidates, where 4 of which are proteins of unknown function. More strikingly, all 6 hypothetical proteins in the list of the 45 top-ranking antigens are immunopotent T-cell candidates (Rv0079, Rv1813c, Rv2030c and Rv2627c are part of the 29 top best T-cell antigens, while Rv1738 and Rv2628 are documented in literature sources on individual antigens). The distribution of the remaining immunopotent T-cell antigens is comparable, ~2–3 antigens per functional category. In addition, among the 29 immunopotent antigens are also included 3 out of the 8 antigens classified as Intermediary metabolism and respiration in the list of 45 top hits, as well as 2 of 4 Lipid metabolism categorized proteins, the well documented classical Ag85A (Rv3804c) and Ag85B (Rv1886c).
The 20 most immunopotent T-cell antigens included in the list of 45 top-hit candidates, can also be examined with respect to their distribution among different classes/phases (as assigned for each ORF product, see Additional file 1: List of 189 selected antigens). Out of the 20 antigens, 11 are DosR regulon members (Rv0079, Rv1196, Rv1813c, Rv2029c, Rv2030, Rv2031c, Rv2032, Rv2627c, Rv3127, Rv3130c, Rv3132); 4 are classical vaccine antigens (Rv1886c, Rv3804c, Rv1980c, Rv2875), one resuscitation antigen (Rv0867c), one putative reactivation antigen (Rv0288) and 3 are listed under "others" (Rv1908c – katG, Rv2780-ald, Rv3873-PPE68, an ESX1 component). Search for T-cell immune response data originating from literature documentation on individual antigens in the list of 189 selected genes identified at least 15 additional immunopotent candidates, 9 of which are included in the top list of 45 antigens (the resuscitation factors – Rv1009, Rv1884c, Rv2389, Rv2450; Rv1174c-a low Mw T-cell antigen and the DosR regulon antigens – Rv2006, Rv1738, Rv1733c, Rv2626c and Rv2628 studied by Leyten and coworkers [24] & Roupie and coworkers [25]). Interestingly, the most potent T-cell antigens, if estimated solely on the in silico predictions of CTL binders conducted herein are ~10 antigens harboring 80–200 epitopes recognized by all 12 supertypes (in decreasing order Rv0101-nrp, Rv2380c-mbtI, Rv0284-ftsk, Rv2383c-mbtB, Rv2006, Rv2590-fadD9, Rv3347c-PPE55, Rv0450c-mmpL4, Rv1997-ctpF, and Rv1736c). It may be of interest to evaluate experimentally these promising antigens for their activity as T-cell antigens.
Conclusion
This study illustrates the process of screening the complete Mtb genome, aiming at identifying and selecting potential vaccine candidates. The screen was implemented to discern genes covering all phases of the infection, which would, in turn, contribute either to construction of a multi-stage vaccine or to the design of a subunit vaccine supplemental to the BCG vaccine, solely directed to the early-phase events of the infection. The strategy designed for rational candidate selection was applied onto a knowledge dataset which compiled numerous sources of relevant published experimental information. The literature-based dataset includes ~1200 antigens, for which evidences on aspects relevant to vaccine and/or virulence traits of the bacteria, existed (classified into 11 categories). An initial reduction to a list of 344 antigens was conducted, based on: (1) a preliminary total score calculated from the number of positive signs in the various categories, resulting in 143 antigens having a relatively high score of 3–8; (2) a subsequent selection from the remaining 201 antigens (scoring 1–2), by manual curation of specific sources of information related either to vaccine or pathogenesis; resulted in additional 46 antigens (interestingly, only 4 of the 46: the classical Rv1908c, Rv1926c, Rv1980c, and the ESAT protein Rv1793 were eventually included among the final list of 45 top-hits antigens (Table 3)). These filtering steps generated the list of 189 selected vaccine candidates, on which further extensive bioinformatic and immunoinformatic analyses were conducted. Of the 189 antigens in the list, only 34 were previously described in the literature as putative vaccine candidates. These 34 known antigens represent over 80% of the total number of such antigens listed in the previous step of the selection process (see Figure. 1). Among these 34 antigens, at least 5 are under clinical trials (the classical antigens: Ag85A (Rv3804c) and Ag85B (Rv1886c), ESAT-6 (Rv3875); mtb72f: Rv0125 and Rv1196), 10 were used in protective studies in animal models and 19 are candidates eliciting a strong immune response.
Examination of the distribution of the 189 selected antigens among whole genome-defined functional categories (Additional file 1: List of 189 selected antigens) discloses an enrichment of proteins related to lipid metabolism as well as virulence and stress proteins, in comparison to their fraction in the genome. Also, the in silico analyses contributed to the updating of the annotation, resulting in a dramatic decrease in the number of proteins of unknown function in comparison to their fraction in the whole genome. The comprehensive dataset was then employed to rank the selected antigens, by applying a ranking scheme based on the assignment of both qualitative and quantitative scores to each of the antigens, in the 14 criteria employed. This ranking enabled to down select a list of the best 45 candidates, denoted as top-hit antigens (Table 3).
Inspection of the proposed list of 189 antigens provided in Additional file 1: List of 189 selected antigens reveals representatives of all classes/phases of the infection, including early phase well-documented classical antigens as well as 14 other early phase antigens. It is worth noting that all five known resuscitation antigens (RpfA-E) are included and furthermore, all 5 are among the 45 top-hit antigens (Table 3). The DosR regulon comprises of ~50 genes [52, 53, 235] (1.3% of the genome), 36 of which are represented in the list of 189 antigens, and 20 in the list of 45 top-ranking antigens (Additional file 1: List of 189 selected antigens and Table 3). Overall, an enrichment of DosR-regulated and reactivation/resuscitation antigens is observed in the list of 45 top-ranking antigens (74% as compared to 36% in the list of 189 selected antigens). This enrichment is obviously in part a consequence of the criteria used for selection, which emphasize late stage phases of the bacteria life cycle. In light of this observation, the result that early stage classical antigens (such as Rv1886 (Ag85B), Rv3804 (Ag85A) and Rv0288 (esxH)) are found among the highest scoring candidates (GroupI and GroupII of the top-ranking 45 antigens, Table 3), provides some validity to the methodology used in this study to map antigens pertinent to all phases of the disease. Murphy and coworkers [18] recently suggested a list of 118 putative dormancy drug targets; interestingly, 27 of these overlap with our 189 selected candidates. Out of these 27 antigens, 16 are part of the DosR regulon, and 11 are included in the list of 45 top-hit antigens. This extent of overlap is a reflection of the facts that: (a) our datasets are not identical; (b) in our analysis, considerable weight was assigned to the immunogenic and virulence potential of each ORF in the genome.
The list of top-ranking 45 antigens (Table 3) could therefore provide a platform for choosing combinations of representatives from the late-stage antigens, which, together with the classical antigens, may contribute to an improved protection as compared to current vaccines based on the early-phase directed BCG. Inclusion of DosR antigens in a future vaccine may be essential in view of the apparent limited immune response induced by the BCG to the DosR regulon proteins, indicating that the vaccine strain probably does not express these late stage antigens [118] moreover the DosR regulon was shown to be constitutively expressed in the Beijing epidemic strain [168], further emphasizing the relevance of these late stage proteins.
Guided by the analysis described in this study, and based on the list of 45 top-ranking antigens (Table 3), we have recently generated a novel rBCG vaccine denoted AERAS-407, which allows over expression of the following selected antigens from this list: (1) the classical antigens Ag85A and Ag85B; (2) DosR regulon genes, the expression of which was induced via overexpression of dosR; (3) the resuscitation antigens Rv0867c, Rv1884c, and Rv2389c. In addition the AERAS-407 vaccine includes the reactivation antigen Rv3407 [95]. This novel rBCG vaccine candidate has been produced and is currently under experimental evaluation.
References
Kaufmann SH: Envisioning future strategies for vaccination against tuberculosis. Nat Rev Immunol. 2006, 6 (9): 699-704. 10.1038/nri1920.
Andersen P: Vaccine strategies against latent tuberculosis infection. Trends Microbiol. 2007, 15 (1): 7-13. 10.1016/j.tim.2006.11.008.
Fine PE: Variation in protection by BCG: implications of and for heterologous immunity. Lancet. 1995, 346 (8986): 1339-1345. 10.1016/S0140-6736(95)92348-9.
Andersen P, Doherty TM: The success and failure of BCG implications – for a novel tuberculosis vaccine. Nat Rev Microbiol. 2005, 3 (8): 656-662. 10.1038/nrmicro1211.
Andersen P, Doherty TM: TB subunit vaccines – putting the pieces together. Microbes Infect. 2005, 7 (5–6): 911-921. 10.1016/j.micinf.2005.03.013.
Skeiky YA, Sadoff JC: Advances in tuberculosis vaccine strategies. Nat Rev Microbiol. 2006, 4 (6): 469-476. 10.1038/nrmicro1419.
Baumann S, Nasser Eddine A, Kaufmann SH: Progress in tuberculosis vaccine development. Curr Opin Immunol. 2006, 18 (4): 438-448. 10.1016/j.coi.2006.05.016.
Dietrich J, Lundberg CV, Andersen P: TB vaccine strategies – what is needed to solve a complex problem?. Tuberculosis (Edinb). 2006, 86 (3–4): 163-168. 10.1016/j.tube.2006.01.009.
Dietrich J, Weldingh K, Andersen P: Prospects for a novel vaccine against tuberculosis. Vet Microbiol. 2006, 112 (2–4): 163-169. 10.1016/j.vetmic.2005.11.030.
Gupta UD, Katoch VM, McMurray DN: Current status of TB vaccines. Vaccine. 2007, 25 (19): 3742-3751. 10.1016/j.vaccine.2007.01.112.
Rachman H, Kaufmann SH: Exploring functional genomics for the development of novel intervention strategies against tuberculosis. Int J Med Microbiol. 2007, 297 (7–8): 559-567. 10.1016/j.ijmm.2007.03.003.
Sable SB, Kalra M, Verma I, Khuller GK: Tuberculosis subunit vaccine design: the conflict of antigenicity and immunogenicity. Clin Immunol. 2007, 122 (3): 239-251. 10.1016/j.clim.2006.10.010.
Sable SB, Plikaytis BB, Shinnick TM: Tuberculosis subunit vaccine development: impact of physicochemical properties of mycobacterial test antigens. Vaccine. 2007, 25 (9): 1553-1566. 10.1016/j.vaccine.2006.11.014.
Sable SB, Verma I, Behera D, Khuller GK: Human immune recognition-based multicomponent subunit vaccines against tuberculosis. Eur Respir J. 2005, 25 (5): 902-910. 10.1183/09031936.05.00105404.
Sable SB, Verma I, Khuller GK: Multicomponent antituberculous subunit vaccine based on immunodominant antigens of Mycobacterium tuberculosis. Vaccine. 2005, 23 (32): 4175-4184. 10.1016/j.vaccine.2005.03.040.
Kaufmann SH, Cole ST, Mizrahi V, Rubin E, Nathan C: Mycobacterium tuberculosis and the host response. J Exp Med. 2005, 201 (11): 1693-1697. 10.1084/jem.20050842.
Rachman H, Strong M, Ulrichs T, Grode L, Schuchhardt J, Mollenkopf H, Kosmiadi GA, Eisenberg D, Kaufmann SH: Unique transcriptome signature of Mycobacterium tuberculosis in pulmonary tuberculosis. Infect Immun. 2006, 74 (2): 1233-1242. 10.1128/IAI.74.2.1233-1242.2006.
Murphy DJ, Brown JR: Identification of gene targets against dormant phase Mycobacterium tuberculosis infections. BMC Infect Dis. 2007, 7: 84-10.1186/1471-2334-7-84.
Palendira U, Spratt JM, Britton WJ, Triccas JA: Expanding the antigenic repertoire of BCG improves protective efficacy against aerosol Mycobacterium tuberculosis infection. Vaccine. 2005, 23 (14): 1680-1685. 10.1016/j.vaccine.2004.10.007.
Kalra M, Grover A, Mehta N, Singh J, Kaur J, Sable SB, Behera D, Sharma P, Verma I, Khuller GK: Supplementation with RD antigens enhances the protective efficacy of BCG in tuberculous mice. Clin Immunol. 2007, 125 (2): 173-183. 10.1016/j.clim.2007.07.007.
Chan J, Flynn J: The immunological aspects of latency in tuberculosis. Clin Immunol. 2004, 110 (1): 2-12. 10.1016/S1521-6616(03)00210-9.
Kaufmann SH: How can immunology contribute to the control of tuberculosis?. Nat Rev Immunol. 2001, 1 (1): 20-30. 10.1038/35095558.
van Pinxteren LA, Cassidy JP, Smedegaard BH, Agger EM, Andersen P: Control of latent Mycobacterium tuberculosis infection is dependent on CD8 T cells. Eur J Immunol. 2000, 30 (12): 3689-3698. 10.1002/1521-4141(200012)30:12<3689::AID-IMMU3689>3.0.CO;2-4.
Leyten EM, Lin MY, Franken KL, Friggen AH, Prins C, van Meijgaarden KE, Voskuil MI, Weldingh K, Andersen P, Schoolnik GK, et al: Human T-cell responses to 25 novel antigens encoded by genes of the dormancy regulon of Mycobacterium tuberculosis. Microbes Infect. 2006, 8 (8): 2052-2060. 10.1016/j.micinf.2006.03.018.
Roupie V, Romano M, Zhang L, Korf H, Lin MY, Franken KL, Ottenhoff TH, Klein MR, Huygen K: Immunogenicity of eight dormancy regulon-encoded proteins of Mycobacterium tuberculosis in DNA-vaccinated and tuberculosis-infected mice. Infect Immun. 2007, 75 (2): 941-949. 10.1128/IAI.01137-06.
Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, Gordon SV, Eiglmeier K, Gas S, Barry CE, et al: Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998, 393 (6685): 537-544. 10.1038/31159.
Hammond AS, Klein MR, Corrah T, Fox A, Jaye A, McAdam KP, Brookes RH: Mycobacterium tuberculosis genome-wide screen exposes multiple CD8 T cell epitopes. Clin Exp Immunol. 2005, 140 (1): 109-116. 10.1111/j.1365-2249.2005.02751.x.
McMurry J, Sbai H, Gennaro ML, Carter EJ, Martin W, DeGroot AS: Analyzing Mycobacterium tuberculosis proteomes for candidate vaccine epitopes. Tuberculosis (Edinb). 2005, 85 (1–2): 95-105. 10.1016/j.tube.2004.09.005.
McMurry JA, Kimball S, Lee JH, Rivera D, Martin W, Weiner DB, Kutzler M, Sherman DR, Kornfeld H, De Groot AS: Epitope-driven TB vaccine development: a streamlined approach using immuno-informatics, ELISpot assays, and HLA transgenic mice. Curr Mol Med. 2007, 7 (4): 351-368. 10.2174/156652407780831584.
Mustafa AS, Shaban FA: ProPred analysis and experimental evaluation of promiscuous T-cell epitopes of three major secreted antigens of Mycobacterium tuberculosis. Tuberculosis (Edinb). 2006, 86 (2): 115-124. 10.1016/j.tube.2005.05.001.
Vani J, Shaila MS, Chandra NR, Nayak R: A combined immuno-informatics and structure-based modeling approach for prediction of T cell epitopes of secretory proteins of Mycobacterium tuberculosis. Microbes Infect. 2006, 8 (3): 738-746. 10.1016/j.micinf.2005.09.012.
Vordermeier M, Whelan AO, Hewinson RG: Recognition of mycobacterial epitopes by T cells across mammalian species and use of a program that predicts human HLA-DR binding peptides to predict bovine epitopes. Infect Immun. 2003, 71 (4): 1980-1987. 10.1128/IAI.71.4.1980-1987.2003.
The NCBI ftp site of bacterial genomes. [ftp://ftp.ncbi.nlm.nih.gov/genomes/Bacteria]
Marchler-Bauer A, Anderson JB, Cherukuri PF, DeWeese-Scott C, Geer LY, Gwadz M, He S, Hurwitz DI, Jackson JD, Ke Z, et al: CDD: a Conserved Domain Database for protein classification. Nucleic Acids Res. 2005, D192-196. 33 Database
Schultz J, Milpetz F, Bork P, Ponting CP: SMART, a simple modular architecture research tool: identification of signaling domains. Proc Natl Acad Sci USA. 1998, 95 (11): 5857-5864. 10.1073/pnas.95.11.5857.
Finn RD, Mistry J, Schuster-Bockler B, Griffiths-Jones S, Hollich V, Lassmann T, Moxon S, Marshall M, Khanna A, Durbin R, et al: Pfam: clans, web tools and services. Nucleic Acids Res. 2006, D247-251. 10.1093/nar/gkj149. 34 Database
Mulder NJ, Apweiler R, Attwood TK, Bairoch A, Bateman A, Binns D, Bork P, Buillard V, Cerutti L, Copley R, et al: New developments in the InterPro database. Nucleic Acids Res. 2007, D224-228. 10.1093/nar/gkl841. 35 Database
Prosite Database – of protein domains, families and functional sites. [http://www.ebi.ac.uk/ppsearch/]
Bendtsen JD, Nielsen H, von Heijne G, Brunak S: Improved prediction of signal peptides: SignalP 3.0. J Mol Biol. 2004, 340 (4): 783-795. 10.1016/j.jmb.2004.05.028.
Nielsen H, Engelbrecht J, Brunak S, von Heijne G: A neural network method for identification of prokaryotic and eukaryotic signal peptides and prediction of their cleavage sites. Int J Neural Syst. 1997, 8 (5–6): 581-599. 10.1142/S0129065797000537.
Rose RW, Bruser T, Kissinger JC, Pohlschroder M: Adaptation of protein secretion to extremely high-salt conditions by extensive use of the twin-arginine translocation pathway. Mol Microbiol. 2002, 45 (4): 943-950. 10.1046/j.1365-2958.2002.03090.x.
Hofmann K, Stoffel W: TMbase A – database of membrane spanning proteins segments. Biol Chem Hoppe-Seyler. 1993, 374: 166.
Madan Babu M, Sankaran K: DOLOP – database of bacterial lipoproteins. Bioinformatics. 2002, 18 (4): 641-643. 10.1093/bioinformatics/18.4.641.
Larsen MV, Lundegaard C, Lamberth K, Buus S, Brunak S, Lund O, Nielsen M: An integrative approach to CTL epitope prediction: a combined algorithm integrating MHC class I binding, TAP transport efficiency, and proteasomal cleavage predictions. Eur J Immunol. 2005, 35 (8): 2295-2303. 10.1002/eji.200425811.
Peters B, Sidney J, Bourne P, Bui HH, Buus S, Doh G, Fleri W, Kronenberg M, Kubo R, Lund O, et al: The immune epitope database and analysis resource: from vision to blueprint. PLoS Biol. 2005, 3 (3): e91-10.1371/journal.pbio.0030091.
TB-sgc The-TB Structural Genomics Consortium. [http://www.doe-mbi.ucla.edu/TB/]
Tuberculist – the database on Mycobacterium tuberculosis genetics. [http://genolist.pasteur.fr/TubercuList]
TBDB – an integrated platform for TB drug discovery. [http://www.tbdb.org]
MTBreg-The Database of Conditionally Regulated Proteins in Mycobacterium tuberculosis. [http://www.doe-mbi.ucla.edu/Services/MTBreg/]
BioHealthBase – the Biodefense and Public Health Database. [http://www.biohealthbase.org/GSearch]
Camus JC, Pryor MJ, Medigue C, Cole ST: Re-annotation of the genome sequence of Mycobacterium tuberculosis H37Rv. Microbiology. 2002, 148 (Pt 10): 2967-2973.
Voskuil MI, Schnappinger D, Visconti KC, Harrell MI, Dolganov GM, Sherman DR, Schoolnik GK: Inhibition of respiration by nitric oxide induces a Mycobacterium tuberculosis dormancy program. J Exp Med. 2003, 198 (5): 705-713. 10.1084/jem.20030205.
Voskuil MI, Visconti KC, Schoolnik GK: Mycobacterium tuberculosis gene expression during adaptation to stationary phase and low-oxygen dormancy. Tuberculosis (Edinb). 2004, 84 (3–4): 218-227. 10.1016/j.tube.2004.02.003.
Talaat AM, Ward SK, Wu CW, Rondon E, Tavano C, Bannantine JP, Lyons R, Johnston SA: Mycobacterial bacilli are metabolically active during chronic tuberculosis in murine lungs: insights from genome-wide transcriptional profiling. J Bacteriol. 2007, 189 (11): 4265-4274. 10.1128/JB.00011-07.
Zhu W, Plikaytis BB, Shinnick TM: Resuscitation factors from mycobacteria: homologs of Micrococcus luteus proteins. Tuberculosis (Edinb). 2003, 83 (4): 261-269. 10.1016/S1472-9792(03)00052-0.
Cappelli G, Volpe E, Grassi M, Liseo B, Colizzi V, Mariani F: Profiling of Mycobacterium tuberculosis gene expression during human macrophage infection: upregulation of the alternative sigma factor G, a group of transcriptional regulators, and proteins with unknown function. Res Microbiol. 2006, 157 (5): 445-455. 10.1016/j.resmic.2005.10.007.
Fisher MA, Plikaytis BB, Shinnick TM: Microarray analysis of the Mycobacterium tuberculosis transcriptional response to the acidic conditions found in phagosomes. J Bacteriol. 2002, 184 (14): 4025-4032. 10.1128/JB.184.14.4025-4032.2002.
Monahan IM, Betts J, Banerjee DK, Butcher PD: Differential expression of mycobacterial proteins following phagocytosis by macrophages. Microbiology. 2001, 147 (Pt 2): 459-471.
Rengarajan J, Bloom BR, Rubin EJ: Genome-wide requirements for Mycobacterium tuberculosis adaptation and survival in macrophages. Proc Natl Acad Sci USA. 2005, 102 (23): 8327-8332. 10.1073/pnas.0503272102.
Rosas-Magallanes V, Stadthagen-Gomez G, Rauzier J, Barreiro LB, Tailleux L, Boudou F, Griffin R, Nigou J, Jackson M, Gicquel B, et al: Signature-tagged transposon mutagenesis identifies novel Mycobacterium tuberculosis genes involved in the parasitism of human macrophages. Infect Immun. 2007, 75 (1): 504-507. 10.1128/IAI.00058-06.
Sassetti CM, Boyd DH, Rubin EJ: Genes required for mycobacterial growth defined by high density mutagenesis. Mol Microbiol. 2003, 48 (1): 77-84. 10.1046/j.1365-2958.2003.03425.x.
Schnappinger D, Ehrt S, Voskuil MI, Liu Y, Mangan JA, Monahan IM, Dolganov G, Efron B, Butcher PD, Nathan C, et al: Transcriptional Adaptation of Mycobacterium tuberculosis within Macrophages: Insights into the Phagosomal Environment. J Exp Med. 2003, 198 (5): 693-704. 10.1084/jem.20030846.
Stewart GR, Patel J, Robertson BD, Rae A, Young DB: Mycobacterial mutants with defective control of phagosomal acidification. PLoS Pathog. 2005, 1 (3): 269-278. 10.1371/journal.ppat.0010033.
Talaat AM, Lyons R, Howard ST, Johnston SA: The temporal expression profile of Mycobacterium tuberculosis infection in mice. Proc Natl Acad Sci USA. 2004, 101 (13): 4602-4607. 10.1073/pnas.0306023101.
He H, Hovey R, Kane J, Singh V, Zahrt TC: MprAB is a stress-responsive two-component system that directly regulates expression of sigma factors SigB and SigE in Mycobacterium tuberculosis. J Bacteriol. 2006, 188 (6): 2134-2143. 10.1128/JB.188.6.2134-2143.2006.
Wayne LG, Sohaskey CD: Nonreplicating persistence of mycobacterium tuberculosis. Annu Rev Microbiol. 2001, 55: 139-163. 10.1146/annurev.micro.55.1.139.
Sherman DR, Voskuil M, Schnappinger D, Liao R, Harrell MI, Schoolnik GK: Regulation of the Mycobacterium tuberculosis hypoxic response gene encoding alpha-crystallin. Proc Natl Acad Sci USA. 2001, 98 (13): 7534-7539. 10.1073/pnas.121172498.
Flynn JL, Chan J: Tuberculosis: latency and reactivation. Infect Immun. 2001, 69 (7): 4195-4201. 10.1128/IAI.69.7.4195-4201.2001.
Scanga CA, Mohan VP, Joseph H, Yu K, Chan J, Flynn JL: Reactivation of latent tuberculosis: variations on the Cornell murine model. Infect Immun. 1999, 67 (9): 4531-4538.
Tufariello JM, Jacobs WR, Chan J: Individual Mycobacterium tuberculosis resuscitation-promoting factor homologues are dispensable for growth in vitro and in vivo. Infect Immun. 2004, 72 (1): 515-526. 10.1128/IAI.72.1.515-526.2004.
Tufariello JM, Mi K, Xu J, Manabe YC, Kesavan AK, Drumm J, Tanaka K, Jacobs WR, Chan J: Deletion of the Mycobacterium tuberculosis resuscitation-promoting factor Rv1009 gene results in delayed reactivation from chronic tuberculosis. Infect Immun. 2006, 74 (5): 2985-2995. 10.1128/IAI.74.5.2985-2995.2006.
Starck J, Kallenius G, Marklund BI, Andersson DI, Akerlund T: Comparative proteome analysis of Mycobacterium tuberculosis grown under aerobic and anaerobic conditions. Microbiology. 2004, 150 (Pt 11): 3821-3829. 10.1099/mic.0.27284-0.
Dubnau E, Chan J, Mohan VP, Smith I: responses of mycobacterium tuberculosis to growth in the mouse lung. Infect Immun. 2005, 73 (6): 3754-3757. 10.1128/IAI.73.6.3754-3757.2005.
Fenhalls G, Stevens L, Moses L, Bezuidenhout J, Betts JC, Helden Pv P, Lukey PT, Duncan K: In situ detection of Mycobacterium tuberculosis transcripts in human lung granulomas reveals differential gene expression in necrotic lesions. Infect Immun. 2002, 70 (11): 6330-6338. 10.1128/IAI.70.11.6330-6338.2002.
Jain SK, Hernandez-Abanto SM, Cheng QJ, Singh P, Ly LH, Klinkenberg LG, Morrison NE, Converse PJ, Nuermberger E, Grosset J, et al: Accelerated detection of Mycobacterium tuberculosis genes essential for bacterial survival in guinea pigs, compared with mice. J Infect Dis. 2007, 195 (11): 1634-1642. 10.1086/517526.
Lamichhane G, Tyagi S, Bishai WR: Designer arrays for defined mutant analysis to detect genes essential for survival of Mycobacterium tuberculosis in mouse lungs. Infect Immun. 2005, 73 (4): 2533-2540. 10.1128/IAI.73.4.2533-2540.2005.
Shi L, Jung YJ, Tyagi S, Gennaro ML, North RJ: Expression of Th1-mediated immunity in mouse lungs induces a Mycobacterium tuberculosis transcription pattern characteristic of nonreplicating persistence. Proc Natl Acad Sci USA. 2003, 100 (1): 241-246. 10.1073/pnas.0136863100.
Shi L, North R, Gennaro ML: Effect of growth state on transcription levels of genes encoding major secreted antigens of Mycobacterium tuberculosis in the mouse lung. Infect Immun. 2004, 72 (4): 2420-2424. 10.1128/IAI.72.4.2420-2424.2004.
Purkayastha A, McCue LA, McDonough KA: Identification of a Mycobacterium tuberculosis putative classical nitroreductase gene whose expression is coregulated with that of the acr aene within macrophages, in standing versus shaking cultures, and under low oxygen conditions. Infect Immun. 2002, 70 (3): 1518-1529. 10.1128/IAI.70.3.1518-1529.2002.
Yuan Y, Crane DD, Barry CE: Stationary phase-associated protein expression in Mycobacterium tuberculosis: function of the mycobacterial alpha-crystallin homolog. J Bacteriol. 1996, 178 (15): 4484-4492.
Yuan Y, Crane DD, Simpson RM, Zhu YQ, Hickey MJ, Sherman DR, Barry CE: The 16-kDa alpha-crystallin (Acr) protein of Mycobacterium tuberculosis is required for growth in macrophages. Proc Natl Acad Sci USA. 1998, 95 (16): 9578-9583. 10.1073/pnas.95.16.9578.
Florczyk MA, McCue LA, Purkayastha A, Currenti E, Wolin MJ, McDonough KA: A family of acr-coregulated Mycobacterium tuberculosis genes shares a common DNA motif and requires Rv3133c (dosR or devR) for expression. Infect Immun. 2003, 71 (9): 5332-5343. 10.1128/IAI.71.9.5332-5343.2003.
Gomez M, Johnson S, Gennaro ML: Identification of secreted proteins of Mycobacterium tuberculosis by a bioinformatic approach. Infect Immun. 2000, 68 (4): 2323-2327. 10.1128/IAI.68.4.2323-2327.2000.
Brusasca PN, Colangeli R, Lyashchenko KP, Zhao X, Vogelstein M, Spencer JS, McMurray DN, Gennaro ML: Immunological characterization of antigens encoded by the RD1 region of the Mycobacterium tuberculosis genome. Scand J Immunol. 2001, 54 (5): 448-452. 10.1046/j.1365-3083.2001.00975.x.
Cockle PJ, Gordon SV, Lalvani A, Buddle BM, Hewinson RG, Vordermeier HM: Identification of novel Mycobacterium tuberculosis antigens with potential as diagnostic reagents or subunit vaccine candidates by comparative genomics. Infect Immun. 2002, 70 (12): 6996-7003. 10.1128/IAI.70.12.6996-7003.2002.
Vekemans J, Ota MO, Sillah J, Fielding K, Alderson MR, Skeiky YA, Dalemans W, McAdam KP, Lienhardt C, Marchant A: Immune responses to mycobacterial antigens in the Gambian population: implications for vaccines and immunodiagnostic test design. Infect Immun. 2004, 72 (1): 381-388. 10.1128/IAI.72.1.381-388.2004.
Weldingh K, Rosenkrands I, Okkels LM, Doherty TM, Andersen P: Assessing the serodiagnostic potential of 35 Mycobacterium tuberculosis proteins and identification of four novel serological antigens. J Clin Microbiol. 2005, 43 (1): 57-65. 10.1128/JCM.43.1.57-65.2005.
Yeremeev VV, Kondratieva TK, Rubakova EI, Petrovskaya SN, Kazarian KA, Telkov MV, Biketov SF, Kaprelyants AS, Apt AS: Proteins of the Rpf family: immune cell reactivity and vaccination efficacy against tuberculosis in mice. Infect Immun. 2003, 71 (8): 4789-4794. 10.1128/IAI.71.8.4789-4794.2003.
Ben Amor Y, Shashkina E, Johnson S, Bifani PJ, Kurepina N, Kreiswirth B, Bhattacharya S, Spencer J, Rendon A, Catanzaro A, et al: Immunological characterization of novel secreted antigens of Mycobacterium tuberculosis. Scand J Immunol. 2005, 61 (2): 139-146. 10.1111/j.0300-9475.2005.01557.x.
Rodriguez GM: Control of iron metabolism in Mycobacterium tuberculosis. Trends Microbiol. 2006, 14 (7): 320-327. 10.1016/j.tim.2006.05.006.
Rodriguez GM, Smith I: Mechanisms of iron regulation in mycobacteria: role in physiology and virulence. Mol Microbiol. 2003, 47 (6): 1485-1494. 10.1046/j.1365-2958.2003.03384.x.
Schaible UE, Kaufmann SH: Iron and microbial infection. Nat Rev Microbiol. 2004, 2 (12): 946-953. 10.1038/nrmicro1046.
Gold B, Rodriguez GM, Marras SA, Pentecost M, Smith I: The Mycobacterium tuberculosis IdeR is a dual functional regulator that controls transcription of genes involved in iron acquisition, iron storage and survival in macrophages. Mol Microbiol. 2001, 42 (3): 851-865. 10.1046/j.1365-2958.2001.02684.x.
Rodriguez GM, Voskuil MI, Gold B, Schoolnik GK, Smith I: ideR, An essential gene in mycobacterium tuberculosis: role of IdeR in iron-dependent gene expression, iron metabolism, and oxidative stress response. Infect Immun. 2002, 70 (7): 3371-3381. 10.1128/IAI.70.7.3371-3381.2002.
Mollenkopf HJ, Grode L, Mattow J, Stein M, Mann P, Knapp B, Ulmer J, Kaufmann SH: Application of mycobacterial proteomics to vaccine design: improved protection by Mycobacterium bovis BCG prime-Rv3407 DNA boost vaccination against tuberculosis. Infect Immun. 2004, 72 (11): 6471-6479. 10.1128/IAI.72.11.6471-6479.2004.
Vipond J, Clark SO, Hatch GJ, Vipond R, Marie Agger E, Tree JA, Williams A, Marsh PD: Re-formulation of selected DNA vaccine candidates and their evaluation as protein vaccines using a guinea pig aerosol infection model of tuberculosis. Tuberculosis (Edinb). 2006, 86 (3–4): 218-224. 10.1016/j.tube.2006.01.014.
Vipond J, Vipond R, Allen-Vercoe E, Clark SO, Hatch GJ, Gooch KE, Bacon J, Hampshire T, Shuttleworth H, Minton NP, et al: Selection of novel TB vaccine candidates and their evaluation as DNA vaccines against aerosol challenge. Vaccine. 2006, 24 (37–39): 6340-6350. 10.1016/j.vaccine.2006.05.025.
Al-Attiyah R, Madi N, El-Shamy AS, Wiker H, Andersen P, Mustafa A: Cytokine profiles in tuberculosis patients and healthy subjects in response to complex and single antigens of Mycobacterium tuberculosis. FEMS Immunol Med Microbiol. 2006, 47 (2): 254-261. 10.1111/j.1574-695X.2006.00110.x.
Alderson MR, Bement T, Day CH, Zhu L, Molesh D, Skeiky YA, Coler R, Lewinsohn DM, Reed SG, Dillon DC: Expression cloning of an immunodominant family of Mycobacterium tuberculosis antigens using human CD4(+) T cells. J Exp Med. 2000, 191 (3): 551-560. 10.1084/jem.191.3.551.
Brandt L, Elhay M, Rosenkrands I, Lindblad EB, Andersen P: ESAT-6 subunit vaccination against Mycobacterium tuberculosis. Infect Immun. 2000, 68 (2): 791-795. 10.1128/IAI.68.2.791-795.2000.
Brandt L, Skeiky YA, Alderson MR, Lobet Y, Dalemans W, Turner OC, Basaraba RJ, Izzo AA, Lasco TM, Chapman PL, et al: The protective effect of the Mycobacterium bovis BCG vaccine is increased by coadministration with the Mycobacterium tuberculosis 72-kilodalton fusion polyprotein Mtb72F in M. tuberculosis-infected guinea pigs. Infect Immun. 2004, 72 (11): 6622-6632. 10.1128/IAI.72.11.6622-6632.2004.
Brodin P, Majlessi L, Marsollier L, de Jonge MI, Bottai D, Demangel C, Hinds J, Neyrolles O, Butcher PD, Leclerc C, et al: Dissection of ESAT-6 system 1 of Mycobacterium tuberculosis and impact on immunogenicity and virulence. Infect Immun. 2006, 74 (1): 88-98. 10.1128/IAI.74.1.88-98.2006.
Chaitra MG, Nayak R, Shaila MS: Modulation of immune responses in mice to recombinant antigens from PE and PPE families of proteins of Mycobacterium tuberculosis by the Ribi adjuvant. Vaccine. 2007, 25 (41): 7168-7176. 10.1016/j.vaccine.2007.07.026.
Choudhary RK, Pullakhandam R, Ehtesham NZ, Hasnain SE: Expression and characterization of Rv2430c, a novel immunodominant antigen of Mycobacterium tuberculosis. Protein Expr Purif. 2004, 36 (2): 249-253. 10.1016/j.pep.2004.03.016.
Coler RN, Campos-Neto A, Ovendale P, Day FH, Fling SP, Zhu L, Serbina N, Flynn JL, Reed SG, Alderson MR: Vaccination with the T cell antigen Mtb 8.4 protects against challenge with Mycobacterium tuberculosis. J Immunol. 2001, 166 (10): 6227-6235.
Demangel C, Brodin P, Cockle PJ, Brosch R, Majlessi L, Leclerc C, Cole ST: Cell envelope protein PPE68 contributes to Mycobacterium tuberculosis RD1 immunogenicity independently of a 10-kilodalton culture filtrate protein and ESAT-6. Infect Immun. 2004, 72 (4): 2170-2176. 10.1128/IAI.72.4.2170-2176.2004.
Dietrich J, Aagaard C, Leah R, Olsen AW, Stryhn A, Doherty TM, Andersen P: Exchanging ESAT6 with TB10.4 in an Ag85B fusion molecule-based tuberculosis subunit vaccine: efficient protection and ESAT6-based sensitive monitoring of vaccine efficacy. J Immunol. 2005, 174 (10): 6332-6339.
Fan X, Gao Q, Fu R: Differential immunogenicity and protective efficacy of DNA vaccines expressing proteins of Mycobacterium tuberculosis in a mouse model. Microbiol Res. 2007.
Feng CG, Blanchard TJ, Smith GL, Hill AV, Britton WJ: Induction of CD8+ T-lymphocyte responses to a secreted antigen of Mycobacterium tuberculosis by an attenuated vaccinia virus. Immunol Cell Biol. 2001, 79 (6): 569-575. 10.1046/j.1440-1711.2001.01042.x.
Ferraz JC, Stavropoulos E, Yang M, Coade S, Espitia C, Lowrie DB, Colston MJ, Tascon RE: A heterologous DNA priming-Mycobacterium bovis BCG boosting immunization strategy using mycobacterial Hsp70, Hsp65, and Apa antigens improves protection against tuberculosis in mice. Infect Immun. 2004, 72 (12): 6945-6950. 10.1128/IAI.72.12.6945-6950.2004.
Geluk A, Lin MY, van Meijgaarden KE, Leyten EM, Franken KL, Ottenhoff TH, Klein MR: T-cell recognition of the HspX protein of Mycobacterium tuberculosis correlates with latent M. tuberculosis infection but not with M. bovis BCG vaccination. Infect Immun. 2007, 75 (6): 2914-2921. 10.1128/IAI.01990-06.
Geluk A, van Meijgaarden KE, Franken KL, Wieles B, Arend SM, Faber WR, Naafs B, Ottenhoff TH: Immunological crossreactivity of the Mycobacterium leprae CFP-10 with its homologue in Mycobacterium tuberculosis. Scand J Immunol. 2004, 59 (1): 66-70. 10.1111/j.0300-9475.2004.01358.x.
Giri PK, Verma I, Khuller GK: Enhanced immunoprotective potential of Mycobacterium tuberculosis Ag85 complex protein based vaccine against airway Mycobacterium tuberculosis challenge following intranasal administration. FEMS Immunol Med Microbiol. 2006, 47 (2): 233-241. 10.1111/j.1574-695X.2006.00087.x.
Grover A, Ahmed MF, Singh B, Verma I, Sharma P, Khuller GK: A multivalent combination of experimental antituberculosis DNA vaccines based on Ag85B and regions of difference antigens. Microbes Infect. 2006, 8 (9–10): 2390-2399. 10.1016/j.micinf.2006.04.025.
Hervas-Stubbs S, Majlessi L, Simsova M, Morova J, Rojas MJ, Nouze C, Brodin P, Sebo P, Leclerc C: High frequency of CD4+ T cells specific for the TB10.4 protein correlates with protection against Mycobacterium tuberculosis infection. Infect Immun. 2006, 74 (6): 3396-3407. 10.1128/IAI.02086-05.
Horwitz MA, Lee BW, Dillon BJ, Harth G: Protective immunity against tuberculosis induced by vaccination with major extracellular proteins of Mycobacterium tuberculosis. Proc Natl Acad Sci USA. 1995, 92 (5): 1530-1534. 10.1073/pnas.92.5.1530.
Khera A, Singh R, Shakila H, Rao V, Dhar N, Narayanan PR, Parmasivan CN, Ramanathan VD, Tyagi AK: Elicitation of efficient, protective immune responses by using DNA vaccines against tuberculosis. Vaccine. 2005, 23 (48–49): 5655-5665. 10.1016/j.vaccine.2005.03.056.
Lin MY, Geluk A, Smith SG, Stewart AL, Friggen AH, Franken KL, Verduyn MJ, van Meijgaarden KE, Voskuil MI, Dockrell HM, et al: Lack of immune responses to Mycobacterium tuberculosis DosR regulon proteins following Mycobacterium bovis BCG vaccination. Infect Immun. 2007, 75 (7): 3523-3530. 10.1128/IAI.01999-06.
Liu XQ, Dosanjh D, Varia H, Ewer K, Cockle P, Pasvol G, Lalvani A: Evaluation of T-cell responses to novel RD1-and RD2-encoded Mycobacterium tuberculosis gene products for specific detection of human tuberculosis infection. Infect Immun. 2004, 72 (5): 2574-2581. 10.1128/IAI.72.5.2574-2581.2004.
Majlessi L, Brodin P, Brosch R, Rojas MJ, Khun H, Huerre M, Cole ST, Leclerc C: Influence of ESAT-6 secretion system 1 (RD1) of Mycobacterium tuberculosis on the interaction between mycobacteria and the host immune system. J Immunol. 2005, 174 (6): 3570-3579.
McShane H, Pathan AA, Sander CR, Goonetilleke NP, Fletcher HA, Hill AV: Boosting BCG with MVA85A: the first candidate subunit vaccine for tuberculosis in clinical trials. Tuberculosis (Edinb). 2005, 85 (1–2): 47-52. 10.1016/j.tube.2004.09.015.
Morris S, Kelley C, Howard A, Li Z, Collins F: The immunogenicity of single and combination DNA vaccines against tuberculosis. Vaccine. 2000, 18 (20): 2155-2163. 10.1016/S0264-410X(99)00540-X.
Mukamolova GV, Turapov OA, Kazarian K, Telkov M, Kaprelyants AS, Kell DB, Young M: The rpf gene of Micrococcus luteus encodes an essential secreted growth factor. Mol Microbiol. 2002, 46 (3): 611-621. 10.1046/j.1365-2958.2002.03183.x.
Mustafa AS, Cockle PJ, Shaban F, Hewinson RG, Vordermeier HM: Immunogenicity of Mycobacterium tuberculosis RD1 region gene products in infected cattle. Clin Exp Immunol. 2002, 130 (1): 37-42. 10.1046/j.1365-2249.2002.01937.x.
Mustafa AS, Skeiky YA, Al-Attiyah R, Alderson MR, Hewinson RG, Vordermeier HM: Immunogenicity of Mycobacterium tuberculosis antigens in Mycobacterium bovis BCG-vaccinated and M. bovis-infected cattle. Infect Immun. 2006, 74 (8): 4566-4572. 10.1128/IAI.01660-05.
Nagai S, Wiker HG, Harboe M, Kinomoto M: Isolation and partial characterization of major protein antigens in the culture fluid of Mycobacterium tuberculosis. Infect Immun. 1991, 59 (1): 372-382.
Okkels LM, Brock I, Follmann F, Agger EM, Arend SM, Ottenhoff TH, Oftung F, Rosenkrands I, Andersen P: PPE protein (Rv3873) from DNA segment RD1 of Mycobacterium tuberculosis: strong recognition of both specific T-cell epitopes and epitopes conserved within the PPE family. Infect Immun. 2003, 71 (11): 6116-6123. 10.1128/IAI.71.11.6116-6123.2003.
Olsen AW, Hansen PR, Holm A, Andersen P: Efficient protection against Mycobacterium tuberculosis by vaccination with a single subdominant epitope from the ESAT-6 antigen. Eur J Immunol. 2000, 30 (6): 1724-1732. 10.1002/1521-4141(200006)30:6<1724::AID-IMMU1724>3.0.CO;2-A.
Radosevic K, Wieland CW, Rodriguez A, Weverling GJ, Mintardjo R, Gillissen G, Vogels R, Skeiky YA, Hone DM, Sadoff JC, et al: Protective immune responses to a recombinant adenovirus type 35 tuberculosis vaccine in two mouse strains: CD4 and CD8 T-cell epitope mapping and role of gamma interferon. Infect Immun. 2007, 75 (8): 4105-4115. 10.1128/IAI.00004-07.
Rao V, Dhar N, Shakila H, Singh R, Khera A, Jain R, Naseema M, Paramasivan CN, Narayanan PR, Ramanathan VD, et al: Increased expression of Mycobacterium tuberculosis 19 kDa lipoprotein obliterates the protective efficacy of BCG by polarizing host immune responses to the Th2 subtype. Scand J Immunol. 2005, 61 (5): 410-417. 10.1111/j.1365-3083.2005.01569.x.
Rao V, Dhar N, Tyagi AK: Modulation of host immune responses by overexpression of immunodominant antigens of Mycobacterium tuberculosis in bacille Calmette-Guerin. Scand J Immunol. 2003, 58 (4): 449-461. 10.1046/j.1365-3083.2003.01321.x.
Romano M, Roupie V, Wang XM, Denis O, Jurion F, Adnet PY, Laali R, Huygen K: Immunogenicity and protective efficacy of tuberculosis DNA vaccines combining mycolyl-transferase Ag85A and phosphate transport receptor PstS-3. Immunology. 2006, 118 (3): 321-332. 10.1111/j.1365-2567.2006.02373.x.
Santosuosso M, McCormick S, Zhang X, Zganiacz A, Xing Z: Intranasal boosting with an adenovirus-vectored vaccine markedly enhances protection by parenteral Mycobacterium bovis BCG immunization against pulmonary tuberculosis. Infect Immun. 2006, 74 (8): 4634-4643. 10.1128/IAI.00517-06.
Scandurra GM, Williams RB, Triccas JA, Pinto R, Gicquel B, Slobedman B, Cunningham A, Britton WJ: Effect of phthiocerol dimycocerosate deficiency on the transcriptional response of human macrophages to Mycobacterium tuberculosis. Microbes Infect. 2007, 9 (1): 87-95. 10.1016/j.micinf.2006.10.013.
Skeiky YA, Lodes MJ, Guderian JA, Mohamath R, Bement T, Alderson MR, Reed SG: Cloning, expression, and immunological evaluation of two putative secreted serine protease antigens of Mycobacterium tuberculosis. Infect Immun. 1999, 67 (8): 3998-4007.
Skjot RL, Brock I, Arend SM, Munk ME, Theisen M, Ottenhoff TH, Andersen P: Epitope mapping of the immunodominant antigen TB10.4 and the two homologous proteins TB10.3 and TB12.9, which constitute a subfamily of the esat-6 gene family. Infect Immun. 2002, 70 (10): 5446-5453. 10.1128/IAI.70.10.5446-5453.2002.
Stewart GR, Wilkinson KA, Newton SM, Sullivan SM, Neyrolles O, Wain JR, Patel J, Pool KL, Young DB, Wilkinson RJ: Effect of deletion or overexpression of the 19-kilodalton lipoprotein Rv3763 on the innate response to Mycobacterium tuberculosis. Infect Immun. 2005, 73 (10): 6831-6837. 10.1128/IAI.73.10.6831-6837.2005.
Sugawara I, Udagawa T, Taniyama T: Protective efficacy of recombinant (Ag85A) BCG Tokyo with Ag85A peptide boosting against Mycobacterium tuberculosis-infected guinea pigs in comparison with that of DNA vaccine encoding Ag85A. Tuberculosis (Edinb). 2007, 87 (2): 94-101. 10.1016/j.tube.2006.05.001.
Tsenova L, Harbacheuski R, Moreira AL, Ellison E, Dalemans W, Alderson MR, Mathema B, Reed SG, Skeiky YA, Kaplan G: Evaluation of the Mtb72F polyprotein vaccine in a rabbit model of tuberculous meningitis. Infect Immun. 2006, 74 (4): 2392-2401. 10.1128/IAI.74.4.2392-2401.2006.
Turner J, Dobos KM, Keen MA, Frank AA, Ehlers S, Orme IM, Belisle JT, Cooper AM: A limited antigen-specific cellular response is sufficient for the early control of Mycobacterium tuberculosis in the lung but is insufficient for long-term survival. Infect Immun. 2004, 72 (7): 3759-3768. 10.1128/IAI.72.7.3759-3768.2004.
Wang BL, Xu Y, Li ZM, Xu YM, Weng XH, Wang HH: Antibody response to four secretory proteins from Mycobacterium tuberculosis and their complex antigen in TB patients. Int J Tuberc Lung Dis. 2005, 9 (12): 1327-1334.
Agger EM, Brock I, Okkels LM, Arend SM, Aagaard CS, Weldingh KN, Andersen P: Human T-cell responses to the RD1-encoded protein TB27.4 (Rv3878) from Mycobacterium tuberculosis. Immunology. 2003, 110 (4): 507-512. 10.1111/j.1365-2567.2003.01763.x.
Brock I, Weldingh K, Leyten EM, Arend SM, Ravn P, Andersen P: Specific T-cell epitopes for immunoassay-based diagnosis of Mycobacterium tuberculosis infection. J Clin Microbiol. 2004, 42 (6): 2379-2387. 10.1128/JCM.42.6.2379-2387.2004.
Mustafa AS: Recombinant and synthetic peptides to identify Mycobacterium tuberculosis antigens and epitopes of diagnostic and vaccine relevance. Tuberculosis (Edinb). 2005, 85 (5–6): 367-376. 10.1016/j.tube.2005.08.006.
Huet G, Castaing JP, Fournier D, Daffe M, Saves I: Protein splicing of SufB is crucial for the functionality of the Mycobacterium tuberculosis SUF machinery. J Bacteriol. 2006, 188 (9): 3412-3414. 10.1128/JB.188.9.3412-3414.2006.
Huet G, Daffe M, Saves I: Identification of the Mycobacterium tuberculosis SUF machinery as the exclusive mycobacterial system of [Fe-S] cluster assembly: evidence for its implication in the pathogen's survival. J Bacteriol. 2005, 187 (17): 6137-6146. 10.1128/JB.187.17.6137-6146.2005.
Munoz-Elias EJ, McKinney JD: Carbon metabolism of intracellular bacteria. Cell Microbiol. 2006, 8 (1): 10-22. 10.1111/j.1462-5822.2005.00648.x.
Bloch H, Segal W: Biochemical differentiation of Mycobacterium tuberculosis grown in vivo and in vitro. J Bacteriol. 1956, 72 (2): 132-141.
Bhatt K, Gurcha SS, Bhatt A, Besra GS, Jacobs WR: Two polyketide-synthase-associated acyltransferases are required for sulfolipid biosynthesis in Mycobacterium tuberculosis. Microbiology. 2007, 153 (Pt 2): 513-520. 10.1099/mic.0.2006/003103-0.
Kremer L, Dover LG, Carrere S, Nampoothiri KM, Lesjean S, Brown AK, Brennan PJ, Minnikin DE, Locht C, Besra GS: Mycolic acid biosynthesis and enzymic characterization of the beta-ketoacyl-ACP synthase A-condensing enzyme from Mycobacterium tuberculosis. Biochem J. 2002, 364 (Pt 2): 423-430. 10.1042/BJ20011628.
Takayama K, Wang C, Besra GS: Pathway to synthesis and processing of mycolic acids in Mycobacterium tuberculosis. Clin Microbiol Rev. 2005, 18 (1): 81-101. 10.1128/CMR.18.1.81-101.2005.
Hotter GS, Wards BJ, Mouat P, Besra GS, Gomes J, Singh M, Bassett S, Kawakami P, Wheeler PR, de Lisle GW, et al: Transposon mutagenesis of Mb0100 at the ppe1-nrp locus in Mycobacterium bovis disrupts phthiocerol dimycocerosate (PDIM) and glycosylphenol-PDIM biosynthesis, producing an avirulent strain with vaccine properties at least equal to those of M. bovis BCG. J Bacteriol. 2005, 187 (7): 2267-2277. 10.1128/JB.187.7.2267-2277.2005.
Camacho LR, Constant P, Raynaud C, Laneelle MA, Triccas JA, Gicquel B, Daffe M, Guilhot C: Analysis of the phthiocerol dimycocerosate locus of Mycobacterium tuberculosis. Evidence that this lipid is involved in the cell wall permeability barrier. J Biol Chem. 2001, 276 (23): 19845-19854. 10.1074/jbc.M100662200.
Infante E, Aguilar LD, Gicquel B, Pando RH: Immunogenicity and protective efficacy of the Mycobacterium tuberculosis fadD26 mutant. Clin Exp Immunol. 2005, 141 (1): 21-28. 10.1111/j.1365-2249.2005.02832.x.
McKinney JD, Honer zu Bentrup K, Munoz-Elias EJ, Miczak A, Chen B, Chan WT, Swenson D, Sacchettini JC, Jacobs WR, Russell DG: Persistence of Mycobacterium tuberculosis in macrophages and mice requires the glyoxylate shunt enzyme isocitrate lyase. Nature. 2000, 406 (6797): 735-738. 10.1038/35021074.
Munoz-Elias EJ, McKinney JD: Mycobacterium tuberculosis isocitrate lyases 1 and 2 are jointly required for in vivo growth and virulence. Nat Med. 2005, 11 (6): 638-644. 10.1038/nm1252.
Munoz-Elias EJ, Upton AM, Cherian J, McKinney JD: Role of the methylcitrate cycle in Mycobacterium tuberculosis metabolism, intracellular growth, and virulence. Mol Microbiol. 2006, 60 (5): 1109-1122. 10.1111/j.1365-2958.2006.05155.x.
Cotes K, Dhouib R, Douchet I, Chahinian H, de Caro A, Carriere F, Canaan S: Characterization of an exported monoglyceride lipase from Mycobacterium tuberculosis possibly involved in the metabolism of host cell membrane lipids. Biochem J. 2007
Mishra KC, de Chastellier C, Narayana Y, Bifani P, Brown AK, Besra GS, Katoch VM, Joshi B, Balaji KN, Kremer L: The Mycobacterium tuberculosis triacylglycerol hydrolase LipY: functional role of the PE domain and immunogenicity. Infect Immun. 2007
Berg S, Kaur D, Jackson M, Brennan PJ: The glycosyltransferases of Mycobacterium tuberculosis roles-in the synthesis of arabinogalactan, lipoarabinomannan, and other glycoconjugates. Glycobiology. 2007, 17 (6): 35-56R. 10.1093/glycob/cwm010.
Brennan PJ, Crick DC: The cell-wall core of Mycobacterium tuberculosis in the context of drug discovery. Curr Top Med Chem. 2007, 7 (5): 475-488. 10.2174/156802607780059763.
Jackson M, Stadthagen G, Gicquel B: Long-chain multiple methyl-branched fatty acid-containing lipids of Mycobacterium tuberculosis: biosynthesis, transport, regulation and biological activities. Tuberculosis (Edinb). 2007, 87 (2): 78-86. 10.1016/j.tube.2006.05.003.
Ryll R, Kumazawa Y, Yano I: Immunological properties of trehalose dimycolate (cord factor) and other mycolic acid-containing glycolipids – a review. Microbiol Immunol. 2001, 45 (12): 801-811.
Rao V, Gao F, Chen B, Jacobs WR, Glickman MS: Trans-cyclopropanation of mycolic acids on trehalose dimycolate suppresses Mycobacterium tuberculosis – induced inflammation and virulence. J Clin Invest. 2006, 116 (6): 1660-1667. 10.1172/JCI27335.
Hunter RL, Olsen M, Jagannath C, Actor JK: Trehalose 6,6'-dimycolate and lipid in the pathogenesis of caseating granulomas of tuberculosis in mice. Am J Pathol. 2006, 168 (4): 1249-1261. 10.2353/ajpath.2006.050848.
Hunter RL, Venkataprasad N, Olsen MR: The role of trehalose dimycolate (cord factor) on morphology of virulent M. tuberculosis in vitro. Tuberculosis (Edinb). 2006, 86 (5): 349-356. 10.1016/j.tube.2005.08.017.
Daniel J, Deb C, Dubey VS, Sirakova TD, Abomoelak B, Morbidoni HR, Kolattukudy PE: Induction of a novel class of diacylglycerol acyltransferases and triacylglycerol accumulation in Mycobacterium tuberculosis as it goes into a dormancy-like state in culture. J Bacteriol. 2004, 186 (15): 5017-5030. 10.1128/JB.186.15.5017-5030.2004.
Reed MB, Gagneux S, Deriemer K, Small PM, Barry CE: The W-Beijing lineage of Mycobacterium tuberculosis overproduces triglycerides and has the DosR dormancy regulon constitutively upregulated. J Bacteriol. 2007, 189 (7): 2583-2589. 10.1128/JB.01670-06.
Nachin L, Nannmark U, Nystrom T: Differential roles of the universal stress proteins of Escherichia coli in oxidative stress resistance, adhesion, and motility. J Bacteriol. 2005, 187 (18): 6265-6272. 10.1128/JB.187.18.6265-6272.2005.
Kvint K, Nachin L, Diez A, Nystrom T: The bacterial universal stress protein: function and regulation. Curr Opin Microbiol. 2003, 6 (2): 140-145. 10.1016/S1369-5274(03)00025-0.
O'Toole R, Williams HD: Universal stress proteins and Mycobacterium tuberculosis. Res Microbiol. 2003, 154 (6): 387-392. 10.1016/S0923-2508(03)00081-0.
Wilkinson KA, Stewart GR, Newton SM, Vordermeier HM, Wain JR, Murphy HN, Horner K, Young DB, Wilkinson RJ: Infection biology of a novel alpha-crystallin of Mycobacterium tuberculosis: Acr2. J Immunol. 2005, 174 (7): 4237-4243.
Pang X, Howard ST: Regulation of the alpha-crystallin gene acr2 by the MprAB two-component system of Mycobacterium tuberculosis. J Bacteriol. 2007, 189 (17): 6213-6221. 10.1128/JB.00492-07.
Stewart GR, Newton SM, Wilkinson KA, Humphreys IR, Murphy HN, Robertson BD, Wilkinson RJ, Young DB: The stress-responsive chaperone alpha-crystallin 2 is required for pathogenesis of Mycobacterium tuberculosis. Mol Microbiol. 2005, 55 (4): 1127-1137. 10.1111/j.1365-2958.2004.04450.x.
De Smet KA, Weston A, Brown IN, Young DB, Robertson BD: Three pathways for trehalose biosynthesis in mycobacteria. Microbiology. 2000, 146 (Pt 1): 199-208.
Murphy HN, Stewart GR, Mischenko VV, Apt AS, Harris R, McAlister MS, Driscoll PC, Young DB, Robertson BD: The OtsAB pathway is essential for trehalose biosynthesis in Mycobacterium tuberculosis. J Biol Chem. 2005, 280 (15): 14524-14529. 10.1074/jbc.M414232200.
Zhang M, Yang Y, Xu Y, Qie Y, Wang J, Zhu B, Wang Q, Jin R, Xu S, Wang H: Trehalose-6-phosphate Phosphatase from Mycobacterium tuberculosis induces humoral and cellular immune responses. FEMS Immunol Med Microbiol. 2007, 49 (1): 68-74. 10.1111/j.1574-695X.2006.00174.x.
Pym AS, Saint-Joanis B, Cole ST: Effect of katG mutations on the virulence of Mycobacterium tuberculosis and the implication for transmission in humans. Infect Immun. 2002, 70 (9): 4955-4960. 10.1128/IAI.70.9.4955-4960.2002.
Sekiguchi J, Miyoshi-Akiyama T, Augustynowicz-Kopec E, Zwolska Z, Kirikae F, Toyota E, Kobayashi I, Morita K, Kudo K, Kato S, et al: Detection of multidrug resistance in Mycobacterium tuberculosis. J Clin Microbiol. 2007, 45 (1): 179-192. 10.1128/JCM.00750-06.
Li Z, Kelley C, Collins F, Rouse D, Morris S: Expression of katG in Mycobacterium tuberculosis is associated with its growth and persistence in mice and guinea pigs. J Infect Dis. 1998, 177 (4): 1030-1035.
Layec S, Decaris B, Leblond-Bourget N: Characterization of Proteins Belonging to the CHAP-Related Superfamily within the Firmicutes. J Mol Microbiol Biotechnol. 2008, 14 (1–3): 31-40. 10.1159/000106080.
Gao LY, Pak M, Kish R, Kajihara K, Brown EJ: A mycobacterial operon essential for virulence in vivo and invasion and intracellular persistence in macrophages. Infect Immun. 2006, 74 (3): 1757-1767. 10.1128/IAI.74.3.1757-1767.2006.
Hett EC, Chao MC, Steyn AJ, Fortune SM, Deng LL, Rubin EJ: A partner for the resuscitation-promoting factors of Mycobacterium tuberculosis. Mol Microbiol. 2007, 66 (3): 658-668. 10.1111/j.1365-2958.2007.05945.x.
Krithika R, Marathe U, Saxena P, Ansari MZ, Mohanty D, Gokhale RS: A genetic locus required for iron acquisition in Mycobacterium tuberculosis. Proc Natl Acad Sci USA. 2006, 103 (7): 2069-2074. 10.1073/pnas.0507924103.
Monfeli RR, Beeson C: Targeting Iron Acquisition by Mycobacterium tuberculosis. Infect Disord Drug Targets. 2007, 7 (3): 213-220. 10.2174/187152607782110031.
De Voss JJ, Rutter K, Schroeder BG, Su H, Zhu Y, Barry CE: The salicylate-derived mycobactin siderophores of Mycobacterium tuberculosis are essential for growth in macrophages. Proc Natl Acad Sci USA. 2000, 97 (3): 1252-1257. 10.1073/pnas.97.3.1252.
Rodriguez GM, Smith I: Identification of an ABC transporter required for iron acquisition and virulence in Mycobacterium tuberculosis. J Bacteriol. 2006, 188 (2): 424-430. 10.1128/JB.188.2.424-430.2006.
Rosenkrands I, Slayden RA, Crawford J, Aagaard C, Barry CE, Andersen P: Hypoxic response of Mycobacterium tuberculosis studied by metabolic labeling and proteome analysis of cellular and extracellular proteins. J Bacteriol. 2002, 184 (13): 3485-3491. 10.1128/JB.184.13.3485-3491.2002.
Covert BA, Spencer JS, Orme IM, Belisle JT: The application of proteomics in defining the T cell antigens of Mycobacterium tuberculosis. Proteomics. 2001, 1 (4): 574-586. 10.1002/1615-9861(200104)1:4<574::AID-PROT574>3.0.CO;2-8.
Sartain MJ, Slayden RA, Singh KK, Laal S, Belisle JT: Disease state differentiation and identification of tuberculosis biomarkers via native antigen array profiling. Mol Cell Proteomics. 2006, 5 (11): 2102-2113. 10.1074/mcp.M600089-MCP200.
Sohaskey CD: Regulation of nitrate reductase activity in Mycobacterium tuberculosis by oxygen and nitric oxide. Microbiology. 2005, 151 (Pt 11): 3803-3810. 10.1099/mic.0.28263-0.
Sohaskey CD, Wayne LG: Role of narK2X and narGHJI in hypoxic upregulation of nitrate reduction by Mycobacterium tuberculosis. J Bacteriol. 2003, 185 (24): 7247-7256. 10.1128/JB.185.24.7247-7256.2003.
Skeiky YA, Alderson MR, Ovendale PJ, Guderian JA, Brandt L, Dillon DC, Campos-Neto A, Lobet Y, Dalemans W, Orme IM, et al: Differential immune responses and protective efficacy induced by components of a tuberculosis polyprotein vaccine, Mtb72F, delivered as naked DNA or recombinant protein. J Immunol. 2004, 172 (12): 7618-7628.
Turner AJ, Brown CD, Carson JA, Barnes K: The neprilysin family in health and disease. Adv Exp Med Biol. 2000, 477: 229-240.
Makinoshima H, Glickman MS: Regulation of Mycobacterium tuberculosis cell envelope composition and virulence by intramembrane proteolysis. Nature. 2005, 436 (7049): 406-409. 10.1038/nature03713.
Lun S, Bishai WR: Characterization of a novel cell wall-anchored protein with carboxylesterase activity required for virulence in Mycobacterium tuberculosis. J Biol Chem. 2007, 282 (25): 18348-18356. 10.1074/jbc.M700035200.
Aagaard C, Govaerts M, Meng Okkels L, Andersen P, Pollock JM: Genomic approach to identification of Mycobacterium bovis diagnostic antigens in cattle. J Clin Microbiol. 2003, 41 (8): 3719-3728. 10.1128/JCM.41.8.3719-3728.2003.
Skjot RL, Oettinger T, Rosenkrands I, Ravn P, Brock I, Jacobsen S, Andersen P: Comparative evaluation of low-molecular-mass proteins from Mycobacterium tuberculosis identifies members of the ESAT-6 family as immunodominant T-cell antigens. Infect Immun. 2000, 68 (1): 214-220.
DiGiuseppe Champion PA, Cox JS: Protein secretion systems in Mycobacteria. Cell Microbiol. 2007, 9 (6): 1376-1384. 10.1111/j.1462-5822.2007.00943.x.
Guinn KM, Hickey MJ, Mathur SK, Zakel KL, Grotzke JE, Lewinsohn DM, Smith S, Sherman DR: Individual RD1-region genes are required for export of ESAT-6/CFP-10 and for virulence of Mycobacterium tuberculosis. Mol Microbiol. 2004, 51 (2): 359-370. 10.1046/j.1365-2958.2003.03844.x.
McLaughlin B, Chon JS, MacGurn JA, Carlsson F, Cheng TL, Cox JS, Brown EJ: A mycobacterium ESX-1-secreted virulence factor with unique requirements for export. PLoS Pathog. 2007, 3 (8): e105-10.1371/journal.ppat.0030105.
Gey Van Pittius NC, Gamieldien J, Hide W, Brown GD, Siezen RJ, Beyers AD: The ESAT-6 gene cluster of Mycobacterium tuberculosis and other high G+C Gram-positive bacteria. Genome Biol. 2001, 2 (10): RESEARCH0044-10.1186/gb-2001-2-10-research0044.
MacGurn JA, Cox JS: A genetic screen for Mycobacterium tuberculosis mutants defective for phagosome maturation arrest identifies components of the ESX-1 secretion system. Infect Immun. 2007, 75 (6): 2668-2678. 10.1128/IAI.01872-06.
Keep NH, Ward JM, Cohen-Gonsaud M, Henderson B: Wake up! Peptidoglycan lysis and bacterial non-growth states. Trends Microbiol. 2006, 14 (6): 271-276. 10.1016/j.tim.2006.04.003.
Mukamolova GV, Murzin AG, Salina EG, Demina GR, Kell DB, Kaprelyants AS, Young M: Muralytic activity of Micrococcus luteus Rpf and its relationship to physiological activity in promoting bacterial growth and resuscitation. Mol Microbiol. 2006, 59 (1): 84-98. 10.1111/j.1365-2958.2005.04930.x.
Mukamolova GV, Kormer SS, Kell DB, Kaprelyants AS: Stimulation of the multiplication of Micrococcus luteus by an autocrine growth factor. Arch Microbiol. 1999, 172 (1): 9-14. 10.1007/s002030050733.
Mukamolova GV, Turapov OA, Young DI, Kaprelyants AS, Kell DB, Young M: A family of autocrine growth factors in Mycobacterium tuberculosis. Mol Microbiol. 2002, 46 (3): 623-635. 10.1046/j.1365-2958.2002.03184.x.
Downing KJ, Mischenko VV, Shleeva MO, Young DI, Young M, Kaprelyants AS, Apt AS, Mizrahi V: Mutants of Mycobacterium tuberculosis lacking three of the five rpf-like genes are defective for growth in vivo and for resuscitation in vitro. Infect Immun. 2005, 73 (5): 3038-3043. 10.1128/IAI.73.5.3038-3043.2005.
Rezwan M, Grau T, Tschumi A, Sander P: Lipoprotein synthesis in mycobacteria. Microbiology. 2007, 153 (Pt 3): 652-658. 10.1099/mic.0.2006/000216-0.
Diaz-Silvestre H, Espinosa-Cueto P, Sanchez-Gonzalez A, Esparza-Ceron MA, Pereira-Suarez AL, Bernal-Fernandez G, Espitia C, Mancilla R: The 19-kDa antigen of Mycobacterium tuberculosis is a major adhesin that binds the mannose receptor of THP-1 monocytic cells and promotes phagocytosis of mycobacteria. Microb Pathog. 2005, 39 (3): 97-107. 10.1016/j.micpath.2005.06.002.
Bigi F, Gioffre A, Klepp L, Santangelo MP, Alito A, Caimi K, Meikle V, Zumarraga M, Taboga O, Romano MI, et al: The knockout of the lprG-Rv1410 operon produces strong attenuation of Mycobacterium tuberculosis. Microbes Infect. 2004, 6 (2): 182-187. 10.1016/j.micinf.2003.10.010.
Hovav AH, Mullerad J, Davidovitch L, Fishman Y, Bigi F, Cataldi A, Bercovier H: The Mycobacterium tuberculosis recombinant 27-kilodalton lipoprotein induces a strong Th1-type immune response deleterious to protection. Infect Immun. 2003, 71 (6): 3146-3154. 10.1128/IAI.71.6.3146-3154.2003.
Gehring AJ, Dobos KM, Belisle JT, Harding CV, Boom WH: Mycobacterium tuberculosis LprG (Rv1411c): a novel TLR-2 ligand that inhibits human macrophage class II MHC antigen processing. J Immunol. 2004, 173 (4): 2660-2668.
Wang B, Henao-Tamayo M, Harton M, Ordway D, Shanley C, Basaraba RJ, Orme IM: A Toll-like receptor-2-directed fusion protein vaccine against tuberculosis. Clin Vaccine Immunol. 2007, 14 (7): 902-906. 10.1128/CDLI.00077-07.
Bottai D, Batoni G, Esin S, Maisetta G, Pardini M, Florio W, Rindi L, Garzelli C, Campa M: Expression of SA5K, a secretion antigen of Mycobacterium tuberculosis, inside human macrophages and in sputum from tuberculosis patients. FEMS Microbiol Lett. 2003, 226 (2): 229-235. 10.1016/S0378-1097(03)00602-5.
Bottai D, Esin S, Batoni G, Pardini M, Maisetta G, Donati V, Favilli F, Florio W, Campa M: Disruption of the gene encoding for secretion antigen SA5K affects growth of Mycobacterium bovis bacillus Calmette-Guerin in human macrophages and in mice. Res Microbiol. 2005, 156 (3): 393-402. 10.1016/j.resmic.2004.11.002.
Bottai D, Batoni G, Esin S, Florio W, Brancatisano FL, Favilli F, Maisetta G, Campa M: The secretion antigen SA5K has a role in the adaptation of Mycobacterium bovis bacillus Calmette-Guerin to intracellular stress and hypoxia. Microbes Infect. 2006, 8 (8): 2254-2261. 10.1016/j.micinf.2006.04.020.
Zhang Y, Yang Y, Woods A, Cotter RJ, Sun Z: Resuscitation of dormant Mycobacterium tuberculosis by phospholipids or specific peptides. Biochem Biophys Res Commun. 2001, 284 (2): 542-547. 10.1006/bbrc.2001.4993.
Banu S, Honore N, Saint-Joanis B, Philpott D, Prevost MC, Cole ST: Are the PE-PGRS proteins of Mycobacterium tuberculosis variable surface antigens?. Mol Microbiol. 2002, 44 (1): 9-19. 10.1046/j.1365-2958.2002.02813.x.
Brennan MJ, Delogu G, Chen Y, Bardarov S, Kriakov J, Alavi M, Jacobs WR: Evidence that mycobacterial PE_PGRS proteins are cell surface constituents that influence interactions with other cells. Infect Immun. 2001, 69 (12): 7326-7333. 10.1128/IAI.69.12.7326-7333.2001.
Gey van Pittius NC, Sampson SL, Lee H, Kim Y, van Helden PD, Warren RM: Evolution and expansion of the Mycobacterium tuberculosis PE and PPE multigene families and their association with the duplication of the ESAT-6 (esx) gene cluster regions. BMC Evol Biol. 2006, 6: 95-10.1186/1471-2148-6-95.
Voskuil MI, Schnappinger D, Rutherford R, Liu Y, Schoolnik GK: Regulation of the Mycobacterium tuberculosis PE/PPE genes. Tuberculosis (Edinb). 2004, 84 (3–4): 256-262. 10.1016/j.tube.2003.12.014.
Brennan MJ, Delogu G: The PE multigene family: a 'molecular mantra' for mycobacteria. Trends Microbiol. 2002, 10 (5): 246-249. 10.1016/S0966-842X(02)02335-1.
Delogu G, Sanguinetti M, Pusceddu C, Bua A, Brennan MJ, Zanetti S, Fadda G: PE_PGRS proteins are differentially expressed by Mycobacterium tuberculosis in host tissues. Microbes Infect. 2006, 8 (8): 2061-2067. 10.1016/j.micinf.2006.03.015.
Sreenu VB, Kumar P, Nagaraju J, Nagarajaram HA: Microsatellite polymorphism across the M. tuberculosis and M. bovis genomes: implications on genome evolution and plasticity. BMC Genomics. 2006, 7: 78-10.1186/1471-2164-7-78.
Ramakrishnan L, Federspiel NA, Falkow S: Granuloma-specific expression of Mycobacterium virulence proteins from the glycine-rich PE-PGRS family. Science. 2000, 288 (5470): 1436-1439. 10.1126/science.288.5470.1436.
Delogu G, Brennan MJ: Comparative immune response to PE and PE_PGRS antigens of Mycobacterium tuberculosis. Infect Immun. 2001, 69 (9): 5606-5611. 10.1128/IAI.69.9.5606-5611.2001.
Basu S, Pathak SK, Banerjee A, Pathak S, Bhattacharyya A, Yang Z, Talarico S, Kundu M, Basu J: Execution of macrophage apoptosis by PE_PGRS33 of Mycobacterium tuberculosis is mediated by Toll-like receptor 2-dependent release of tumor necrosis factor-alpha. J Biol Chem. 2007, 282 (2): 1039-1050. 10.1074/jbc.M604379200.
Li Y, Miltner E, Wu M, Petrofsky M, Bermudez LE: A Mycobacterium avium PPE gene is associated with the ability of the bacterium to grow in macrophages and virulence in mice. Cell Microbiol. 2005, 7 (4): 539-548. 10.1111/j.1462-5822.2004.00484.x.
Chakhaiyar P, Nagalakshmi Y, Aruna B, Murthy KJ, Katoch VM, Hasnain SE: Regions of high antigenicity within the hypothetical PPE major polymorphic tandem repeat open-reading frame, Rv show a differential humoral response and a low T cell response in various categories of patients with tuberculosis. J Infect Dis. 2004, 190 (7): 1237-1244. 10.1086/423938.
Choudhary RK, Mukhopadhyay S, Chakhaiyar P, Sharma N, Murthy KJ, Katoch VM, Hasnain SE: PPE antigen Rv2430c of Mycobacterium tuberculosis induces a strong B-cell response. Infect Immun. 2003, 71 (11): 6338-6343. 10.1128/IAI.71.11.6338-6343.2003.
Zhang H, Wang J, Lei J, Zhang M, Yang Y, Chen Y, Wang H: PPE protein (Rv3425) from DNA segment RD11 of Mycobacterium tuberculosis: a potential B-cell antigen used for serological diagnosis to distinguish vaccinated controls from tuberculosis patients. Clin Microbiol Infect. 2007, 13 (2): 139-145. 10.1111/j.1469-0691.2006.01561.x.
Singh KK, Dong Y, Patibandla SA, McMurray DN, Arora VK, Laal S: Immunogenicity of the Mycobacterium tuberculosis PPE55 (Rv3347c) protein during incipient and clinical tuberculosis. Infect Immun. 2005, 73 (8): 5004-5014. 10.1128/IAI.73.8.5004-5014.2005.
Narayana Y, Joshi B, Katoch VM, Mishra KC, Balaji KN: Differential B-cell responses are induced by Mycobacterium tuberculosis PE antigens Rv1169c, Rv0978c, and Rv1818c. Clin Vaccine Immunol. 2007, 14 (10): 1334-1341. 10.1128/CVI.00181-07.
Voskuil MI: Mycobacterium tuberculosis gene expression during environmental conditions associated with latency. Tuberculosis (Edinb). 2004, 84 (3–4): 138-143. 10.1016/j.tube.2003.12.008.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1755-8794/1/18/prepub
Acknowledgements
This work was supported by a grant from AERAS Global TB Vaccine Foundation.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
AZ and NA performed most of the analysis and participated in writing the draft ms. JF participated in discussions during the study and was responsible for constructing the rBCG based on the analysis. JCS participated in discussions at all stages of the study and co-wrote the final ms. AS conceived the study, participated in the analysis and co-wrote the manuscript. All authors read and approved the final manuscript.
Anat Zvi, Naomi Ariel contributed equally to this work.
Electronic supplementary material
12920_2007_18_MOESM1_ESM.xls
Additional file 1: List of 189 selected antigens. (a) In bold and underlined: top-ranking antigens included in the list of 45 candidate genes. (b) The Gene name and annotation are based on the data deposited at the NCBI [GenBank: AL123456]. In square brackets: updated gene name, annotation, and functional category, resulting from the analyses conducted in this study. (c) The functional categories are as provided by Cole and his coworkers [26], and the assignment of a functional category to each ORF product is according to the Tuberculist database [47], unless an updated category is assigned (in square brackets), according to the re-annotation performed in this study (see (b)). (d) The classes/phases of Mtb infection assigned to each selected ORF (see "Methods"). (XLS 54 KB)
12920_2007_18_MOESM2_ESM.xls
Additional file 2: Raw data of the 189 selected antigens. The data (a '+' sign or an experimental value) extracted from global and particular analyses are provided, for each category and references therein (see Table 1). In addition, the qualitative and quantitative scores for each antigen in each of the categories are given. (XLS 190 KB)
12920_2007_18_MOESM3_ESM.xls
Additional file 3: Qualitative scores of the 189 selected antigens. The qualitative score for each of the 189 selected ORF in each of the categories is provided. A "+" sign is indicated if data exists at least in one study in that particular category (see Table 1 for data resources), and the arithmetic sum of all "+" for a particular ORF is given in the right column as the total qualitative score (Qual_Total). (a) The Gene name and annotation are based on the data deposited at the NCBI, [GenBank: AL123456]. In square brackets: updated gene name and/or annotation, resulting from the analyses conducted in this study. For the complete data values of the 189 selected antigens in each of the categories, see Additional file 2: "Raw data of the 189 selected antigens". (XLS 80 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Zvi, A., Ariel, N., Fulkerson, J. et al. Whole genome identification of Mycobacterium tuberculosisvaccine candidates by comprehensive data mining and bioinformatic analyses. BMC Med Genomics 1, 18 (2008). https://doi.org/10.1186/1755-8794-1-18
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1755-8794-1-18