
Abstract
Interstitial deletions of the short arm of chromosome 6 are rare and have been associated with developmental delay, hypotonia, congenital anomalies, and dysmorphic features. We used array comparative genomic hybridization in a South Carolina Autism Project (SCAP) cohort of 97 subjects with autism spectrum disorders (ASDs) and identified an ~ 5.4 Mb deletion on chromosome 6p22.3-p23 in a 15-year-old patient with intellectual disability and ASDs. Subsequent database queries revealed five additional individuals with overlapping submicroscopic deletions and presenting with developmental and speech delay, seizures, behavioral abnormalities, heart defects, and dysmorphic features. The deletion found in the SCAP patient harbors ATXN1, DTNBP1, JARID2, and NHLRC1 that we propose may be responsible for ASDs and developmental delay.
Similar content being viewed by others


Background
Deletions involving the distal part of the short arm of chromosome 6 are relatively rare. Terminal deletions of 6p24-pter have been associated with developmental delay, brain malformations (including Dandy-Walker malformation), anterior eye chamber abnormalities, hearing loss, ear abnormalities, micrognathia, and heart defects [1–6]. Patients with larger sized deletions of 6p23-pter also presented with microcephaly, genital anomalies, language impairment, and delayed motor development [1, 3, 5, 7–14]. The identified ocular developmental abnormalities are caused by deficiency of the dosage sensitive FOXC1 gene (MIM 01090) [15–20]. In addition, deletions and duplications involving FOXC1 have been shown recently to be responsible for Dandy-Walker malformation [21].
Interstitial deletions of 6p22-p24 have been reported even less often and are generally associated with psychomotor and growth delay, hypotonia as well as several congenital abnormalities, including hydrocephalus, microcephaly, structural eye abnormalities, hypertelorism, low set and rotated ears, nasal anomalies, micrognathia, palatal abnormalities, short folded neck, defects of heart, kidney, and feet, abnormal genitalia, and abnormal fingers with hypoplastic nails [4, 5, 13, 22–26].
Here, we describe six individuals, five of whom have overlapping interstitial deletions in chromosome 6p22.3-p24.3 encompassing ATXN1. The majority of patients had neurological or behavioral abnormalities, including developmental and speech delay, autism spectrum disorders (ASDs), attention deficit hyperactivity disorder (ADHD), repetitive behaviors, and various dysmorphic features.
Clinical reports
Patient 1
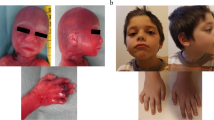
This 15-year-old male proband was enrolled in the South Carolina Autism Project (SCAP) study at the J.C. Self Research Institute of Human Genetics at the Greenwood Genetics Center in Greenwood, South Carolina. He was the second child of healthy parents. The pregnancy and delivery were uneventful. He was developmentally delayed, used a few words until 3 years of age, and remained nonverbal until 7-8 years of age. At 15 years of age, he used about six words but no phrases or sentences. The proband was also socially inappropriate and was classified in the autism spectrum according to ADOS and ADI-R testing. He had narrow facies, flat midface, overbite, slight prominence and jutting of the tissue of chin, and pits on the skin at the base of the nasal septum. His eyes appeared to be recessed due to flat midface, and his neck was long (Figure 1a). There were some flaring or winging of the scapulae. Wood's lamp exam revealed confluent minimally hypopigmented blotches about the size of a half dollar over his sacrum. Thumb tissues were somewhat broad distally. He had short thumbnails, broad and short feet, short and broad great toes, long second toes, and short third toes. Deep tendon reflexes were 1+ in the upper and lower limbs, and plantar reflexes were flexor. The father was unavailable for testing. This patient died at the age of 26 years following intestinal rupture.

Patients 1 (A), 4 (B-C), and 6 (D).
Patient 2
This four-year-old male proband had developmental and speech delay, repetitive movements, hyperactivity, and possible ASDs; however, no formal ASDs testing was performed. Metabolic studies did not reveal any abnormalities. There were no dysmorphic features. A 2/6 systolic murmur was observed. Reportedly, the mother and the older brother of this patient are mentally disabled. DNA from the proband's older brother was not available.
Patient 3
This newborn female patient passed away soon after birth due to multiple congenital defects. The autopsy report described low set ears, bilateral partial aniridia, incomplete palpebral fissures bilaterally, micrognathia, shortened philtrum, redundant nuchal folds, two supernumerary nipples, hypoplastic toe nails, enlarged heart with multiple congenital defects (dilated right atrium, interrupted aortic arch, dilated pulmonary trunk with superiorly placed left pulmonary artery take-off, patent foramen ovale, two left and one right pulmonary veins draining to left atrium, perimembranous ventricular septal defect with portion of tricuspid valve extending through VSD, patent ductus arteriosus, and narrowed aortic and mitral valves), incomplete lobation of the right upper and middle lung lobes, bicornuate uterus, diastasis recti, bilateral simian creases of the hands, and anteriorly placed anus. The parents were not available for further studies.
Patient 4
This male patient was first seen at the age of six years. He was born by spontaneous vaginal delivery. His mother reported that the fetus was exposed to cigarette smoke and a large amount of alcohol. He presented with developmental delay, seizure disorder, attention defficit hyperactivity disorder (ADHD) diagnosed at 24 months, behavioral problems, anger and impulse control problems, speech problems, difficulties expressing himself, craniofacial dysmorphology, long face, almond shaped eyes, low set ears with forward facing lobules, thick malaligned philtrum, tall chin with retrognathia and overbite, pectus carinatum, toe abnormalities, and clubbed foot (Figure 1b, c). His father was described by the mother as having speech problems. Metabolic screening and fragile X testing of the proband were normal. Blood specimens from the parents were not available. He required right tibial and proximal fibular epiphysiodesis for leg length discrepancy at the age of 15. Currently, he is 17 years old with significant developmental delays and behavioral issues.
Patient 5
A 7-year-old-girl was described as being nondysmorphic and having developmental and speech delay, bilateral strabismus (corrected at four years of age), and three generalized seizures without fever at four months of age. Her EEG, MRI and CT were normal. The patient also had an ongoing iron deficiency, for which she was being treated. The mother had an uncomplicated pregnancy and an induced vaginal delivery. The parents are healthy, as are the older two full sisters and half sister.
Patient 6
This three-year-old male patient was born after an induced vaginal delivery due to maternal hypertension. There were no complications during the pregnancy. He was described as having stocky appearance, frontal upsweep, wide spaced and deep set eyes, broad forehead, mild micrognathia (status post tracheostomy), and small thumbs (Figure 1d). Other physical findings included biparietal bossing, small ears, narrow alveolar archis, missing teeth, tracheostomy scar, flared ribs, and bilateral clinodactyly. He had tiny frontal lobe hemorrhages on an MRI performed a few days after his birth. CT showed an arachnoid cyst. Additionally, he had hypotonia, a heart murmur, atrial septal defect, and bicuspid aortic valve, mandibular hypoplasia, a history of jaw distraction, heel cord release, asthma and recurrent aspiration pneumonia, a history of G-tube (removed), eczema, possible submucous cleft palate, speech problems, mild hearing loss on Auditory Brainstem Response Evaluation, fatigue, a history of febrile seizures, lack of coordination, and global developmental and learning delays. At the time of his visit, he was very hyperactive and did not speak any words. He was diagnosed with sensory processing disorder with developmental dyspraxia. The parents reported that he used two signs, had better receptive than expressive language (with a lot of body language), was easily distracted, destructive, and wild. Blood specimens from the parents were not available.
Results
Using array comparative genomic hybridization (array CGH), in patient 1 with ASDs from the SCAP collection, we identified an ~ 5.4 Mb deletion on chromosome 6p22.3-p23, harboring 21 RefSeq genes (Figure 2a) and sequenced its breakpoints. The distal breakpoint (chr6:13,662,096) maps within a LINE element (L1MEe) and the proximal breakpoint (chr6:19,042,218) maps within a unique sequence. There was no microhomology between the deletion breakpoints (Figure 2c). G-banded chromosome slides were retrospectively reviewed at the Greenwood Genetics Center and an interstitial deletion 6p22.3p23 was detected (data not shown). Polymerase chain reaction (PCR) in the proband's mother showed that she was not a carrier for the deletion (Figure 2b).

Chromosome 6p deletion in patients 1-6 - a) Array CGH probe plots in patients 1-6. X axis, chromosome 6 position; Y axis, log2 ratio. Semitransparent filled boxes on CGH plots highlight the region of aberration. b) Patient 1-specific junction fragment is not present in his unaffected mother (Mo) or unaffected control (Co). M, marker. c) Chromatogram showing the breakpoint fusion in the patient 1-specific junction fragment. d) Metaphase FISH showing a deletion at 6p22.3 in patient 5. Chromosome 6p22.3-specific BAC clone RP11-140A3 is labeled in red, and the chromosome 6 centromere control probe D6Z1 is labeled in green. The presence of one red signal indicates deletion of 6p22.3 on one homologue (arrow). e) Interphase FISH showing a deletion at 6p24.1-p22.3 in patient 6. Chromosome 6p23-specific BAC clone RP11-127P7 is labeled in red, and the chromosome 6 centromere probe is labeled in green as a control. The presence of one red signal indicates deletion on one homologue (del 6). Nl6, normal chromosome 6.
Although the father of patient 1 was not available for analysis, we attempted to determine the deletion origin by CAG repeats polymorphism in ATXN1, a gene located within the deletion, and independently by SNP array analysis. The results of ATXN1 polyglutamine expansion were inconclusive (data not shown). SNP analysis showed that all 1,193 patient's SNPs in the deleted region (on the normal chromosome) matched the mother's SNPs for that region, and that 85% (33,541/39,459) of the patient SNPs in the non-deleted region of chromosome 6 matched the mother's SNPs. Thus, these results indicate that it is highly likely that the normal allele was inherited from the patient's mother and that the allele with the deletion arose on the paternal chromosome 6 (either inherited or de novo).
Array CGH images for deletions in patients 2-6 are shown in Figure 2a. Patient 2 had a 6p22.3 deletion of approximately 1 Mb that involves the ATXN1, FLJ23152, RBM24, and CAP2 genes. The deletion was confirmed by fluorescence in situ hybridization (FISH) analysis and was also found in the patient's mother, who was reported to have some degree of mental disability. Patient 3, who passed away soon after birth, had a deletion of approximately 14.6 Mb on chromosome 6p22.3-p24.3 that involves 58 RefSeq genes. Retrospective high-resolution partial karyotype analysis confirmed the interstitial deletion of 6p22.3-p24.3 (data not shown). Patient 4 had a deletion on chromosome 6p23-p24.3 of approximately 3.6 Mb that involves 28 RefSeq genes. Patient 5 had a 6p22.3 deletion of approximately 5.2 Mb that involves 18 RefSeq genes (Figure 2a, d). Parental FISH studies revealed that the deletion was de novo. Patient 6 had a deletion on chromosome bands 6p22.3-p24.1 of approximately 8.8 Mb that involves 34 RefSeq genes (Figure 2a, e). Patient 6 also had a 2.7 Mb gain on chromosome 15q11.2 (chr15:20,565,530-23,300,182), including the BP1-BP2 region, which has been observed in other probands and their normal parents and therefore most likely represents a familial copy-number variant.
Deletion frequency in control populations
A query of the Database of Genomic Variants revealed no CNVs larger than 200 kb within the largest deletion reported in this study (patient 3). No exonic deletions involving ATXN1 were found in six control groups consisting of 2792 individuals [27], 2493 individuals [28], 2026 individuals [29], 1152 individuals [30], 450 individuals [31], and 270 individuals [32].
Discussion
ASDs ([MIM 209850]) embody a group of behavioral abnormalities, including restricted and repetitive behaviors, and/or defects in social interaction and/or communication. Cytogenetic abnormalities were initially reported in 25-30% of individuals with ASDs and dysmorphic features [33], whereas pathological CNVs have been found in 5-10% of patients with milder ASDs through the use of oligonucleotide microarrays [34–38].
Using array CGH to search for CNVs that may be associated with ASDs in the patients from the SCAP collection, we identified a deletion at chromosome 6p22.3-p23 in a 15 year-old boy with ASDs. Further database [Medical Genetics Laboratories (MGL) and Signature Genomic Laboratories (SGL) diagnostic laboratories] and literature queries revealed additional five and 12 overlapping interstitial deletions, respectively. Patients 1, 2, and 4-6 had developmental and speech delays, features commonly seen in the published patients with the deletions in chromosome 6p22.3-p24.3 (Table 1). Patients 4-6 also had seizures, ADHD or hyperactivity, or repetitive behaviors. In addition, eight out of the 10 patients (pts A, C, E-H, J, and K) with interstitial deletions involving 6p22.3-p24.3 described in the literature (Table 1) had speech delay and one had hyperactivity. Patient 3, who passed away shortly after birth, and patients B and D [23, 24], evaluated at 9 and 13 months of age, respectively, were too young to receive a diagnosis of ASDs and to display some of the behavioral abnormalities described above.
Patient 2 also presented with ASDs, suggesting that one or more loci within chromosome 6p22.3-24.3 may play a role in the development of ASDs. In support of this notion, a DECIPHER (Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources) [39] patient (ID 249613) with an ~ 4.7 Mb interstitial deletion at chromosome 6p22.3 had autistic behavior in addition to hypotonia, developmental delay/intellectual disability, downslanting palpebral fissures, and strabismus (Table 1). Moreover, a patient with ASDs was reported to have a de novo ~ 1.3 Mb interstitial deletion at 6p23 (Table 1) [34]. However, this patient also had a de novo 2 Mb deletion in chromosome 13q14.12-q14.13.
Sixteen deletions (pts 2-6, A-G, and I-L) overlap with the deletion found in patient 1, who had a diagnosis of ASDs (Table 1). Thirteen (pts 2, 4-6, A, C, E-G, and I-L) out of those 16 patients (Table 1) had ASDs and/or some of the ASDs associated features: speech delay, ADHD or hyperactivity, or behavioral abnormalities [40, 41]. We propose that some of the following genes may be responsible for the ASDs features and should be considered in further research testing in patients with ASDs: ATXN1 (deleted in 10 patients), JARID2 (deleted in eight patients), DTNBP1 (deleted in eight patients), and NHLRC1 (deleted in eight patients) (Table 2).
Ten out of 13 patients with ATXN1 deletion (pts 1-3, 5, 6, A-E, G, J, and K) had ASDs, speech delay, ADHD or hyperactivity, or other behavioral abnormalities (Table 1). CAG trinucleotide extensions of 41-81 repeats within the coding region of ATXN1 are responsible for the autosomal dominant spinocerebellar ataxia 1 (SCA1 [MIM 164400]), a neurodegenerative disorder with progressive cerebellar degeneration. ATXN1 is also proposed to function as a regulator of gene expression [42]. Interestingly, Atxn1 homozygous knockout mice were shown to share aberrations with a knock-in mouse model of SCA1 that contained the polyglutamine extension [43, 44]. Despite the fact that the knockout mice did not develop SCA1 or progressive cerebellar degeneration, both models had abnormalities in spatial learning and memory, motor learning and coordination, and in cerebellar gene expression [43, 44]. Additionally, a meta-analysis has suggested that SNPs within ATXN1 are associated with intelligence quotient in the background of ADHD [45] and Bremer et al. [22] proposed that haploinsufficiency of ATXN1 may therefore contribute to the learning difficulties observed in the patients harboring a 6p22 deletion. Given its importance in brain function and behavior abnormalities in mouse models, we speculate that heterozygous deletions that affect ATXN1 function may be involved with the outcomes of developmental delay and ASDs, either alone or in combination with other gene deletions.
Homozygous missense point mutations in DTNBP1 have been found in patients with Hermansky-Pudlak syndrome (HPS [MIM 203300]), an autosomal recessive disease with features that include albinism, pulmonary fibrosis, and bleeding [46]. DTNBP1 has been proposed to function in organelle biogenesis [46], presynaptic glutamate release in rat neurons [47], neural plasticity in Drosophila [48], and to localize to axons in mouse cerebellum and hippocampus [49]. Additionally, a meta-analysis study found an association of SNPs in DNTBP1 with schizophrenia (SCZD [MIM 181500]) [50, 51]. Finally, DTNBP1 has been connected with autistic features observed sometimes in patients with Duchenne muscular dystrophy (DMD [MIM 310200]) [52–54]. DTNBP1 binds to alpha and beta dystrobrevins, which compose the dystrophin-associated protein complex (DPC) [49].
JARID2 is expressed in embryonic and adult human neurons [55] and may function as a transcriptional repressor [56]; its mouse homolog Jmj is necessary for proper neural tube formation and cardiac development [57]. Recently, a significant association was found between a JARID2 SNP (rs7766973) and autism [58], making this gene another candidate for ASDs.
Homozygous deletions, insertions, missense, or nonsense mutations in NHLRC1 have been found in individuals with myoclonic epilepsy of Lafora (MELF [MIM 254780]), an autosomal recessive disorder characterized by rapid and progressive adolescent-onset epilepsy, mental and motor deterioration, and short survival. NHLRC1 functions as an E3 ubiquitin ligase that mediates degradation of EPM2A (or LAFORIN). Mutations that disturb EPM2A degradation, leading to the accumulation of Lafora bodies, are also causative for MELF [59].
Although most individuals with deletions in chromosome 6p22.3-p24.3 display developmental delay, speech delay, and ASDs or other behavioral abnormalities, there is no single locus that is deleted in all patients with 6p22-p24 deletion (Figure 3). Penetrance of the phenotype caused by mutations in specific genes in this region is likely incomplete and may depend on the presence of modifiers found in the non-deleted alleles, regulatory regions, or other genes elsewhere in the genome. In addition, under-ascertainment with the lack of use of proper ASDs diagnostic tools such as ADOS and ADI-R may confuse the phenotype-genotype correlations.

Extent and gene (RefSeq) content of chromosome 6p22-p24 deletions. Labeling of horizontal bars representing deletions described in the literature are as follows: A, [25]; B, [24]; C, P1 from [23]; D, P2 from [23]; E, 91-145 from [4]; F, 95-800 from [4]; G, PF from [4]; H, [13]; I, AU010604 from [34]; J, [26]; K, [22]; and L, DECIPHER patient ID 249613 [39]. Blue regions depict the maximum extents of the deletions. Longest gene isoforms are shown. Histone gene cluster on 6p22.1-p22.2 is not depicted due to space limitations. Gene names are positioned below or to the left of genes. Green bars represent individuals from the literature, who were reported to have ASDs. Gene names in blue represent strong candidates for ASDs or autistic features given their role in the brain function and development or involvement in neurological disorders (see Table 2). Red vertical bar depicts the smallest deletion overlap, involving 13 deletions (patients 1-3, 5, 6, A-E, G, J, and K). There are four RefSeq genes in this region: ATXN1, FLJ23152, RBM24, and CAP 2. See Table 2 for gene functions. Pt, patient.
Finally, four of the six patients described in this study had variable dysmorphic features, including craniofacial dysmorphisms, structural ear defects, and hands and feet abnormalities, which are commonly found in individuals described in the literature. Similar to the behavioral, developmental, and intellectual abnormalities described above, there is no single locus that is deleted in all individuals with 6p22-p24 deletion that may explain these dysmorphic features. Of note, other features that are commonly seen in patients described in the literature, such as congenital heart defects and hypotonia, were not found in all patients described in this report.
Methods
Consents
Written informed consents were approved by the Institutional Review Board of Self Regional Hospital at Greenwood, SC, for the J.C. Self Research Institute of Human Genetics (IRB 24), the Institutional Review Board for Human Subject Research at Baylor College of Medicine (H-25466), and by the Institutional Review Board of Spokane (study number 1500) and were obtained for patients 1, 4 and 6, and patient 5, respectively.
Chromosomal microarray analysis
For patient 1, DNA was extracted from peripheral blood and lymphoblastoid cell line. DNA from the cell line was used for initial array CGH analysis at Baylor College of Medicine (BCM). DNA from peripheral blood was used to confirm the array CGH findings. For patients 2-4 and 6, DNA was extracted from whole blood using the Puregene DNA isolation kit (Gentra System, Inc., Minneapolis, MN). For patient 5, a blood specimen was sent to SGL, in Spokane, Washington for clinical array CGH analysis. Parental DNA was only tested by FISH.
All arrays used in this study were designed and analyzed based on genome build hg18 (NCBI Build 36, March 2006). The coordinates found in tables and figures were converted to hg19 (GRCh 37), February 2009 using UCSC liftOver [60].
Patient 1 was tested for copy-number variants (CNVs) using array CGH with an Agilent custom designed exon targeted microarray with coverage for 294 ASDs candidate genes (44 K, design ID 019729, Agilent Technologies, Inc., Santa Clara, CA) [61]. Patients 2-4 and 6 were studied using Agilent OLIGO custom clinical microarrays versions 7.4 OLIGO, 6.3 OLIGO, 6.4 OLIGO, and 8.0 OLIGO, respectively, designed at the MGL at BCM [62–65]. Patient 5 was studied using the 105 K-feature SignatureChip Oligo Solution® whole-genome custom microarray manufactured by Agilent for SGL [66]. Additionally, the Agilent catalog SurePrint G3 Human CGH Microarray 1 × 1 M of design ID 021529 was used to confirm CNVs in patients 1, 3, and 4.
The protocols for DNA digestion, labeling, purification, hybridization, array scan, and analysis of the clinical arrays followed the manufacturer's instructions with some modifications as previously described [63, 66, 67].
SNP array analysis
SNP genotyping for chromosome 6 was done in patient 1 and his mother using the commercially available Illumina Human 610-Quad BeadChip Kit (Illumina, Inc., San Diego, CA). Arrays were scanned using the Illumina Iscan with Autoloader2. SNP genotyping and absence of heterozygosity analyses were performed using the Illumina GenomeStudio data analysis software.
FISH analysis
Confirmatory FISH analyses were performed in patients 2 and 4-6 using standard cytogenetic procedures with bacterial artificial chromosome (BAC) clones RP11-346 F18, RP11-637019, RP11-140A3, and RP11-127P7, respectively. The parental samples were tested for patients 2 and 5.
PCR and sequence analysis
PCR to confirm the 6p22.3-p23 deletion and to amplify the junction fragment in patient 1 was performed using Takara LA PCR kit (Takara Bio, Inc., Shiga, Japan) with the forward primer 5' - TGGTGTAGTTAAGGGGAAAGAGAGAGGAG - 3' and the reverse primer 5' - CTGCAGTATAAGCATACTACTACCCACTTAGGG - 3' (Sigma-Aldrich Corp., St. Louis, MO). To test the patient's mother for a low level somatic mosaicism, which might have been missed by the array CGH assay, PCR was performed using GoTaq® Flexi DNA Polymerase kit (Promega Corporation, Madison, WI) with the forward primer 5' - TTTGGATTGGGAGGAATGAA - 3' and reverse primer 5' - GGGGAAAGAAACGGAACATC - 3' (Sigma-Aldrich Corp.). PCR products were analyzed using 1% agarose gel electrophoresis, purified from the agarose gel using the PCR Purification Kit (Qiagen, Valencia, CA), and then sent for Sanger dideoxy sequencing (SeqWright and Lone Star, Houston, TX).
ATXN1(ATAXIN1) STR extension analysis
To determine the size of the polyglutamine track in the ATXN1 gene, we followed the previously published procedures with minor modifications [68–70]. PCR was performed using Applied Biosystems Amplitaq's PCR kit (Applied Biosystems, Foster City, CA) with forward primer 5' - AACTGGAAATGTGGACGTAC - 3' and reverse primer 5' - CAACATGGGCAGTCTGAG - 3' (Integrated DNA Technologies, Inc., Coralville, IA). PCR products were analyzed by electrophoresis on a sequencing polyacrylamide gel. The gel was visualized by autoradiography at -70°C overnight.
Abbreviations
- ADHD:
-
Attention deficit hyperactivity disorder
- ASDs:
-
Autism spectrum disorders
- BAC:
-
Bacterial artificial chromosome
- BCM:
-
Baylor College of Medicine
- CGH:
-
Comparative genomic hybridization
- CNV:
-
Copy-number variant
- DECIPHER:
-
Database of chromosomal imbalance and phenotype in humans using ensembl resources
- FISH:
-
Fluorescence in situ hybridization
- MGL:
-
Medical Genetics Laboratories
- PCR:
-
Polymerase chain reaction
- SCAP:
-
South Carolina autism project
- SGL:
-
Signature Genomic Laboratories.
References
Reid CS, Stamberg J, Phillips JA: Monosomy for distal segment 6p: clinical description and use is localising a region important for expression of Hageman factor [abstract]. Pediatr Res 1983, 40: 3127.
Zurcher VL, Golden WL, Zinn AB: Distal deletion of the short arm of chromosome 6. Am J Med Genet 1990, 35: 261–265. 10.1002/ajmg.1320350223
Law CJ, Fisher AM, Temple IK: Distal 6p deletion syndrome: a report of a case with anterior chamber eye anomaly and review of published reports. J Med Genet 1998, 35: 685–689. 10.1136/jmg.35.8.685
Davies AF, Mirza G, Sekhon G, Turnpenny P, Leroy F, Speleman F, Law C, van Regemorter N, Vamos E, Flinter F, Ragoussis J: Delineation of two distinct 6p deletion syndromes. Hum Genet 1999, 104: 64–72. 10.1007/s004390050911
Mirza G, Williams RR, Mohammed S, Clark R, Newbury-Ecob R, Baldinger S, Flinter F, Ragoussis J: Refined genotype-phenotype correlations in cases of chromosome 6p deletion syndromes. Eur J Hum Genet 2004, 12: 718–728. 10.1038/sj.ejhg.5201194
Lin RJ, Cherry AM, Chen KC, Lyons M, Hoyme HE, Hudgins L: Terminal deletion of 6p results in a recognizable phenotype. Am J Med Genet A 2005, 136: 162–168.
Sachs ES, Hoogeboom AJ, Niermeijer MF, Schreuder GM: Clinical evidence for localisation of HLA proximal of chromosome 6p22. Lancet 1983, 1: 659.
Jalal SM, Macias VR, Roop H, Morgan F, King P: Two rare cases of 6p partial deletion. Clin Genet 1989, 36: 196–199.
Kelly PC, Blake WW, Davis JR: Tandem Y/6 translocation with partial deletion 6 (p23--pter). Clin Genet 1989, 36: 204–207.
Kormann-Bortolotto MH, Farah LM, Soares D, Corbani M, Muller R, Adell AC: Terminal deletion 6p23: a case report. Am J Med Genet 1990, 37: 475–477. 10.1002/ajmg.1320370410
Palmer CG, Bader P, Slovak ML, Comings DE, Pettenati MJ: Partial deletion of chromosome 6p: delineation of the syndrome. Am J Med Genet 1991, 39: 155–160. 10.1002/ajmg.1320390208
Plaja A, Vidal R, Soriano D, Bou X, Vendrell T, Mediano C, Pueyo JM, Labrana X, Sarret E: Terminal deletion of 6p: report of a new case. Ann Genet 1994, 37: 196–199.
Davies AF, Mirza G, Flinter F, Ragoussis J: An interstitial deletion of 6p24-p25 proximal to the FKHL locus and including AP-2 that affects anterior eye chamber development. J Med Genet 1999, 36: 708–710.
Anderlid BM, Schoumans J, Hallqvist A, Stahl Y, Wallin A, Blennow E, Nordenskjold M: Cryptic subtelomeric 6p deletion in a girl with congenital malformations and severe language impairment. Eur J Hum Genet 2003, 11: 89–92. 10.1038/sj.ejhg.5200907
Mears AJ, Jordan T, Mirzayans F, Dubois S, Kume T, Parlee M, Ritch R, Koop B, Kuo WL, Collins C, Marshall J, Gould DB, Pearce W, Carlsson P, Enerback S, Morissette J, Bhattacharya S, Hogan B, Raymond V, Walter MA: Mutations of the forkhead/winged-helix gene, FKHL , in patients with Axenfeld-Rieger anomaly. Am J Hum Genet 1998, 63: 1316–1328. 10.1086/302109
Nishimura DY, Swiderski RE, Alward WL, Searby CC, Patil SR, Bennet SR, Kanis AB, Gastier JM, Stone EM, Sheffield VC: The forkhead transcription factor gene FKHL is responsible for glaucoma phenotypes which map to 6p25. Nat Genet 1998, 19: 140–147. 10.1038/493
Lehmann OJ, Ebenezer ND, Jordan T, Fox M, Ocaka L, Payne A, Leroy BP, Clark BJ, Hitchings RA, Povey S, Khaw PT, Bhattacharya SS: Chromosomal duplication involving the forkhead transcription factor gene FOXC causes iris hypoplasia and glaucoma. Am J Hum Genet 2000, 67: 1129–1135.
Mirzayans F, Gould DB, Heon E, Billingsley GD, Cheung JC, Mears AJ, Walter MA: Axenfeld-Rieger syndrome resulting from mutation of the FKHL gene on chromosome 6p25. Eur J Hum Genet 2000, 8: 71–74. 10.1038/sj.ejhg.5200354
Nishimura DY, Searby CC, Alward WL, Walton D, Craig JE, Mackey DA, Kawase K, Kanis AB, Patil SR, Stone EM, Sheffield VC: A spectrum of FOXC mutations suggests gene dosage as a mechanism for developmental defects of the anterior chamber of the eye. Am J Hum Genet 2001, 68: 364–372. 10.1086/318183
Lehmann OJ, Ebenezer ND, Ekong R, Ocaka L, Mungall AJ, Fraser S, McGill JI, Hitchings RA, Khaw PT, Sowden JC, Povey S, Walter MA, Bhattacharya SS, Jordan T: Ocular developmental abnormalities and glaucoma associated with interstitial 6p25 duplications and deletions. Invest Ophthalmol Vis Sci 2002, 43: 1843–1849.
Aldinger KA, Lehmann OJ, Hudgins L, Chizhikov VV, Bassuk AG, Ades LC, Krantz ID, Dobyns WB, Millen KJ: FOXC is required for normal cerebellar development and is a major contributor to chromosome 6p25.3 Dandy-Walker malformation. Nat Genet 2009, 41: 1037–1042. 10.1038/ng.422
Bremer A, Schoumans J, Nordenskjold M, Anderlid BM, Giacobini M: An interstitial deletion of 7.1 Mb in chromosome band 6p22.3 associated with developmental delay and dysmorphic features including heart defects, short neck, and eye abnormalities. Eur J Med Genet 2009, 52: 358–362. 10.1016/j.ejmg.2009.06.002
Davies AF, Olavesen MG, Stephens RJ, Davidson R, Delneste D, van Regemorter N, Vamos E, Flinter F, Abusaad I, Ragoussis J: A detailed investigation of two cases exhibiting characteristics of the 6p deletion syndrome. Hum Genet 1996, 98: 454–459. 10.1007/s004390050239
Moriarty AP, Kerr-Muir MG: Sclerocornea and interstitial deletion of the short arm of chromosome 6-(46XY del[6] [p22 p24]). J Pediatr Ophthalmol Strabismus 1992, 29: 177–179.
van Swaay E, Beverstock GC, van de Kamp JJ: A patient with an interstitial deletion of the short arm of chromosome 6. Clin Genet 1988, 33: 95–101.
Zirn B, Hempel M, Hahn A, Neubauer B, Wagenstaller J, Rivera-Brugues N, Strom TM, Kohler A: Polyneuropathy, scoliosis, tall stature, and oligodontia represent novel features of the interstitial 6p deletion phenotype. Am J Med Genet A 2008, 146A: 2960–2965. 10.1002/ajmg.a.32536
Kirov G, Grozeva D, Norton N, Ivanov D, Mantripragada KK, Holmans P, International Schizophrenia Consortium, Wellcome Trust Case Control Consortium, Craddock N, Owen MJ, O'Donovan MC: Support for the involvement of large copy number variants in the pathogenesis of schizophrenia. Hum Mol Genet 2009, 18: 1497–1503. 10.1093/hmg/ddp043
Itsara A, Cooper GM, Baker C, Girirajan S, Li J, Absher D, Krauss RM, Myers RM, Ridker PM, Chasman DI, Mefford H, Ying P, Nickerson DA, Eichler EE: Population analysis of large copy number variants and hotspots of human genetic disease. Am J Hum Genet 2009, 84: 148–161. 10.1016/j.ajhg.2008.12.014
Shaikh TH, Gai X, Perin JC, Glessner JT, Xie H, Murphy K, O'Hara R, Casalunovo T, Conlin LK, D'Arcy M, Frackelton EC, Geiger EA, Haldeman-Englert C, Imielinski M, Kim CE, Medne L, Annaiah K, Bradfield JP, Dabaghyan E, Eckert A, Onyiah CC, Ostapenko S, Otieno FG, Santa E, Shaner JL, Skraban R, Smith RM, Elia J, Goldmuntz E, Spinner NB, et al.: High-resolution mapping and analysis of copy number variations in the human genome: a data resource for clinical and research applications. Genome Res 2009, 19: 1682–1690. 10.1101/gr.083501.108
Zogopoulos G, Ha KC, Naqib F, Moore S, Kim H, Montpetit A, Robidoux F, Laflamme P, Cotterchio M, Greenwood C, Scherer SW, Zanke B, Hudson TJ, Bader GD, Gallinger S: Germ-line DNA copy number variation frequencies in a large North American population. Hum Genet 2007, 122: 345–353. 10.1007/s00439-007-0404-5
Conrad DF, Pinto D, Redon R, Feuk L, Gokcumen O, Zhang Y, Aerts J, Andrews TD, Barnes C, Campbell P, Fitzgerald T, Hu M, Ihm CH, Kristiansson K, Macarthur DG, Macdonald JR, Onyiah I, Pang AW, Robson S, Stirrups K, Valsesia A, Walter K, Wei J, Wellcome Trust Case Control Consortium, Tyler-Smith C, Carter NP, Lee C, Scherer SW, Hurles ME: Origins and functional impact of copy number variation in the human genome. Nature 2010, 464: 704–712. 10.1038/nature08516
Redon R, Ishikawa S, Fitch KR, Feuk L, Perry GH, Andrews TD, Fiegler H, Shapero MH, Carson AR, Chen W, Cho EK, Dallaire S, Freeman JL, González JR, Gratacòs M, Huang J, Kalaitzopoulos D, Komura D, MacDonald JR, Marshall CR, Mei R, Montgomery L, Nishimura K, Okamura K, Shen F, Somerville MJ, Tchinda J, Valsesia A, Woodwark C, Yang F, et al.: Global variation in copy number in the human genome. Nature 2006, 444: 444–454. 10.1038/nature05329
Jacquemont ML, Sanlaville D, Redon R, Raoul O, Cormier-Daire V, Lyonnet S, Amiel J, Le Merrer M, Heron D, de Blois MC, Prieur M, Vekemans M, Carter NP, Munnich A, Colleaux L, Philippe A: Array-based comparative genomic hybridisation identifies high frequency of cryptic chromosomal rearrangements in patients with syndromic autism spectrum disorders. J Med Genet 2006, 43: 843–849. 10.1136/jmg.2006.043166
Sebat J, Lakshmi B, Malhotra D, Troge J, Lese-Martin C, Walsh T, Yamrom B, Yamrom B, Yoon S, Krasnitz A, Kendall J, Leotta A, Pai D, Zhang R, Lee YH, Hicks J, Spence SJ, Lee AT, Puura K, Lehtimaki T, Ledbetter D, Gregersen PK, Bregman J, Sutcliffe JS, Jobanputra V, Chung W, Warburton D, King MC, Skuse D, Geschwind DH, Gilliam TC, Ye K, Wigler M: Strong association of de novo copy number mutations with autism. Science 2007, 316: 445–449. 10.1126/science.1138659
Marshall CR, Noor A, Vincent JB, Lionel AC, Feuk L, Skaug J, Shago M, Moessner R, Pinto D, Ren Y, Thiruvahindrapduram B, Fiebig A, Schreiber S, Friedman J, Ketelaars CE, Vos YJ, Ficicioglu C, Kirkpatrick S, Nicolson R, Sloman L, Summers A, Gibbons CA, Teebi A, Chitayat D, Weksberg R, Thompson A, Vardy C, Crosbie V, Luscombe S, Baatjes R, Zwaigenbaum L, Roberts W, Fernandez B, Szatmari P, Scherer SW: Structural variation of chromosomes in autism spectrum disorder. Am J Hum Genet 2008, 82: 477–488. 10.1016/j.ajhg.2007.12.009
Itsara A, Wu H, Smith JD, Nickerson DA, Romieu I, London SJ, Eichler EE: De novo rates and selection of large copy number variation. Genome Res 2010, 20: 1469–1481. 10.1101/gr.107680.110
Pinto D, Pagnamenta AT, Klei L, Anney R, Merico D, Regan R, Conroy J, Magalhaes TR, Correia C, Abrahams BS, Almeida J, Bacchelli E, Bader GD, Bailey AJ, Baird G, Battaglia A, Berney T, Bolshakova N, Bölte S, Bolton PF, Bourgeron T, Brennan S, Brian J, Bryson SE, Carson AR, Casallo G, Casey J, Chung BH, Cochrane L, Corsello C, et al.: Functional impact of global rare copy number variation in autism spectrum disorders. Nature 2010, 466: 368–372. 10.1038/nature09146
Sanders SJ, Ercan-Sencicek AG, Hus V, Luo R, Murtha MT, Moreno-De-Luca D, Chu SH, Moreau MP, Gupta AR, Thomson SA, Mason CE, Bilguvar K, Celestino-Soper PB, Choi M, Crawford EL, Davis L, Wright NR, Dhodapkar RM, DiCola M, DiLullo NM, Fernandez TV, Fielding-Singh V, Fishman DO, Frahm S, Garagaloyan R, Goh GS, Kammela S, Klei L, Lowe JK, Lund SC, et al.: Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron 2011, 70: 863–885. 10.1016/j.neuron.2011.05.002
DECIPHER (Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources)[http://decipher.sanger.ac.uk/]
Tuchman R, Rapin I: Epilepsy in autism. Lancet Neurol 2002, 1: 352–358. 10.1016/S1474-4422(02)00160-6
Goldstein S, Schwebach AJ: The comorbidity of Pervasive Developmental Disorder and Attention Deficit Hyperactivity Disorder: results of a retrospective chart review. J Autism Dev Disord 2004, 34: 329–339.
Gehrking KM, Andresen JM, Duvick L, Lough J, Zoghbi HY, Orr HT: Partial loss of Tip60 slows mid-stage neurodegeneration in a spinocerebellar ataxia type 1 (SCA1) mouse model. Hum Mol Genet 2011, 20: 2204–2212. 10.1093/hmg/ddr108
Matilla A, Roberson ED, Banfi S, Morales J, Armstrong DL, Burright EN, Orr HT, Sweatt JD, Zoghbi HY, Matzuk MM: Mice lacking ataxin-1 display learning deficits and decreased hippocampal paired-pulse facilitation. J Neurosci 1998, 18: 5508–5516.
Crespo-Barreto J, Fryer JD, Shaw CA, Orr HT, Zoghbi HY: Partial loss of ataxin-1 function contributes to transcriptional dysregulation in spinocerebellar ataxia type 1 pathogenesis. PLoS Genet 2010, 6: e1001021. 10.1371/journal.pgen.1001021
Rizzi TS, Arias-Vasquez A, Rommelse N, Kuntsi J, Anney R, Asherson P, Buitelaar J, Banaschewski T, Ebstein R, Ruano D, Van der Sluis S, Markunas CA, Garrett ME, Ashley-Koch AE, Kollins SH, Anastopoulos AD, Hansell NK, Wright MJ, Montgomery GW, Martin NG, Harris SE, Davies G, Tenesa A, Porteous DJ, Starr JM, Deary IJ, St Pourcain B, Davey-Smith G, Timpson NJ, Evans DM, et al.: The ATXN and TRIM3 genes are related to intelligence in an ADHD background: evidence from a large collaborative study totaling 4,963 subjects. Am J Med Genet B Neuropsychiatr Genet 2010, 156: 145–157.
Li W, Zhang Q, Oiso N, Novak EK, Gautam R, O'Brien EP, Tinsley CL, Blake DJ, Spritz RA, Copeland NG, Jenkins NA, Amato D, Roe BA, Starcevic M, Dell'Angelica EC, Elliott RW, Mishra V, Kingsmore SF, Paylor RE, Swank RT: Hermansky-Pudlak syndrome type 7 (HPS-7) results from mutant dysbindin, a member of the biogenesis of lysosome-related organelles complex 1 (BLOC-1). Nat Genet 2003, 35: 84–89. 10.1038/ng1229
Numakawa T, Yagasaki Y, Ishimoto T, Okada T, Suzuki T, Iwata N, Ozaki N, Taguchi T, Tatsumi M, Kamijima K, Straub RE, Weinberger DR, Kunugi H, Hashimoto R: Evidence of novel neuronal functions of dysbindin, a susceptibility gene for schizophrenia. Hum Mol Genet 2004, 13: 2699–2708. 10.1093/hmg/ddh280
Dickman DK, Davis GW: The schizophrenia susceptibility gene dysbindin controls synaptic homeostasis. Science 2009, 326: 1127–1130. 10.1126/science.1179685
Benson MA, Newey SE, Martin-Rendon E, Hawkes R, Blake DJ: Dysbindin, a novel coiled-coil-containing protein that interacts with the dystrobrevins in muscle and brain. J Biol Chem 2001, 276: 24232–24241. 10.1074/jbc.M010418200
Straub RE, Jiang Y, MacLean CJ, Ma Y, Webb BT, Myakishev MV, Harris-Kerr C, Wormley B, Sadek H, Kadambi B, Cesare AJ, Gibberman A, Wang X, O'Neill FA, Walsh D, Kendler KS: Genetic variation in the 6p22.3 gene DTNBP , the human ortholog of the mouse dysbindin gene, is associated with schizophrenia. Am J Hum Genet 2002, 71: 337–348. 10.1086/341750
Allen NC, Bagade S, McQueen MB, Ioannidis JP, Kavvoura FK, Khoury MJ, Tanzi RE, Bertram L: Systematic meta-analyses and field synopsis of genetic association studies in schizophrenia: the SzGene database. Nat Genet 2008, 40: 827–834. 10.1038/ng.171
Hendriksen JG, Vles JS: Neuropsychiatric disorders in males with duchenne muscular dystrophy: frequency rate of attention-deficit hyperactivity disorder (ADHD), autism spectrum disorder, and obsessive-compulsive disorder. J Child Neurol 2008, 23: 477–481.
Hinton VJ, Cyrulnik SE, Fee RJ, Batchelder A, Kiefel JM, Goldstein EM, Kaufmann P, De Vivo DC: Association of autistic spectrum disorders with dystrophinopathies. Pediatr Neurol 2009, 41: 339–346. 10.1016/j.pediatrneurol.2009.05.011
Pagnamenta AT, Holt R, Yusuf M, Pinto D, Wing K, Betancur C, Scherer SW, Volpi EV, Monaco AP: A family with autism and rare copy number variants disrupting the Duchenne/Becker muscular dystrophy gene DM and TRPM . J Neurodev Disord 2011, 3: 124–131. 10.1007/s11689-011-9076-5
Berge-Lefranc JL, Jay P, Massacrier A, Cau P, Mattei MG, Bauer S, Marsollier C, Berta P, Fontes M: Characterization of the human jumonji gene. Hum Mol Genet 1996, 5: 1637–1641. 10.1093/hmg/5.10.1637
Toyoda M, Shirato H, Nakajima K, Kojima M, Takahashi M, Kubota M, Suzuki-Migishima R, Motegi Y, Yokoyama M, Takeuchi T: Jumonji downregulates cardiac cell proliferation by repressing cyclin D1 expression. Dev Cell 2003, 5: 85–97. 10.1016/S1534-5807(03)00189-8
Takahashi M, Kojima M, Nakajima K, Suzuki-Migishima R, Motegi Y, Yokoyama M, Takeuchi T: Cardiac abnormalities cause early lethality of jumonji mutant mice. Biochem Biophys Res Commun 2004, 324: 1319–1323. 10.1016/j.bbrc.2004.09.203
Weiss LA, Arking DE, Daly MJ, Chakravarti A: A genome-wide linkage and association scan reveals novel loci for autism. Nature 2009, 461: 802–808. 10.1038/nature08490
Gentry MS, Worby CA, Dixon JE: Insights into Lafora disease: malin is an E3 ubiquitin ligase that ubiquitinates and promotes the degradation of laforin. Proc Natl Acad Sci USA 2005, 102: 8501–8506. 10.1073/pnas.0503285102
University of California Santa Cruz Genome Browser[http://genome.ucsc.edu/]
Agilent Technologies eArray[https://earray.chem.agilent.com]
Baylor College of Medicine Medical Genetics Laboratories[https://www.bcm.edu/geneticlabs/]
Ou Z, Kang SH, Shaw CA, Carmack CE, White LD, Patel A, Beaudet AL, Cheung SW, Chinault AC: Bacterial artificial chromosome-emulation oligonucleotide arrays for targeted clinical array-comparative genomic hybridization analyses. Genet Med 2008, 10: 278–289. 10.1097/GIM.0b013e31816b4420
El-Hattab AW, Smolarek TA, Walker ME, Schorry EK, Immken LL, Patel G, Abbott MA, Lanpher BC, Ou Z, Kang SH, Patel A, Scaglia F, Lupski JR, Cheung SW, Stankiewicz P: Redefined genomic architecture in 15q24 directed by patient deletion/duplication breakpoint mapping. Hum Genet 2009, 126: 589–602. 10.1007/s00439-009-0706-x
Boone PM, Bacino CA, Shaw CA, Eng PA, Hixson PM, Pursley AN, Kang SH, Yang Y, Wiszniewska J, Nowakowska BA, Del GD, Xia Z, Simpson-Patel G, Immken LL, Gibson JB, Tsai AC, Bowers JA, Reimschisel TE, Schaaf CP, Potocki L, Scaglia F, Gambin T, Sykulski M, Bartnik M, Derwinska K, Wisniowiecka-Kowalnik B, Lalani SR, Probst FJ, Bi W, Beaudet AL, et al.: Detection of clinically relevant exonic copy-number changes by array CGH. Hum Mutat 2010, 31: 1326–1342. 10.1002/humu.21360
Ballif BC, Theisen A, McDonald-McGinn DM, Zackai EH, Hersh JH, Bejjani BA, Shaffer LG: Identification of a previously unrecognized microdeletion syndrome of 16q11.2q12.2. Clin Genet 2008, 74: 469–475. 10.1111/j.1399-0004.2008.01094.x
Cheung SW, Shaw CA, Yu W, Li J, Ou Z, Patel A, Yatsenko SA, Cooper ML, Furman P, Stankiewicz P, Lupski JR, Chinault AC, Beaudet AL: Development and validation of a CGH microarray for clinical cytogenetic diagnosis. Genet Med 2005, 7: 422–432. 10.1097/01.GIM.0000170992.63691.32
Chung MY, Ranum LP, Duvick LA, Servadio A, Zoghbi HY, Orr HT: Evidence for a mechanism predisposing to intergenerational CAG repeat instability in spinocerebellar ataxia type I. Nat Genet 1993, 5: 254–258. 10.1038/ng1193-254
Kwiatkowski TJ Jr, Ranum LPW, Banfi S, McCall AE, Jodice C, Persichetti F, Novelletto A, LeBorgne-Demarquoy F, Frontali M, Subramony SH, Beaudet AL, Terrenato L, Zoghbi HY, Orr HT: The gene for autosomal dominant spinocerebellar ataxia (SCA1) maps centromeric to D6S89 and shows no recombination, in nine large kindreds, with a dinucleotide repeat at the AM10 locus. Am J Hum Genet 1993, 53: 391–400.
Orr HT, Chung M, Banfi S, Kwiatkowski TJ, Servadio A, Beaudet AL, McCall AE, Duvick LA, Ranum LPW, Zoghbi HY: Expansion of an unstable trinucleotide CAG repeat in spinocerebellar ataxia type 1. Nat Genet 1993, 4: 221–226. 10.1038/ng0793-221
Gene Ontology Annotation GOA; http://www.ebi.ac.uk/GOA/
Acknowledgements
We are grateful for the participation of the families and patients in this study. The authors acknowledge the assistance of Dr. John Belmont and Dr. Chad Shaw for help with SNP array analysis interpretation, Dr. Samart Bhatt for technical assistance, Caroline Borgan for critical analysis and editing of the manuscript, and Dr. Huda Zoghbi for helpful discussions. The South Carolina Autism Project was funded by the South Carolina Department of Disabilities and Special Needs and a grant from the National Institute of Mental Health (MH57840 to RES). This study makes use of data generated by the DECIPHER Consortium. A full list of centres who contributed to the generation of the data is available from http://decipher.sanger.ac.uk and via email from decipher@sanger.ac.uk. Funding for the project was provided by the Wellcome Trust.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
Many of the authors are faculty members in the Department of Molecular and Human Genetics at BCM, which offers extensive genetic laboratory testing including use of arrays for genomic copy-number analysis, and the Department derives revenue from this activity. JAR is an employee of Signature Genomics, a subsidiary of PerkinElmer, Inc.
Authors' contributions
ALB and PBSC-S designed the experiments. PBSC-S and PE performed the chromosomal microarray analysis. PBSC-S, AW, and JW performed the SNP and ATXN1 STR extension analysis. PBSC-S and PS performed the PCR and sequence analysis. JAR, S-HLK, SWC, and PS signed out the microarray-based hybridization analysis results and analyzed FISH results. CS, RS, JS, MMJN, DT, DC, GSP, LI, RM, and RES clinically examined the patients. PBSC-S and PS drafted the manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Celestino-Soper, P.B., Skinner, C., Schroer, R. et al. Deletions in chromosome 6p22.3-p24.3, including ATXN1, are associated with developmental delay and autism spectrum disorders. Mol Cytogenet 5, 17 (2012). https://doi.org/10.1186/1755-8166-5-17
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1755-8166-5-17