
Abstract
Background
Small supernumerary marker chromosomes (sSMC) can be present in numerically abnormal karyotypes like in a 'Turner-syndrome karyotype' mos 45,X/46,X,+mar.
Results
Here we report the first case of an sSMC found in Turner syndrome karyotypes (sSMCT) derived from chromosome 14 in a Turner syndrome patient. According to cytogenetic and molecular cytogenetic characterization the karyotype was 46,X,+del(14)(q11.1). The present case is the third Turner syndrome case with an sSMCT not derived from the X- or the Y-chromosome.
Conclusion
More comprehensive characterization of such sSMCT might identify them to be more frequent than only ~0.6% in Turner syndrome cases according to available data.
Similar content being viewed by others

Background
Small supernumerary marker chromosomes (sSMC) [1] can be observed in a numerically normal 'basic karyotype', but also in numerically abnormal one like in a 'Turner-syndrome karyotype' (=sSMCT). At present 528 such cases with an sSMCT are reported [2, 3]. sSMCT are very rare in the common population (1:100000 [2]) - however, they can be observed 45 and even 60 times more frequent in infertile and developmentally and/or mentally retarded patients, respectively. The majority of sSMCT(X) form ring-chromosomes, while most sSMCT(Y) are inverted duplicated/isodicentric ones. When a mos 45,X/46,X,der(Y) or 45,X/46,XY is characterized it is important to counsel the patient concerning a possibility of gonadoblastoma and a preventive removal of gonadal tissue. In this connection, the necessity to apply molecular approaches for detection of cryptic 45,X/46,XY mosaicism is discussed, as a direct relationship between percentage of cells exhibiting a 45,X karyotype and patients phenotype does not exist. Additionally, it is a well-known fact that in a karyotype of mos 45,X/46,X,der(X) it is important to test for the ability of the der(X) to be inactivated, i.e. to test for the presence of the XIST-gene [2].
Even though sSMCT derive in >99% of the cases from one of the gonosomes, there are also two previous exceptional reports on sSMCT derived from one of the autosomes [4, 5].
Here we report the third case with an sSMCT originating not from a gonosome but the first one proven to be derived from chromosome 14.
Case presentation
A ten year old girl was studied cytogenetically due to typical features of a Turner syndrome, i.e. short stature, webbing of neck, cubitus valgus, shield chest, congenital dislocation of hip, renal anomalies, clinodactyly, unilateral simian crease on right palm, acyanotic congenital heart disease and small patent ductus arteriosus.
Results
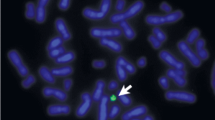
Cytogenetics revealed a karyotype 46,X,+mar in a patient with Turner syndrome. The sSMCT was acquired de novo, as parental chromosome analysis revealed. Array-CGH was done, however no clear imbalance apart from the lack of a second gonosome was observed (see also Fig. 1B). cenM-FISH identified the sSMCT as a derivative of chromosome 14. In cenM-FISH the sSMCT did only show a signal for the probe D14/22Z1, but not for D22Z4 specific for the centromeric region of chromosome 22; thus, the sSMCT could be defined as a der(14). By subcenM-FISH and the array-CGH result was confirmed that the sSMCT did not contain euchromatic material and it could be defined as a del(14)(q11.1) (Fig. 1A).

A) SubcenM-FISH revealed the absence of euchromatic material on the sSMCT reported here. The sSMCT only showed one specific signal, each, for midi 54 (a probe specific for the acrocentric short arms) and the centromeric probe specific for chromosome 14 and 22 (cep 14/22). No specific signals were on the sSMCT for the centromere-near probe RP11-324B11 in 14q11.2 and partial chromosome painting probe of chromosome 14 (the latter not depicted here). Thus, the sSMCT was a del(14)(q11.1). B) After knowing the origin of the sSMC array-CGH was reanalyzed for 14q-proximal region. The first probe on Agilent 4x44K array location is on 19,365,051-19,365,110 at 14q11.2. Thus, only genes from olfactory receptors OR11H12, OR4M1 and OR4Q3, and POTEG and P704P may be involved in the marker chromosome. As it is a region known to be CNV polymorphic and the probe RP11-324B11 at 19,886,099-19,886,646 is not present on the marker according to FISH array-CGH overall is to be considered as non-informative for this sSMC.
Discussion
Here we report the third case of a patient with sSMCT not derived from a gonosome. It is the first such case where the sSMC was characterized in detail by molecular cytogenetics and which turned out to be a de novo derivative of chromosome 14. Previously one case with a der(20) [4] and a not further specified sSMCT, however, proven to be not of gonosomal origin [5] were characterized. Overall, this is an interesting finding as neither chromosome 15 nor 22 were up to now identified as sSMCT, even though these two chromosomes are most frequently involved in sSMC formation [1, 3]. However, this might only be a bias due to only three known cases up to now. Furthermore, the exclusion of a uniparental disomy 14 would have been desirable; unfortunately no paternal material was available for that kind of study.
Among ~3.400 reported sSMC cases studied for their chromosomal origin and subsequently reported [3], by now 528 cases with sSMCT were found. Three of those sSMCT were not of gonosomal origin, i.e. 0.6%. However, the question is, if the percentage of this specific kind of sSMCT is not underestimated. Non-gonosomal sSMCT might be easily missed if they are not further characterized by molecular approaches.
In conclusion, a really comprehensive characterization of all sSMC by different probes, probe sets and approaches could enhance the detection rate of autosomal derived sSMCT.
Materials and methods
Cytogenetics
Metaphase chromosome preparations were obtained from PHA stimulated lymphocyte cultures according to standard procedures. Chromosome analysis was carried out applying GTG banding at a 600 band level according ISCN 2009 [6] in the patient (25 metaphases) and both parents (50 metaphases, each).
Fluorescence in situ hybridization (FISH)
FISH was performed as previously reported [7]. To characterize the sSMC first centromere specific multicolor FISH (cenM-FISH) and then subcentromere-specific M-FISH (subcenM-FISH) was performed; for details see [7]. The here applied probe RP11-324B11 in 14q11.2 is located at 19,886,099-19,886,646 Mb.
Array-CGH
Genomic DNA was extracted from peripheral blood lymphocytes using standard SDS-proteinase K extraction method [8]. DNA concentration was determined with NanoDrop ND-1000 spectrophotometer and software (NanoDrop Technologies, Berlin, Germany). Detection of gene copy number was performed by array-Comparative Genomic Hybridization (array-CGH) experiments following standard and manufacturer's recommendations using 44.000 oligo probes approximately spaced at 40-100 kb intervals across the genome (Human Genome CGH microarray 44B kit, Agilent™). Male genomic DNA (Promega™) was used as reference in sex-match hybridizations which were analyzed with the CGH-analytics software v3.4 by applying Z-score segmentation algorithm with a window size of 10 points to identify chromosome aberrations. Analysis was performed with filter settings: 3-point filter and 0.2 of variation.
Consent section
Written informed consent was obtained from the patient for publication of this case report and accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
References
Liehr T, Claussen U, Starke H: Small supernumerary marker chromosomes (sSMC) in humans. Cytogenet Genome Res 2004,107(1–2):55–67. 10.1159/000079572
Liehr T, Mrasek K, Hinreiner S, Reich D, Ewers E, Bartels I, Seidel J, Manoulakis E, Petersen M, Polityko A, Dufke A, Iourov I, Trifonov V, Vermeesch J, Weise A: Small supernumerary marker chromosomes (sSMC) in patients with a karyotype 45,X/46,X,+mar - 17 new cases and a review of the literature. Sex Dev 2007,1(6):353–362. 10.1159/000111767
Liehr T: sSMC homepage. 2009. [http://www.med.uni-jena.de/fish/sSMC/00START.htm]
Gray BA, Bent-Williams A, Wolff DJ, Zori RT: A non-sex chromosome marker in a patient with an atypical Ullrich-Turner phenotype and mosaicism of 46,X,mar/46,XX. Clin Genet 2001,60(1):73–76. 10.1034/j.1399-0004.2001.600112.x
Wiktor A, Van Dyke DL: FISH analysis helps identify low-level mosaicism in Ullrich-Turner syndrome patients. Genet Med 2004,6(3):132–135.
Shaffer L, Slovak ML, Campbell LJ, eds: ISCN 2009, an international system for human cytogenetic nomenclature (2009). S Karger, Basel 2009.
Liehr T, Mrasek K, Weise A, Dufke A, Rodríguez L, Martínez Guardia N, Sanchís A, Vermeesch JR, Ramel C, Polityko A, Haas OA, Anderson J, Claussen U, von Eggeling F, Starke H: Small supernumerary marker chromosomes-progress towards a genotype-phenotype correlation. Cytogenet Genome Res 2006,112(1–2):23–34. 10.1159/000087510
Old JM, Ludlam CA: Antenatal diagnosis. Baillieres Clin Haematol 1991,4(2):391–428. 10.1016/S0950-3536(05)80165-9
Acknowledgements
Supported in parts by the DAAD (D07/00070 and fellowship for ABH) and Prochance 2008 of the Friedrich Schiller University Jena 21007091 and Dept of Biotechnology (DBT) - BT/PR9111/MED/12/337/2007, India.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
FS, JS and MD performed the cytogenetic studies in the present case and collected the data relative to this case report. EE, NK, AW, ABH, MZ and TL did the molecular cytogenetic analysis and interpretations. JA and JV were involved in the array-CGH analysis. TL drafted the paper and all authors contributed to the finalizing of the manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Sheth, F., Ewers, E., Kosyakova, N. et al. A small supernumerary marker chromosome present in a Turner syndrome patient not derived from X- or Y-chromosome: a case report. Mol Cytogenet 2, 22 (2009). https://doi.org/10.1186/1755-8166-2-22
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1755-8166-2-22